
Genotype-to-Phenotype Associations in the Aggressive Variant Prostate Cancer Molecular Profile (AVPC-m) Components

 ,
,  , , , , , and
, , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Tumor-Derived Xenografts (PDXs)
2.2. Immunohistochemistry
2.3. Genomic Analyses (T200 Targeted Sequencing Panel)
2.4. Gene Expression Analysis (Clariom-S Microarray)
2.5. Statistical Analyses
3. Results
3.1. Clinical Course of PDX Donors
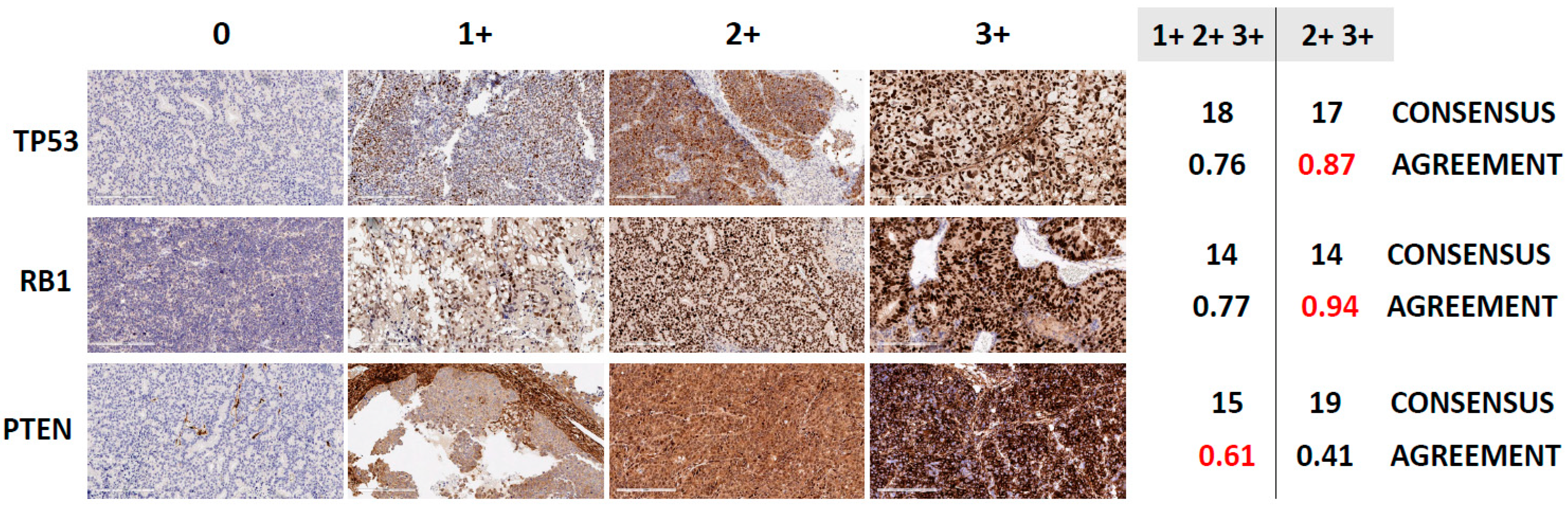
3.2. Assessment of TSP Defects by IHC
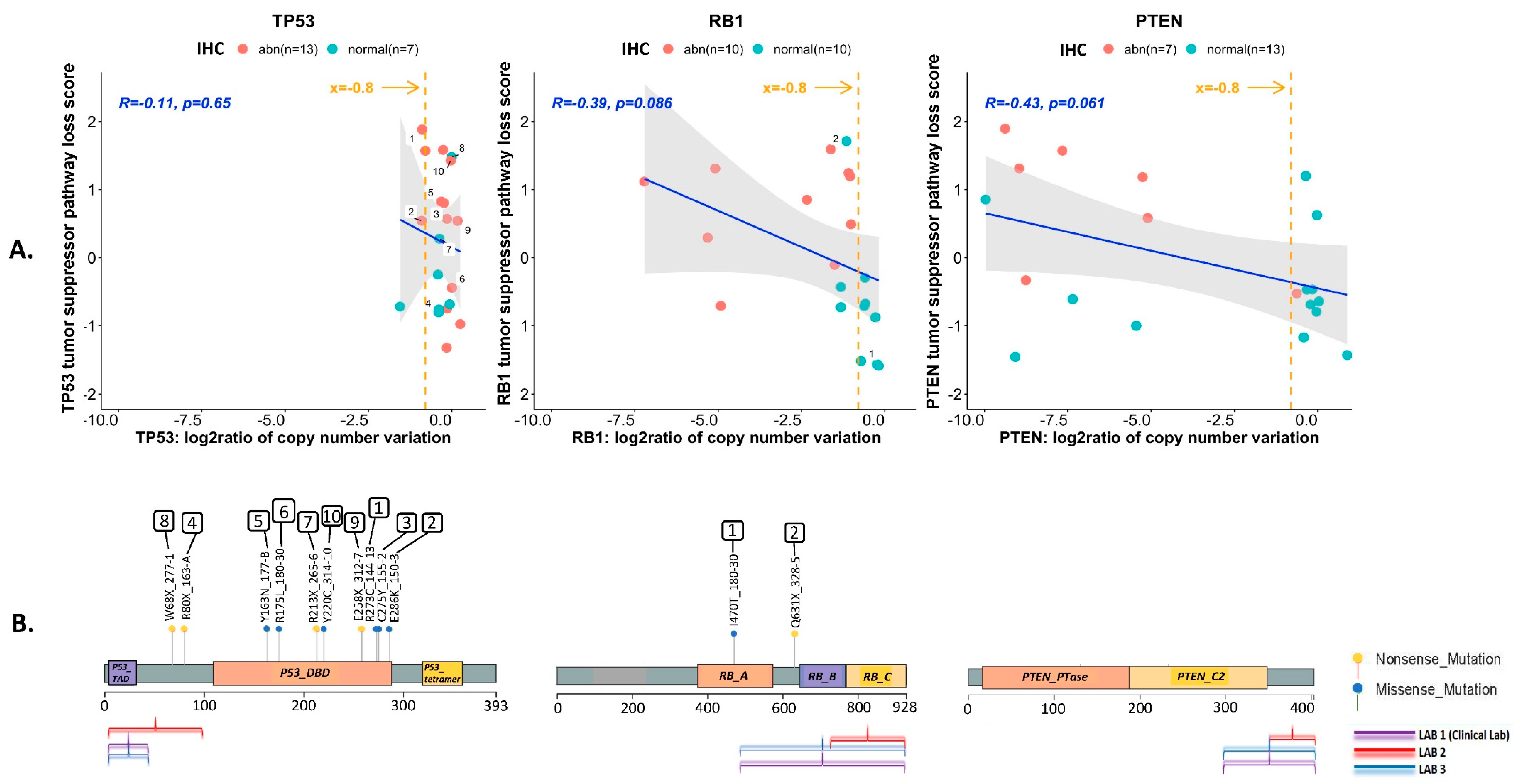
3.3. Correlation between TSP Loss-of-Function Transcriptional Scores and IHC Results
3.4. Assessment of TSP Defects by NGS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AVPC | aggressive variant prostate cancer |
| AVPC-m | AVPC molecular profile |
| TSP | tumor suppressor protein |
| PDX | patient tumor-derived xenograft |
| IHC | immunohistochemistry |
| FFPE | formalin-fixed, paraffin-embedded |
| LOF-TS | loss-of-function transcriptional score |
| NGS | next-generation sequencing |
| SNV | single nucleotide variation |
| CNV | copy number variation |
References
- Aparicio, A.M.; Harzstark, A.L.; Corn, P.G.; Wen, S.; Araujo, J.C.; Tu, S.M.; Pagliaro, L.C.; Kim, J.; Millikan, R.E.; Ryan, C.; et al. Platinum-based chemotherapy for variant castrate-resistant prostate cancer. Clin. Cancer Res. 2013, 19, 3621–3630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aparicio, A.M.; Shen, L.; Tapia, E.L.; Lu, J.F.; Chen, H.C.; Zhang, J.; Wu, G.; Wang, X.; Troncoso, P.; Corn, P.; et al. Combined Tumor Suppressor Defects Characterize Clinically Defined Aggressive Variant Prostate Cancers. Clin. Cancer Res. 2016, 22, 1520–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.C.; Wongvipat, J.; Ku, S.Y.; Gao, D.; Cao, Z.; et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbe, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Corn, P.G.; Heath, E.I.; Zurita, A.; Ramesh, N.; Xiao, L.; Sei, E.; Li-Ning-Tapia, E.; Tu, S.M.; Subudhi, S.K.; Wang, J.; et al. Cabazitaxel plus carboplatin for the treatment of men with metastatic castration-resistant prostate cancers: A randomised, open-label, phase 1-2 trial. Lancet Oncol. 2019, 20, 1432–1443. [Google Scholar] [CrossRef]
- Parolia, A.; Cieslik, M.; Chu, S.C.; Xiao, L.; Ouchi, T.; Zhang, Y.; Wang, X.; Vats, P.; Cao, X.; Pitchiaya, S.; et al. Distinct structural classes of activating FOXA1 alterations in advanced prostate cancer. Nature 2019, 571, 413–418. [Google Scholar] [CrossRef]
- Palanisamy, N.; Yang, J.; Shepherd, P.D.A.; Li-Ning-Tapia, E.M.; Labanca, E.; Manyam, G.C.; Ravoori, M.K.; Kundra, V.; Araujo, J.C.; Efstathiou, E.; et al. The MD Anderson Prostate Cancer Patient-derived Xenograft Series (MDA PCa PDX) Captures the Molecular Landscape of Prostate Cancer and Facilitates Marker-driven Therapy Development. Clin. Cancer Res. 2020, 26, 4933–4946. [Google Scholar] [CrossRef]
- Aparicio, A.; Tzelepi, V.; Araujo, J.C.; Guo, C.C.; Liang, S.; Troncoso, P.; Logothetis, C.J.; Navone, N.M.; Maity, S.N. Neuroendocrine prostate cancer xenografts with large-cell and small-cell features derived from a single patient’s tumor: Morphological, immunohistochemical, and gene expression profiles. Prostate 2011, 71, 846–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guedes, L.B.; Almutairi, F.; Haffner, M.C.; Rajoria, G.; Liu, Z.; Klimek, S.; Zoino, R.; Yousefi, K.; Sharma, R.; De Marzo, A.M.; et al. Analytic, Preanalytic, and Clinical Validation of p53 IHC for Detection of TP53 Missense Mutation in Prostate Cancer. Clin. Cancer Res. 2017, 23, 4693–4703. [Google Scholar] [CrossRef] [Green Version]
- Maughan, B.L.; Guedes, L.B.; Boucher, K.; Rajoria, G.; Liu, Z.; Klimek, S.; Zoino, R.; Antonarakis, E.S.; Lotan, T.L. p53 status in the primary tumor predicts efficacy of subsequent abiraterone and enzalutamide in castration-resistant prostate cancer. Prostate Cancer Prostatic Dis. 2018, 21, 260–268. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Ye, K.; Schulz, M.H.; Long, Q.; Apweiler, R.; Ning, Z. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 2009, 25, 2865–2871. [Google Scholar] [CrossRef]
- Zhang, J.; Fujimoto, J.; Zhang, J.; Wedge, D.C.; Song, X.; Zhang, J.; Seth, S.; Chow, C.W.; Cao, Y.; Gumbs, C.; et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 2014, 346, 256–259. [Google Scholar] [CrossRef] [Green Version]
- Olshen, A.B.; Venkatraman, E.S.; Lucito, R.; Wigler, M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 2004, 5, 557–572. [Google Scholar] [CrossRef] [PubMed]
- Landis, J.R.; Koch, G.G. The measurement of observer agreement for categorical data. Biometrics 1977, 33, 159–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J. A Coefficient of Agreement for Nominal Scales. Educ. Psychol. Meas. 1960, 20, 37–46. [Google Scholar] [CrossRef]
- Tzelepi, V.; Zhang, J.; Lu, J.F.; Kleb, B.; Wu, G.; Wan, X.; Hoang, A.; Efstathiou, E.; Sircar, K.; Navone, N.M.; et al. Modeling a lethal prostate cancer variant with small-cell carcinoma features. Clin. Cancer Res. 2012, 18, 666–677. [Google Scholar] [CrossRef] [Green Version]
- Fumagalli, D.; Blanchet-Cohen, A.; Brown, D.; Desmedt, C.; Gacquer, D.; Michiels, S.; Rothe, F.; Majjaj, S.; Salgado, R.; Larsimont, D.; et al. Transfer of clinically relevant gene expression signatures in breast cancer: From Affymetrix microarray to Illumina RNA-Sequencing technology. BMC Genom. 2014, 15, 1008. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Varn, F.S.; Cai, G.; Xiao, F.; Amos, C.I.; Cheng, C. A P53-Deficiency Gene Signature Predicts Recurrence Risk of Patients with Early-Stage Lung Adenocarcinoma. Cancer Epidemiol. Biomark. Prev. 2018, 27, 86–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.S.; Alshalalfa, M.; Zhao, S.G.; Liu, Y.; Mahal, B.A.; Quigley, D.A.; Wei, T.; Davicioni, E.; Rebbeck, T.R.; Kantoff, P.W.; et al. Novel RB1-Loss Transcriptomic Signature Is Associated with Poor Clinical Outcomes across Cancer Types. Clin. Cancer Res. 2019, 25, 4290–4299. [Google Scholar] [CrossRef] [Green Version]
- Imada, E.L.; Sanchez, D.F.; Dinalankara, W.; Vidotto, T.; Ebot, E.M.; Tyekucheva, S.; Franco, G.R.; Mucci, L.A.; Loda, M.; Schaeffer, E.M.; et al. Transcriptional landscape of PTEN loss in primary prostate cancer. BMC Cancer 2021, 21, 856. [Google Scholar] [CrossRef]
- Lee, Y.R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Wyatt, A.W.; Xue, H.; Wang, Y.; Dong, X.; Haegert, A.; Wu, R.; Brahmbhatt, S.; Mo, F.; Jong, L.; et al. High fidelity patient-derived xenografts for accelerating prostate cancer discovery and drug development. Cancer Res. 2014, 74, 1272–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navone, N.M.; Troncoso, P.; Pisters, L.L.; Goodrow, T.L.; Palmer, J.L.; Nichols, W.W.; von Eschenbach, A.C.; Conti, C.J. p53 protein accumulation and gene mutation in the progression of human prostate carcinoma. J. Natl. Cancer Inst. 1993, 85, 1657–1669. [Google Scholar] [CrossRef] [PubMed]
- Eastham, J.A.; Stapleton, A.M.; Gousse, A.E.; Timme, T.L.; Yang, G.; Slawin, K.M.; Wheeler, T.M.; Scardino, P.T.; Thompson, T.C. Association of p53 mutations with metastatic prostate cancer. Clin. Cancer Res. 1995, 1, 1111–1118. [Google Scholar]
- Tan, H.L.; Sood, A.; Rahimi, H.A.; Wang, W.; Gupta, N.; Hicks, J.; Mosier, S.; Gocke, C.D.; Epstein, J.I.; Netto, G.J.; et al. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin. Cancer Res. 2014, 20, 890–903. [Google Scholar] [CrossRef] [Green Version]
- Lotan, T.L.; Heumann, A.; Rico, S.D.; Hicks, J.; Lecksell, K.; Koop, C.; Sauter, G.; Schlomm, T.; Simon, R. PTEN loss detection in prostate cancer: Comparison of PTEN immunohistochemistry and PTEN FISH in a large retrospective prostatectomy cohort. Oncotarget 2017, 8, 65566–65576. [Google Scholar] [CrossRef]
- Quigley, D.A.; Dang, H.X.; Zhao, S.G.; Lloyd, P.; Aggarwal, R.; Alumkal, J.J.; Foye, A.; Kothari, V.; Perry, M.D.; Bailey, A.M.; et al. Genomic Hallmarks and Structural Variation in Metastatic Prostate Cancer. Cell 2018, 174, 758–769.e9. [Google Scholar] [CrossRef] [Green Version]
- Sirohi, D.; Devine, P.; Grenert, J.P.; van Ziffle, J.; Simko, J.P.; Stohr, B.A. TP53 structural variants in metastatic prostatic carcinoma. PLoS ONE 2019, 14, e0218618. [Google Scholar] [CrossRef] [PubMed]
- Burkhart, D.L.; Morel, K.L.; Sheahan, A.V.; Richards, Z.A.; Ellis, L. The Role of RB in Prostate Cancer Progression. Adv. Exp. Med. Biol. 2019, 1210, 301–318. [Google Scholar] [CrossRef] [PubMed]
- Sabapathy, K.; Lane, D.P. Understanding p53 functions through p53 antibodies. J. Mol. Cell. Biol. 2019, 11, 317–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marei, H.E.; Althani, A.; Afifi, N.; Hasan, A.; Caceci, T.; Pozzoli, G.; Morrione, A.; Giordano, A.; Cenciarelli, C. p53 signaling in cancer progression and therapy. Cancer Cell Int. 2021, 21, 703. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TP53 | Loss No | Loss Yes | Kappa (95% Confidence Interval) | |
|---|---|---|---|---|
| N | N | |||
| IHC Abnormal | 0.381 (−0.021, 0.784) | |||
| No Yes | 5 4 | 2 9 | ||
| CNV | 0.048 (−0.309, 0.405) | |||
| No Yes | 7 2 | 8 3 | ||
| SNV | 0.500 (0.122, 0.878) | |||
| No Yes | 7 2 | 3 8 | ||
| CNV/SNV | 0.381 (−0.021, 0.784) | |||
| No Yes | 5 4 | 2 9 | ||
| CNV/SNV/IHC | 0.239 (−0.056, 0.534) | |||
| No Yes | 2 7 | 0 11 | ||
| SNV/IHC | 0.468 (0.126, 0.810) | |||
| No Yes | 4 5 | 0 11 | ||
| RB1 | ||||
| IHC Abnormal | 0.700 (0.389, 1.000) | |||
| No Yes | 9 2 | 1 8 | ||
| CNV | 0.612 (0.298, 0.925) | |||
| No Yes | 7 4 | 0 9 | ||
| SNV | 0.022 (−0.265, 0.309) | |||
| No Yes | 10 1 | 8 1 | ||
| CNV/SNV | 0.519 (0.199, 0.839) | |||
| No Yes | 6 5 | 0 9 | ||
| CNV/SNV/IHC | 0.519 (0.199, 0.839) | |||
| No Yes | 6 5 | 0 9 | ||
| CNV/IHC | 0.612 (0.298, 0.925) | |||
| No Yes | 7 4 | 0 9 | ||
| PTEN | ||||
| IHC Abnormal | 0.468 (0.071, 0.865) | |||
| No Yes | 10 2 | 3 5 | ||
| CNV | 0.490 (0.106, 0.874) | |||
| No Yes | 9 3 | 2 6 | ||
| CNV/IHC | 0.400 (0.006, 0.794) | |||
| No Yes | 8 4 | 2 6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soundararajan, R.; Viscuse, P.; Pilie, P.; Liu, J.; Logotheti, S.; Laberiano Fernández, C.; Lorenzini, D.; Hoang, A.; Lu, W.; Soto, L.M.S.; et al. Genotype-to-Phenotype Associations in the Aggressive Variant Prostate Cancer Molecular Profile (AVPC-m) Components. Cancers 2022, 14, 3233. https://doi.org/10.3390/cancers14133233
Soundararajan R, Viscuse P, Pilie P, Liu J, Logotheti S, Laberiano Fernández C, Lorenzini D, Hoang A, Lu W, Soto LMS, et al. Genotype-to-Phenotype Associations in the Aggressive Variant Prostate Cancer Molecular Profile (AVPC-m) Components. Cancers. 2022; 14(13):3233. https://doi.org/10.3390/cancers14133233
Chicago/Turabian StyleSoundararajan, Rama, Paul Viscuse, Patrick Pilie, Jingjing Liu, Souzana Logotheti, Caddie Laberiano Fernández, Daniele Lorenzini, Anh Hoang, Wei Lu, Luisa Maren Solis Soto, and et al. 2022. "Genotype-to-Phenotype Associations in the Aggressive Variant Prostate Cancer Molecular Profile (AVPC-m) Components" Cancers 14, no. 13: 3233. https://doi.org/10.3390/cancers14133233
APA StyleSoundararajan, R., Viscuse, P., Pilie, P., Liu, J., Logotheti, S., Laberiano Fernández, C., Lorenzini, D., Hoang, A., Lu, W., Soto, L. M. S., Wistuba, I. I., Xu, M., Song, X., Shepherd, P. D. A., Navone, N. M., Tidwell, R. S. S., Lozano, G., Logothetis, C., Zhang, J., ... Aparicio, A. M. (2022). Genotype-to-Phenotype Associations in the Aggressive Variant Prostate Cancer Molecular Profile (AVPC-m) Components. Cancers, 14(13), 3233. https://doi.org/10.3390/cancers14133233