
Genome-Wide CRISPR Screening Identifies DCK and CCNL1 as Genes That Contribute to Gemcitabine Resistance in Pancreatic Cancer
 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Lentivirus Generation and Transduction
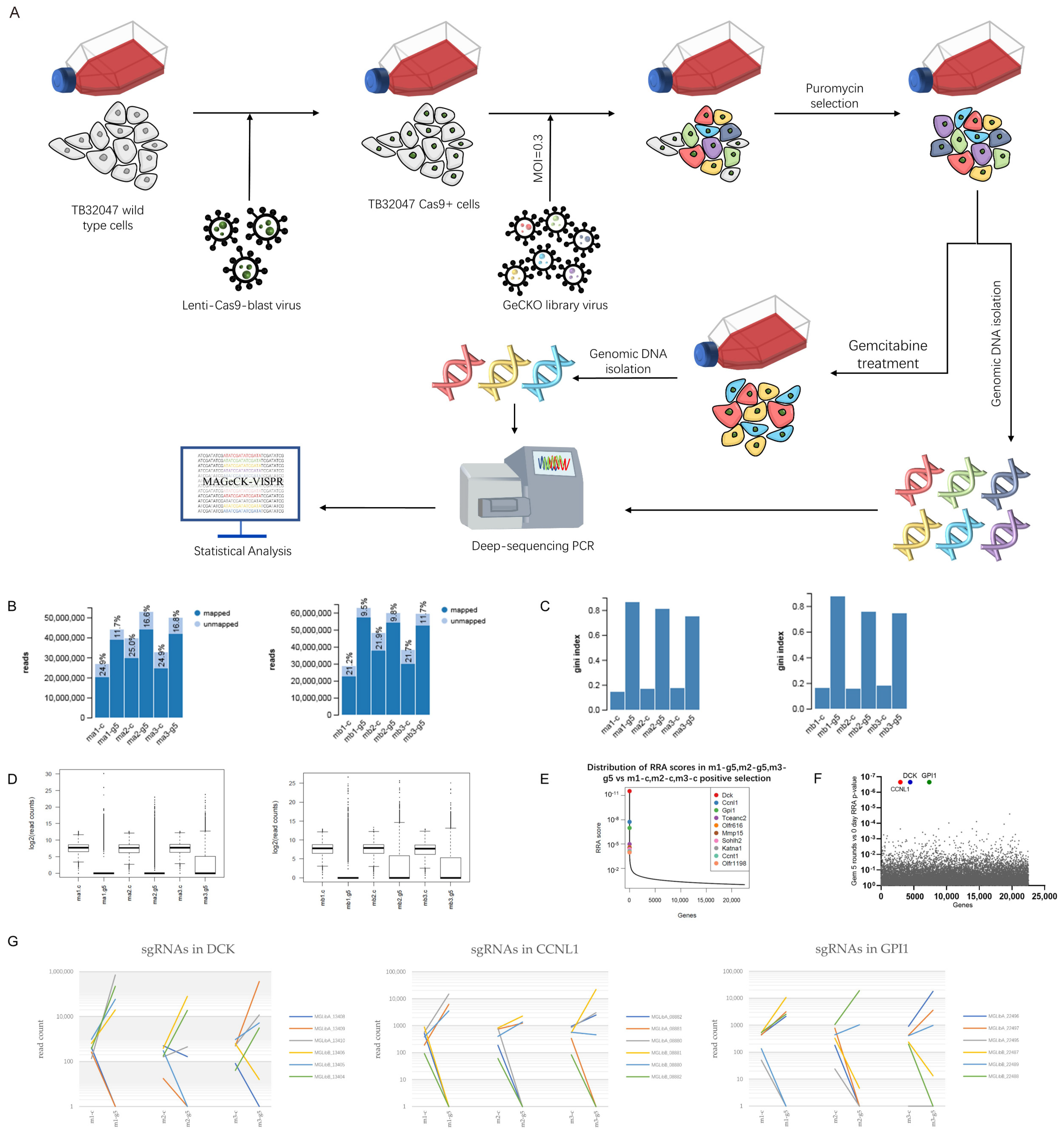
2.3. CRISPR Screening
2.4. Genomic DNA Isolation and PCR Amplification
2.5. CRISPR/Cas9 Gene-Editing Knockout System
2.6. Western Blot and Antibodies
2.7. Quantitative RT–PCR
2.8. Genome Mutation Confirmation
2.9. Chemotherapeutic Drug Resistance Assay
2.10. Apoptosis Assay
2.11. Statistical Analysis
3. Results
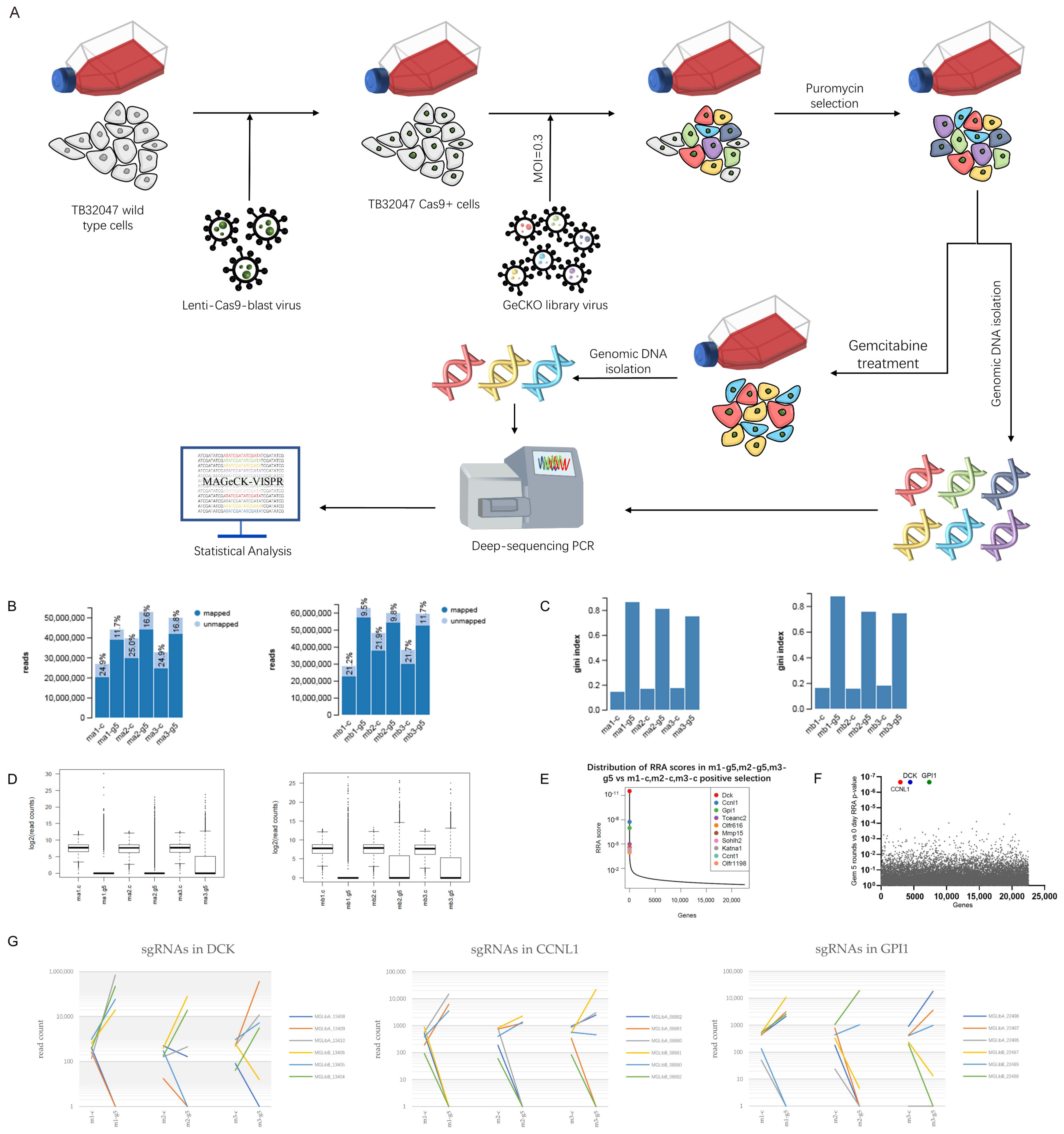
3.1. Genome-Wide CRISPR Screening Identifies DCK and CCNL1 as Genes Involved in Chemoresistance
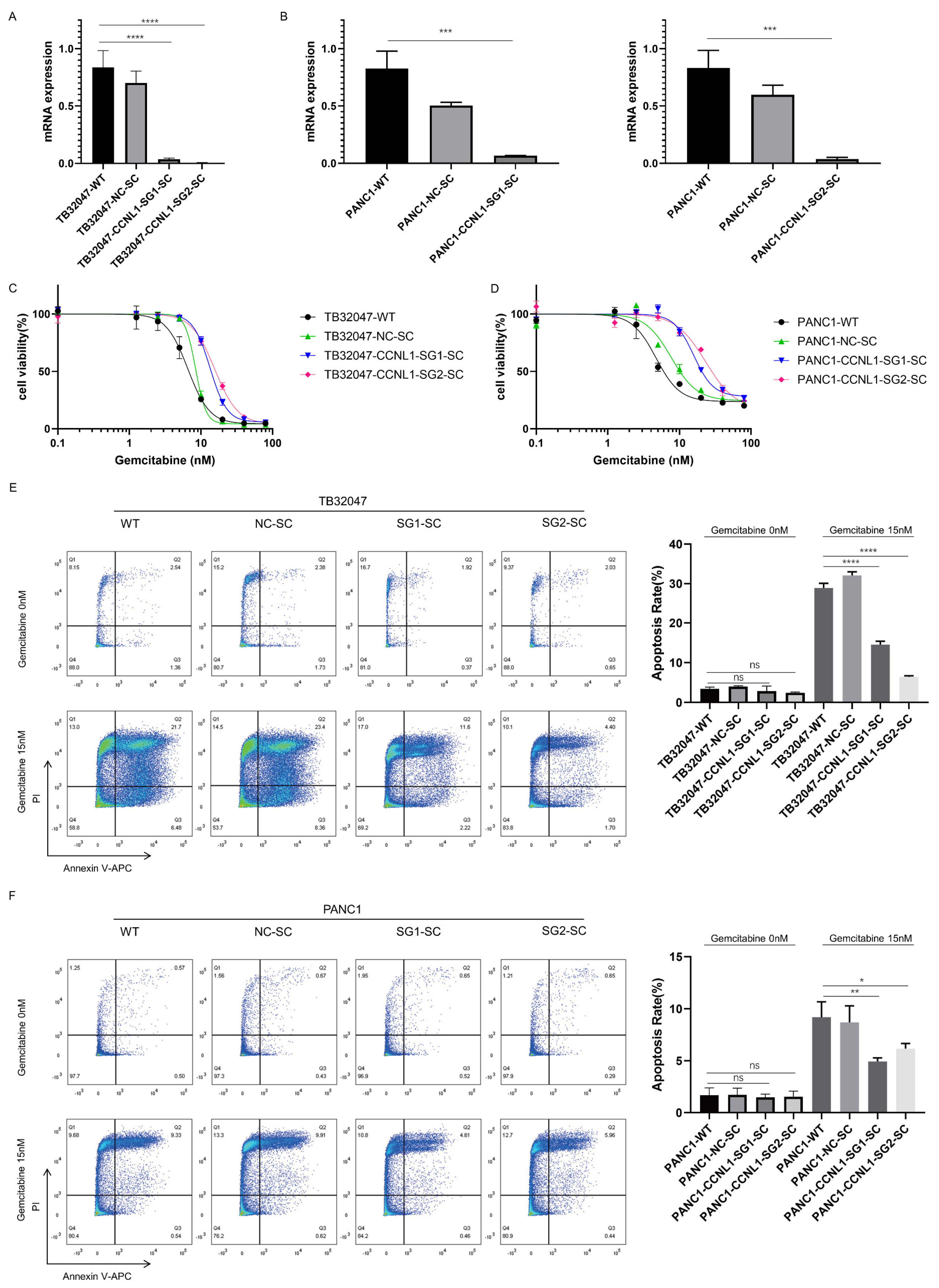
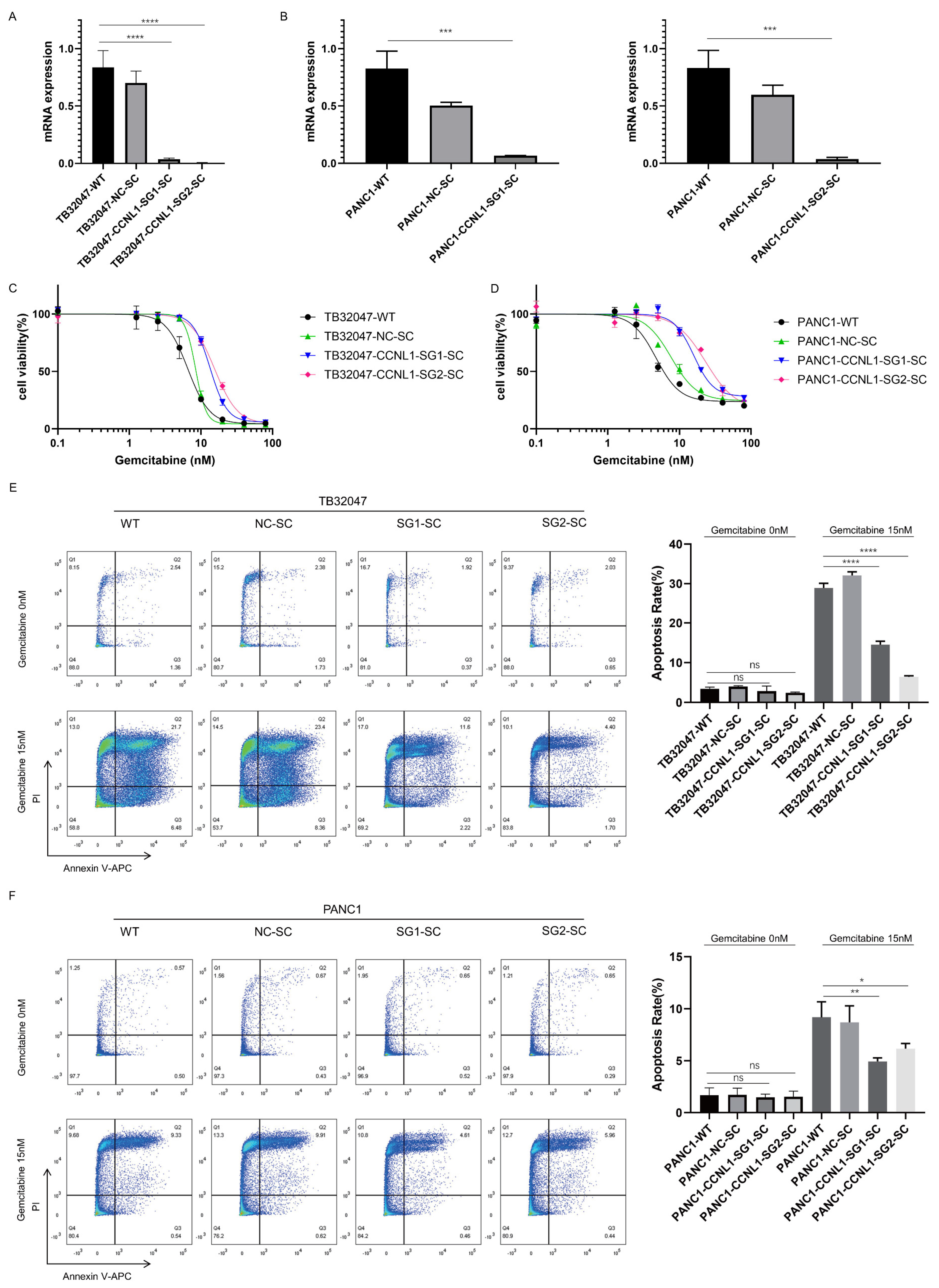
3.2. Loss of DCK and CCNL1 Enhances Resistance to Gemcitabine Treatment in TB32047 and PANC1 Cells
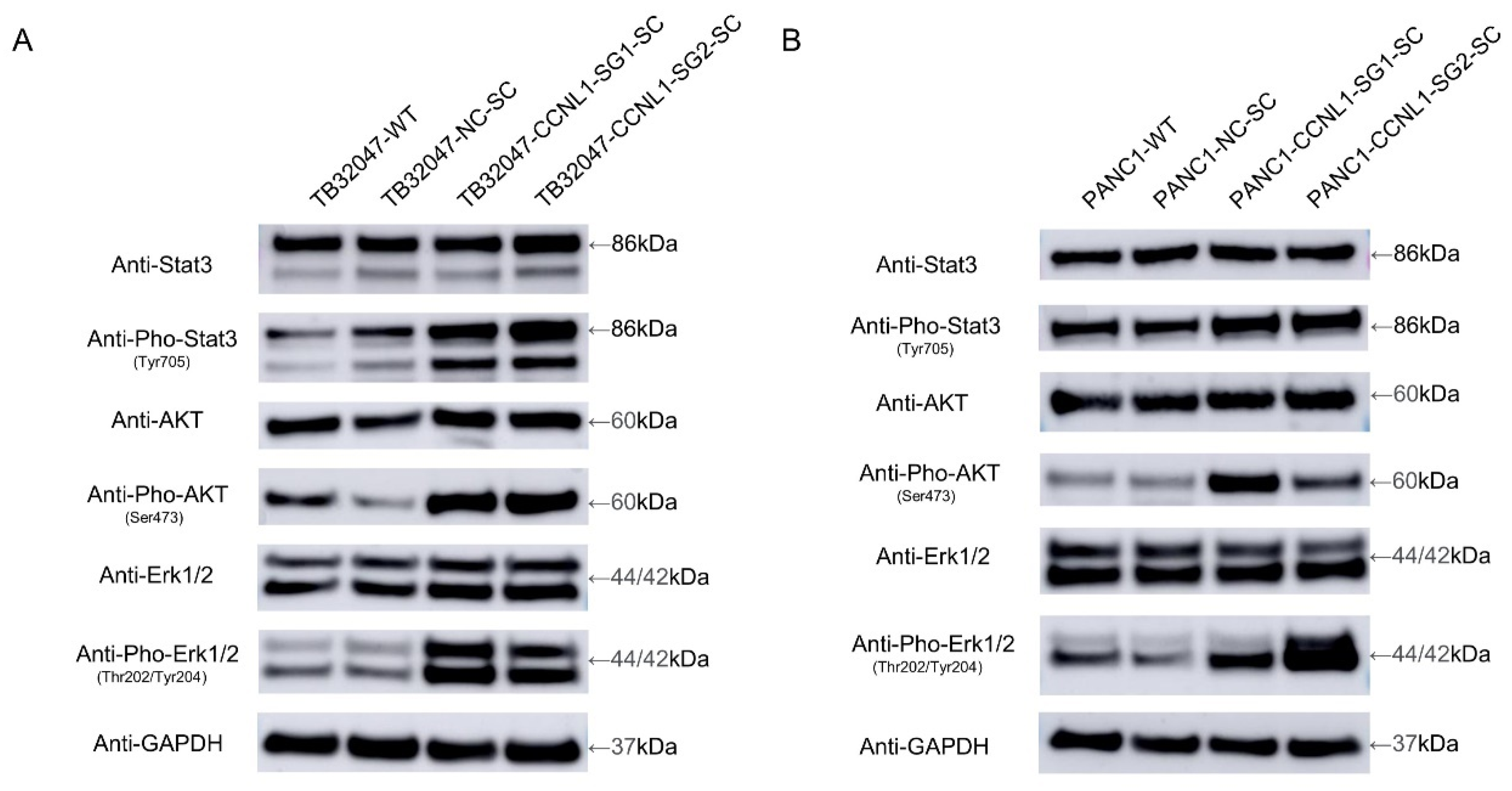
3.3. Knockout of CCNL1 Promotes the Phosphorylation of Erk, Akt, and Stat3 in TB32047 and PANC1 Cells
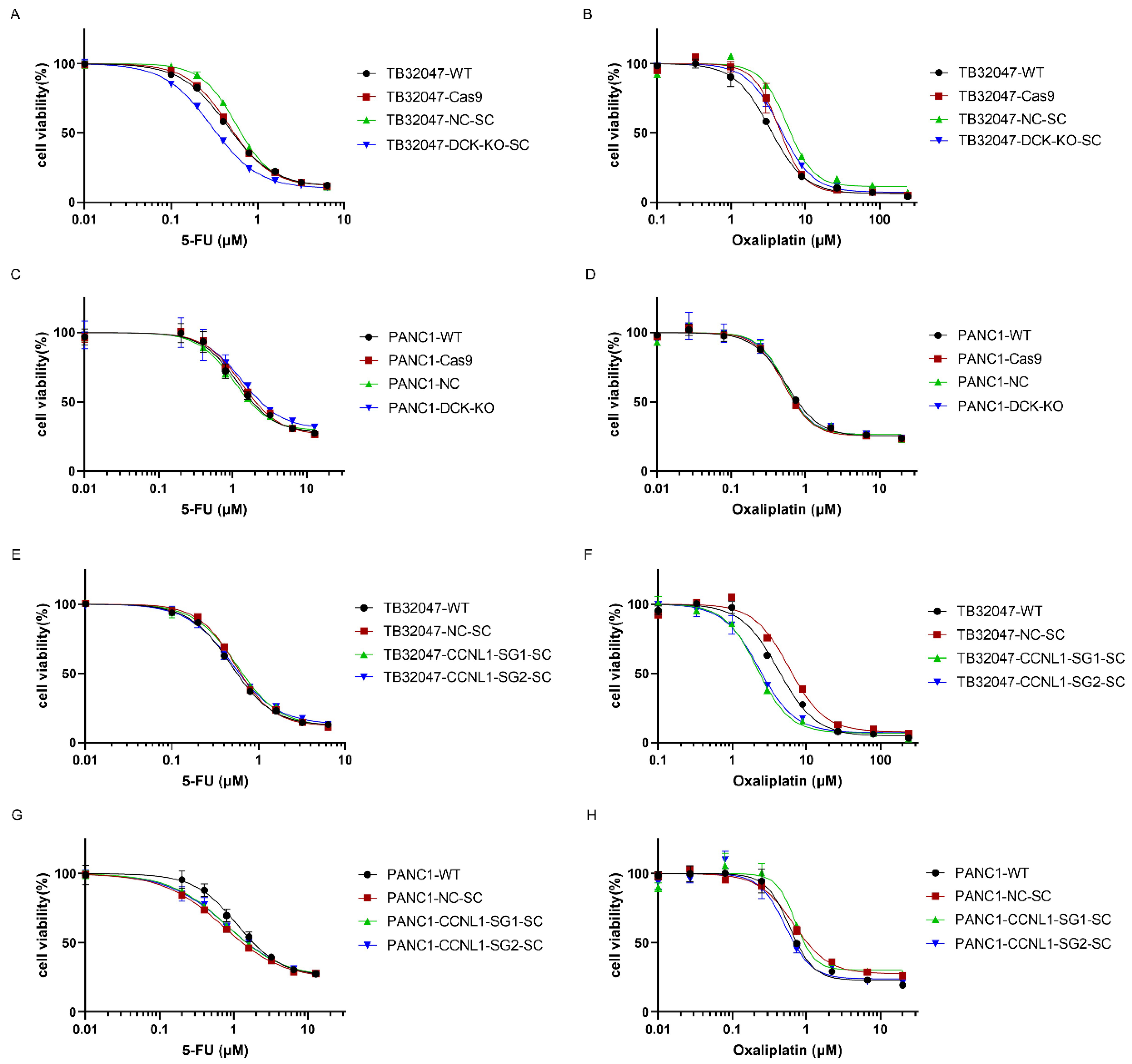
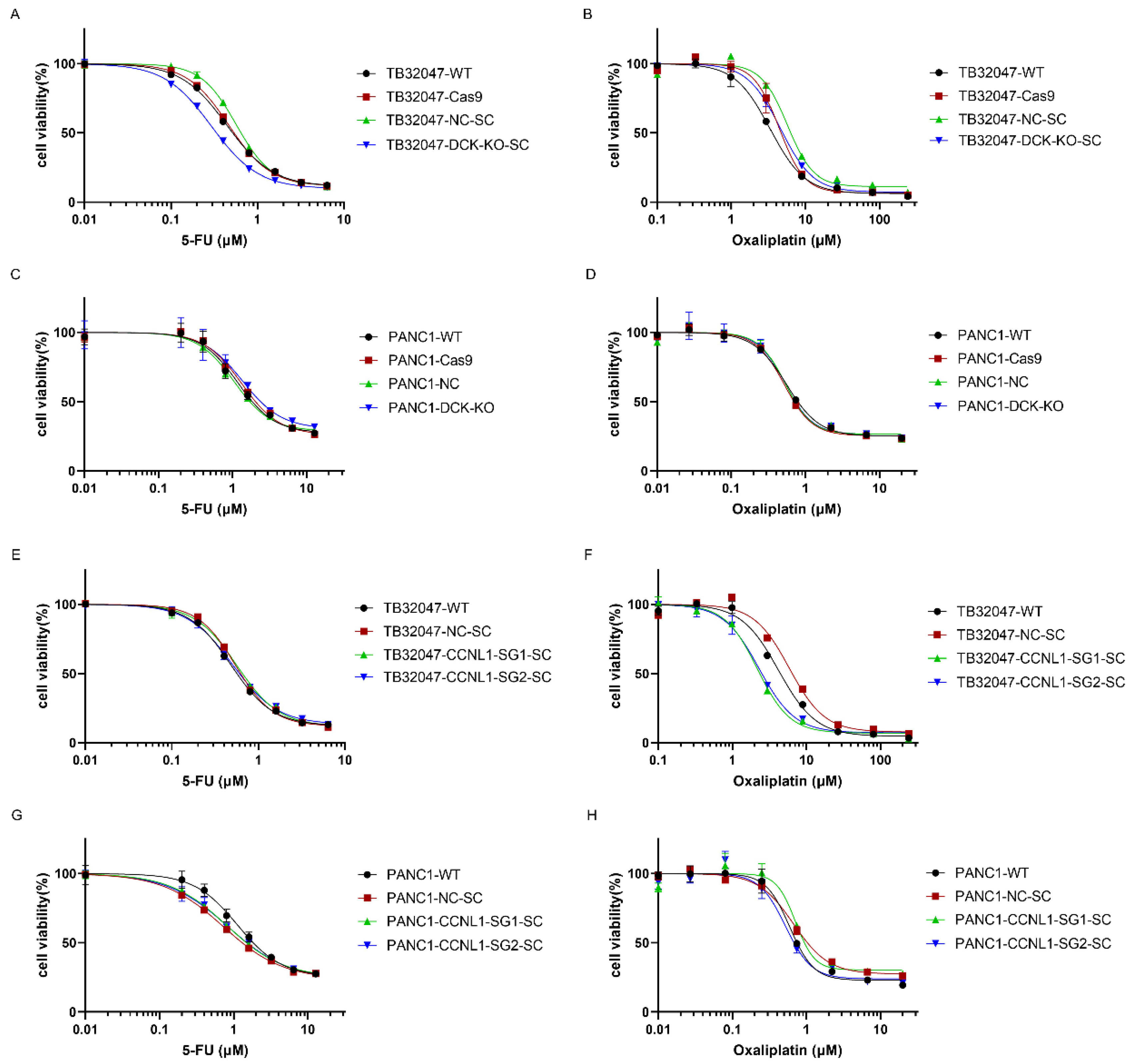
3.4. DCK or CCNL1 Knockout Does Not Confer Cross-Resistance to Other Drugs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Lianos, G.D.; Christodoulou, D.K.; Katsanos, K.H.; Katsios, C.; Glantzounis, G.K. Minimally Invasive Surgical Approaches for Pancreatic Adenocarcinoma: Recent Trends. J. Gastrointest. Cancer 2017, 48, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Sampedro, A.; Gaggia, G.; Ney, A.; Mahamed, I.; Acedo, P. The State-of-the-Art of Phase II/III Clinical Trials for Targeted Pancreatic Cancer Therapies. J. Clin. Med. 2021, 10, 566. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Bhardwaj, V. Therapeutic resistance in pancreatic ductal adenocarcinoma: Current challenges and future opportunities. World J. Gastroenterol. 2021, 27, 6527–6550. [Google Scholar] [CrossRef]
- Chin, V.; Nagrial, A.; Sjoquist, K.; O’Connor, C.A.; Chantrill, L.; Biankin, A.V.; Scholten, R.J.; Yip, D. Chemotherapy and radiotherapy for advanced pancreatic cancer. Cochrane Database Syst. Rev. 2018, 3, CD011044. [Google Scholar] [CrossRef]
- Bergman, A.M.; Pinedo, H.M.; Peters, G.J. Determinants of resistance to 2′,2′-difluorodeoxycytidine (gemcitabine). Drug. Resist. Updat. 2002, 5, 19–33. [Google Scholar] [CrossRef]
- Zeng, S.; Pottler, M.; Lan, B.; Grutzmann, R.; Pilarsky, C.; Yang, H. Chemoresistance in Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 4504. [Google Scholar] [CrossRef] [Green Version]
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Bailey, P.; Pilarsky, C. CRISPR Cas9 in Pancreatic Cancer Research. Front. Cell Dev. Biol. 2019, 7, 239. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.Y.; Park, J.W.; Hwang, Y.; Hahm, J.Y.; Park, J.; Park, K.S.; Seo, S.B. The H3K4 methyltransferase SETD1A is required for proliferation of non-small cell lung cancer cells by promoting S-phase progression. Biochem. Biophys. Res. Commun. 2021, 561, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Chen, J.; Wu, T.; Cheng, Z.; Tian, Y.; Pu, C.; Shi, W.; Suo, X.; Wu, X.; Zhang, K. Genome-scale CRISPR activation screening identifies a role of LRP8 in Sorafenib resistance in Hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2020, 526, 1170–1176. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Choi, A.; Cho, S.Y.; Jun, Y.; Na, D.; Lee, A.; Jang, G.; Kwon, J.Y.; Kim, J.; Lee, S.; et al. Genome-scale CRISPR screening identifies cell cycle and protein ubiquitination processes as druggable targets for erlotinib-resistant lung cancer. Mol. Oncol. 2021, 15, 487–502. [Google Scholar] [CrossRef]
- Dull, T.; Zufferey, R.; Kelly, M.; Mandel, R.J.; Nguyen, M.; Trono, D.; Naldini, L. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 1998, 72, 8463–8471. [Google Scholar] [CrossRef] [Green Version]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Koster, J.; Xu, H.; Chen, C.H.; Xiao, T.; Liu, J.S.; Brown, M.; Liu, X.S. Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol. 2015, 16, 281. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Stemmer, M.; Thumberger, T.; Del Sol Keyer, M.; Wittbrodt, J.; Mateo, J.L. CCTop: An Intuitive, Flexible and Reliable CRISPR/Cas9 Target Prediction Tool. PLoS ONE 2015, 10, e0124633. [Google Scholar] [CrossRef] [Green Version]
- Poddar, S.; Capparelli, E.V.; Rosser, E.W.; Gipson, R.M.; Wei, L.; Le, T.; Jung, M.E.; Radu, C.; Nikanjam, M. Development and preclinical pharmacology of a novel dCK inhibitor, DI-87. Biochem. Pharmacol. 2020, 172, 113742. [Google Scholar] [CrossRef]
- Nakano, T.; Saiki, Y.; Kudo, C.; Hirayama, A.; Mizuguchi, Y.; Fujiwara, S.; Soga, T.; Sunamura, M.; Matsumura, N.; Motoi, F.; et al. Acquisition of chemoresistance to gemcitabine is induced by a loss-of-function missense mutation of DCK. Biochem. Biophys. Res. Commun. 2015, 464, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Saiki, Y.; Yoshino, Y.; Fujimura, H.; Manabe, T.; Kudo, Y.; Shimada, M.; Mano, N.; Nakano, T.; Lee, Y.; Shimizu, S.; et al. DCK is frequently inactivated in acquired gemcitabine-resistant human cancer cells. Biochem. Biophys. Res. Commun. 2012, 421, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Mini, E.; Nobili, S.; Caciagli, B.; Landini, I.; Mazzei, T. Cellular pharmacology of gemcitabine. Ann. Oncol. 2006, 17 (Suppl. S5), v7–v12. [Google Scholar] [CrossRef] [PubMed]
- Ohhashi, S.; Ohuchida, K.; Mizumoto, K.; Fujita, H.; Egami, T.; Yu, J.; Toma, H.; Sadatomi, S.; Nagai, E.; Tanaka, M. Down-regulation of deoxycytidine kinase enhances acquired resistance to gemcitabine in pancreatic cancer. Anticancer Res. 2008, 28, 2205–2212. [Google Scholar] [PubMed]
- Redon, R.; Hussenet, T.; Bour, G.; Caulee, K.; Jost, B.; Muller, D.; Abecassis, J.; du Manoir, S. Amplicon mapping and transcriptional analysis pinpoint cyclin L as a candidate oncogene in head and neck cancer. Cancer Res. 2002, 62, 6211–6217. [Google Scholar] [PubMed]
- Sticht, C.; Hofele, C.; Flechtenmacher, C.; Bosch, F.X.; Freier, K.; Lichter, P.; Joos, S. Amplification of Cyclin L1 is associated with lymph node metastases in head and neck squamous cell carcinoma (HNSCC). Br. J. Cancer 2005, 92, 770–774. [Google Scholar] [CrossRef] [Green Version]
- Loyer, P.; Trembley, J.H.; Grenet, J.A.; Busson, A.; Corlu, A.; Zhao, W.; Kocak, M.; Kidd, V.J.; Lahti, J.M. Characterization of cyclin L1 and L2 interactions with CDK11 and splicing factors: Influence of cyclin L isoforms on splice site selection. J. Biol. Chem. 2008, 283, 7721–7732. [Google Scholar] [CrossRef] [Green Version]
- Dickinson, L.A.; Edgar, A.J.; Ehley, J.; Gottesfeld, J.M. Cyclin L is an RS domain protein involved in pre-mRNA splicing. J. Biol. Chem. 2002, 277, 25465–25473. [Google Scholar] [CrossRef] [Green Version]
- Burotto, M.; Chiou, V.L.; Lee, J.M.; Kohn, E.C. The MAPK pathway across different malignancies: A new perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Mundi, P.S.; Sachdev, J.; McCourt, C.; Kalinsky, K. AKT in cancer: New molecular insights and advances in drug development. Br. J. Clin. Pharmacol. 2016, 82, 943–956. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Jove, R. The STATs of cancer—new molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Shen, C.; Li, Y.; Zhang, T.; Huang, H.; Ren, J.; Hu, Z.; Xu, J.; Xu, B. Oridonin overcomes the gemcitabine resistant PANC-1/Gem cells by regulating GST pi and LRP/1 ERK/JNK signalling. Onco. Targets Ther. 2019, 12, 5751–5765. [Google Scholar] [CrossRef] [Green Version]
- Ryu, W.J.; Han, G.; Lee, S.H.; Choi, K.Y. Suppression of Wnt/beta-catenin and RAS/ERK pathways provides a therapeutic strategy for gemcitabine-resistant pancreatic cancer. Biochem. Biophys. Res. Commun. 2021, 549, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.H.; Hsu, T.W.; Chen, H.A.; Su, C.M.; Huang, M.T.; Chuang, T.H.; Leo Su, J.; Hsieh, C.L.; Chiu, C.F. ERK-mediated transcriptional activation of Dicer is involved in gemcitabine resistance of pancreatic cancer. J. Cell Physiol. 2021, 236, 4420–4434. [Google Scholar] [CrossRef]
- Xiong, Y.; Ju, L.; Yuan, L.; Chen, L.; Wang, G.; Xu, H.; Peng, T.; Luo, Y.; Xiao, Y.; Wang, X. KNSTRN promotes tumorigenesis and gemcitabine resistance by activating AKT in bladder cancer. Oncogene 2021, 40, 1595–1608. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, N.; Saito, Y.; Manabe, H.; Nakaoka, T.; Uchida, R.; Furukawa, R.; Muramatsu, T.; Sugiyama, Y.; Kimura, M.; Saito, H. Glucose Depletion Enhances the Stem Cell Phenotype and Gemcitabine Resistance of Cholangiocarcinoma Organoids through AKT Phosphorylation and Reactive Oxygen Species. Cancers 2019, 11, 1993. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Gu, Y.; Qian, Y.; Hu, B.; Zhu, C.; Wang, G.; Li, J. DNA-PKcs is important for Akt activation and gemcitabine resistance in PANC-1 pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2014, 452, 106–111. [Google Scholar] [CrossRef]
- Gong, J.; Munoz, A.R.; Pingali, S.; Payton-Stewart, F.; Chan, D.E.; Freeman, J.W.; Ghosh, R.; Kumar, A.P. Downregulation of STAT3/NF-kappaB potentiates gemcitabine activity in pancreatic cancer cells. Mol. Carcinog. 2017, 56, 402–411. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Tang, W.; Marquez, R.T.; Li, K.; Highfill, C.A.; He, F.; Lian, J.; Lin, J.; Fuchs, J.R.; Ji, M.; et al. Overcoming chemo/radio-resistance of pancreatic cancer by inhibiting STAT3 signaling. Oncotarget 2016, 7, 11708–11723. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cells | Mutation Number | Mutation Size | Mutation Type |
|---|---|---|---|
| TB32047-CCNL1-SG1-SC | 1 | 2 single nucleotides | 1 bp mutation 1 bp deletion |
| TB32047-CCNL1-SG2-SC | 2 | single nucleotide | deletion |
| 66 bp | 6 bp replaced by 66 bp | ||
| PANC1-CCNL1-SG1-SC | 2 | 102 bp | insertion |
| 359 bp | 4 bp replaced by 359 bp | ||
| PANC1-CCNL1-SG2-SC | 1 | single nucleotide | deletion |
| Cells | Gemcitabine (nM) | Oxaliplatin (µM) | 5-Flourouracil (µM) | |||
|---|---|---|---|---|---|---|
| IC50 | 95% CI | IC50 | 95% CI | IC50 | 95% CI | |
| TB32047-WT | 7.69 | 7.48–7.91 | 3.32 | 3.03–3.64 | 0.44 | 0.42–0.46 |
| TB32047-Cas9 | 8.21 | 7.95–8.48 | 4.42 | 3.92–5.00 | 0.46 | 0.45–0.48 |
| TB32047-NC-SC | 8.85 | 8.37–9.35 | 5.70 | 4.99–6.48 | 0.57 | 0.54–0.60 |
| TB32047-DCK-KO-SC | - | - | 4.54 | 4.28–4.82 | 0.29 | 0.28–0.31 |
| PANC1-WT | 7.75 | 6.52–9.18 | 0.55 | 0.50–0.61 | 1.17 | 0.96–1.46 |
| PANC1-Cas9 | 9.27 | 8.22–10.36 | 0.51 | 0.47–0.56 | 1.33 | 1.17–1.52 |
| PANC1-NC | 10.47 | 9.47–12.55 | 0.53 | 0.46–0.61 | 1.05 | 0.93–1.20 |
| PANC1-DCK-KO | - | - | 0.53 | 0.44–0.63 | 1.35 | 1.01–1.90 |
| TB32047-WT | 6.56 | 5.91–7.29 | 4.21 | 3.74–4.76 | 0.48 | 0.46–0.50 |
| TB32047-NC-SC | 8.42 | 8.08–8.90 | 5.88 | 5.01–6.89 | 0.54 | 0.52–0.57 |
| TB32047-CCNL1-SG1-SC | 13.41 | 12.78–14.08 | 2.17 | 1.97–2.39 | 0.56 | 0.52–0.61 |
| TB32047-CCNL1-SG2-SC | 15.53 | 14.62–16.51 | 2.30 | 2.05–2.56 | 0.50 | 0.46–0.54 |
| PANC1-WT | 4.70 | 4.00–5.66 | 0.60 | 0.53–0.67 | 1.12 | 0.93–1.38 |
| PANC1-NC-SC | 7.67 | 6.23–9.60 | 0.67 | 0.59–0.76 | 0.72 | 0.67–0.77 |
| PANC1-CCNL1-SG1-SC | 15.81 | 14.01–17.89 | 0.71 | 0.61–0.87 | 0.81 | 0.73–0.89 |
| PANC1-CCNL1-SG2-SC | 23.25 | 19.01–32.83 | 0.52 | 0.42–0.64 | 0.83 | 0.68–1.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.; Liu, B.; Liu, D.; Yang, Z.; Zhang, S.; Xu, P.; Xing, Y.; Kutschick, I.; Pfeffer, S.; Britzen-Laurent, N.; et al. Genome-Wide CRISPR Screening Identifies DCK and CCNL1 as Genes That Contribute to Gemcitabine Resistance in Pancreatic Cancer. Cancers 2022, 14, 3152. https://doi.org/10.3390/cancers14133152
Yang H, Liu B, Liu D, Yang Z, Zhang S, Xu P, Xing Y, Kutschick I, Pfeffer S, Britzen-Laurent N, et al. Genome-Wide CRISPR Screening Identifies DCK and CCNL1 as Genes That Contribute to Gemcitabine Resistance in Pancreatic Cancer. Cancers. 2022; 14(13):3152. https://doi.org/10.3390/cancers14133152
Chicago/Turabian StyleYang, Hai, Bin Liu, Dongxue Liu, Zhirong Yang, Shuman Zhang, Pengyan Xu, Yuming Xing, Isabella Kutschick, Susanne Pfeffer, Nathalie Britzen-Laurent, and et al. 2022. "Genome-Wide CRISPR Screening Identifies DCK and CCNL1 as Genes That Contribute to Gemcitabine Resistance in Pancreatic Cancer" Cancers 14, no. 13: 3152. https://doi.org/10.3390/cancers14133152
APA StyleYang, H., Liu, B., Liu, D., Yang, Z., Zhang, S., Xu, P., Xing, Y., Kutschick, I., Pfeffer, S., Britzen-Laurent, N., Grützmann, R., & Pilarsky, C. (2022). Genome-Wide CRISPR Screening Identifies DCK and CCNL1 as Genes That Contribute to Gemcitabine Resistance in Pancreatic Cancer. Cancers, 14(13), 3152. https://doi.org/10.3390/cancers14133152