
The Role of the NLRP3 Inflammasome in HCC Carcinogenesis and Treatment: Harnessing Innate Immunity




Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Epidemiology of Hepatocellular Carcinoma
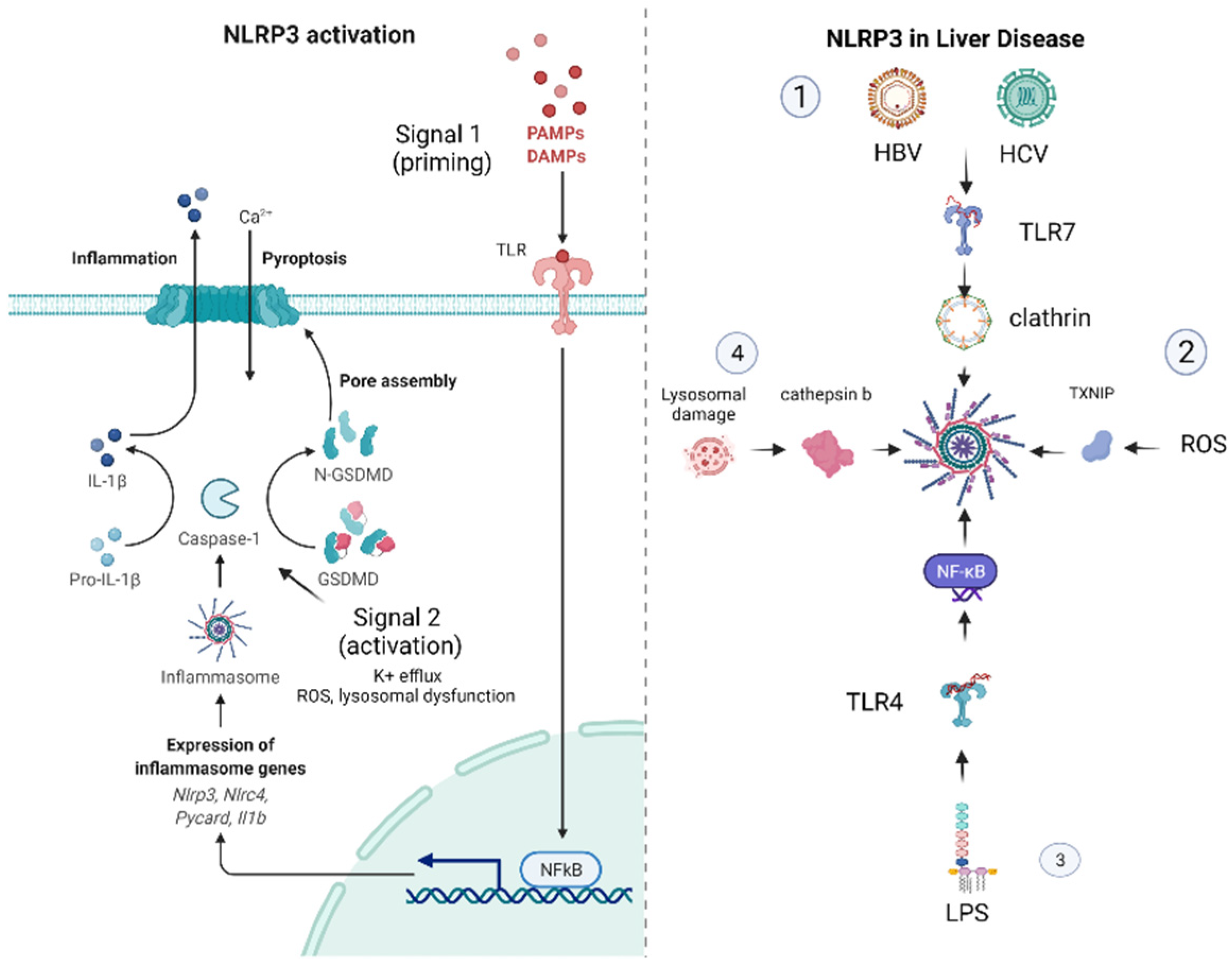
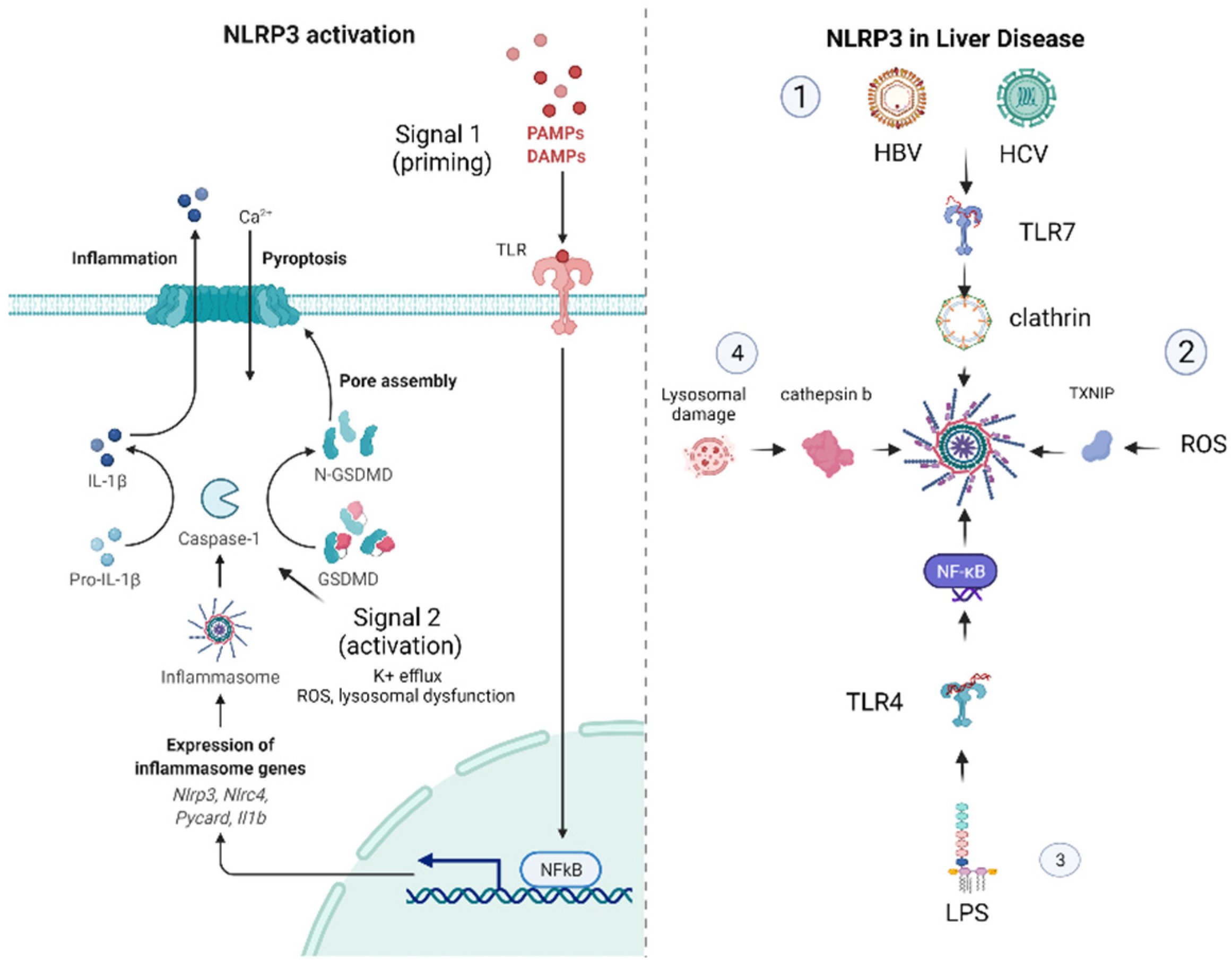
1.2. Mechanisms of NLRP3 Inflammasome Activation
2. The NLRP3 Inflammasome in Liver Disease
2.1. The NLRP3 Inflammasome in NAFLD–NASH
2.2. The Role of NLRP3 in Viral Hepatitis
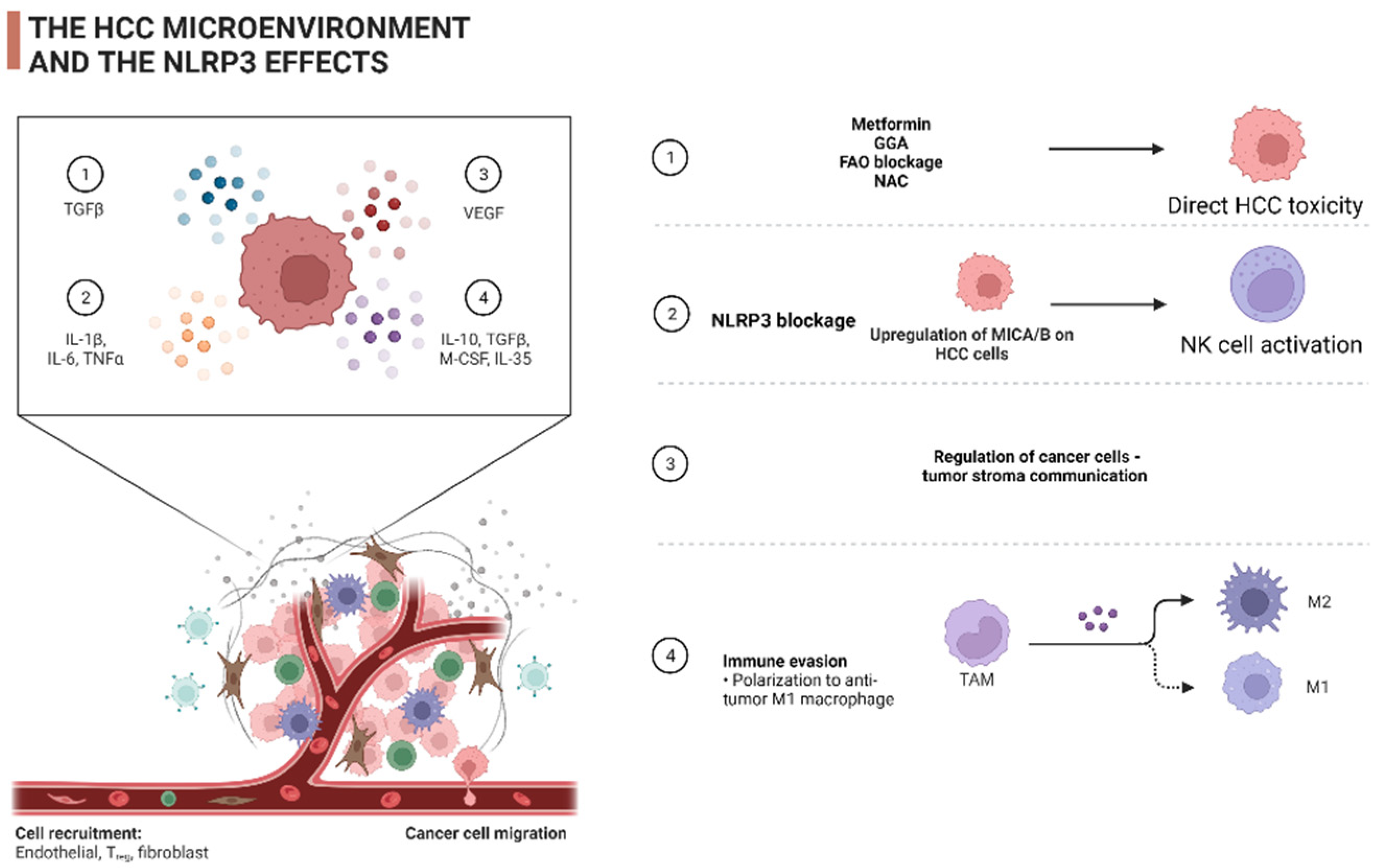
3. The Role of NLRP3 in the Shaping of the HCC Tumor Microenvironment (TME)
4. The Role of the NLRP3 Inflammasome in the Therapeutic Management of HCC
4.1. The NLRP3 as Therapeutic Target
4.2. The NLRP3 as Biomarker
5. Conclusions—Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Galle, P.R.; Forner, A.; Llovet, J.M.; Mazzaferro, V.; Piscaglia, F.; Raoul, J.L.; Schirmacher, P.; Vilgrain, V. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papatheodoridi, A.; Papatheodoridis, G. Hepatocellular carcinoma: The virus or the liver? Liver Int. 2022, 1–9. [Google Scholar] [CrossRef]
- Markakis, G.E.; Koulouris, A.; Tampaki, M.; Cholongitas, E.; Deutsch, M.; Papatheodoridis, G.V.; Koskinas, J. The changing epidemiology of hepatocellular carcinoma in Greece. Ann. Gastroenterol. 2022, 35, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Stepanova, M.; Ong, J.P.; Jacobson, I.M.; Bugianesi, E.; Duseja, A.; Eguchi, Y.; Wong, V.W.; Negro, F.; Yilmaz, Y.; et al. Nonalcoholic Steatohepatitis Is the Fastest Growing Cause of Hepatocellular Carcinoma in Liver Transplant Candidates. Clin. Gastroenterol. Hepatol. 2019, 17, 748–755.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarao, K.; Nozaki, A.; Ikeda, T.; Sato, A.; Komatsu, H.; Komatsu, T.; Taguri, M.; Tanaka, K. Real impact of liver cirrhosis on the development of hepatocellular carcinoma in various liver diseases—Meta-analytic assessment. Cancer Med. 2019, 8, 1054–1065. [Google Scholar] [CrossRef] [Green Version]
- Rebouissou, S.; Nault, J.C. Advances in molecular classification and precision oncology in hepatocellular carcinoma. J. Hepatol. 2020, 72, 215–229. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [Green Version]
- Calderaro, J.; Ziol, M.; Paradis, V.; Zucman-Rossi, J. Molecular and histological correlations in liver cancer. J. Hepatol. 2019, 71, 616–630. [Google Scholar] [CrossRef] [Green Version]
- Calderaro, J.; Couchy, G.; Imbeaud, S.; Amaddeo, G.; Letouzé, E.; Blanc, J.F.; Laurent, C.; Hajji, Y.; Azoulay, D.; Bioulac-Sage, P.; et al. Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. J. Hepatol. 2017, 67, 727–738. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, A.; Ricci, A.D.; Gadaleta-Caldarola, G.; Brandi, G. First-line immune checkpoint inhibitor-based combinations in unresectable hepatocellular carcinoma: Current management and future challenges. Expert Rev. Gastroenterol. Hepatol. 2021, 15, 1245–1251. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.; Ricci, A.D. PD-L1, TMB, and other potential predictors of response to immunotherapy for hepatocellular carcinoma: How can they assist drug clinical trials? Expert Opin. Investig. Drugs 2022, 31, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Choucair, K.; Morand, S.; Stanbery, L.; Edelman, G.; Dworkin, L.; Nemunaitis, J. TMB: A promising immune-response biomarker, and potential spearhead in advancing targeted therapy trials. Cancer Gene Ther. 2020, 27, 841–853. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.G.; Duberg, A.-S.; Aleman, S.; Chung, R.T.; Chan, A.T.; Ludvigsson, J.F. Association of Aspirin with Hepatocellular Carcinoma and Liver-Related Mortality. N. Engl. J. Med. 2020, 382, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Huang, X.; Su, H. The role of the inflammasome and its related pathways in ovarian cancer. Clin. Transl. Oncol. 2022. [Google Scholar] [CrossRef]
- West, A.J.; Deswaerte, V.; West, A.C.; Gearing, L.J.; Tan, P.; Jenkins, B.J. Inflammasome-Associated Gastric Tumorigenesis Is Independent of the NLRP3 Pattern Recognition Receptor. Front. Oncol. 2022, 12, 830350. [Google Scholar] [CrossRef]
- Caruso, S.; O’Brien, D.R.; Cleary, S.P.; Roberts, L.R.; Zucman-Rossi, J. Genetics of Hepatocellular Carcinoma: Approaches to Explore Molecular Diversity. Hepatology 2021, 73, 14–26. [Google Scholar] [CrossRef]
- Wei, Q.; Mu, K.; Li, T.; Zhang, Y.; Yang, Z.; Jia, X.; Zhao, W.; Huai, W.; Guo, P.; Han, L. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab. Investig. 2014, 94, 52–62. [Google Scholar] [CrossRef] [Green Version]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef]
- Zhen, Y.; Zhang, H. NLRP3 inflammasome and inflammatory bowel disease. Front. Immunol. 2019, 10, 276. [Google Scholar] [CrossRef] [Green Version]
- Ranson, N.; Kunde, D.; Eri, R. Regulation and Sensing of Inflammasomes and Their Impact on Intestinal Health. Int. J. Mol. Sci. 2017, 18, 2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirzada, R.H.; Javaid, N.; Choi, S. The Roles of the NLRP3 Inflammasome in Neurodegenerative and Metabolic Diseases and in Relevant Advanced Therapeutic Interventions. Genes 2020, 11, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.Y.; Liu, M.; Wang, F.; Bertin, J.; Núñez, G. A Functional Role for Nlrp6 in Intestinal Inflammation and Tumorigenesis. J. Immunol. 2011, 186, 7187–7194. [Google Scholar] [CrossRef]
- Liao, Y.; Liu, K.; Zhu, L. Emerging Roles of Inflammasomes in Cardiovascular Diseases. Front. Immunol. 2022, 13, 834289. [Google Scholar] [CrossRef] [PubMed]
- Anton-Pampols, P.; Diaz-Requena, C.; Martinez-Valenzuela, L.; Gomez-Preciado, F.; Fulladosa, X.; Vidal-Alabro, A.; Torras, J.; Lloberas, N.; Draibe, J. The Role of Inflammasomes in Glomerulonephritis. Int. J. Mol. Sci. 2022, 23, 4208. [Google Scholar] [CrossRef] [PubMed]
- Saljic, A.; Heijman, J.; Dobrev, D. Emerging Antiarrhythmic Drugs for Atrial Fibrillation. Int. J. Mol. Sci. 2022, 23, 4096. [Google Scholar] [CrossRef]
- Carvalho, A.M.; Bacellar, O.; Carvalho, E.M. Protection and Pathology in Leishmania braziliensis Infection. Pathogens 2022, 11, 466. [Google Scholar] [CrossRef]
- Li, Z.; Chen, X.; Tao, J.; Shi, A.; Zhang, J.; Yu, P. Exosomes Regulate NLRP3 Inflammasome in Diseases. Front. Cell Dev. Biol. 2022, 9, 802509. [Google Scholar] [CrossRef] [PubMed]
- Ohto, U.; Kamitsukasa, Y.; Ishida, H.; Zhang, Z.; Murakami, K.; Hirama, C. Structural basis for the oligomerization-mediated regulation of NLRP3 in fl ammasome activation. Proc. Natl. Acad. Sci. USA 2022, 119, e2121353119. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases: Current perspectives. Nat. Rev. Drug Discov. 2018, 17, 588–606. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.W.; Harmon, C.; O’Farrelly, C. Liver immunology and its role in inflammation and homeostasis. Cell. Mol. Immunol. 2016, 13, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Liaskou, E.; Wilson, D.V.; Oo, Y.H. Innate immune cells in liver inflammation. Mediat. Inflamm. 2012, 2012, 949157. [Google Scholar] [CrossRef] [Green Version]
- Kelly, A.M.; Golden-Mason, L.; Traynor, O.; Geoghegan, J.; McEntee, G.; Hegarty, J.E.; O’Farrelly, C. Changes in hepatic immunoregulatory cytokines in patients with metastatic colorectal carcinoma: Implications for hepatic anti-tumour immunity. Cytokine 2006, 35, 171–179. [Google Scholar] [CrossRef]
- Al Mamun, A.; Akter, A.; Hossain, S.; Sarker, T.; Safa, S.A.; Mustafa, Q.G.; Muhammad, S.A.; Munir, F. Role of NLRP3 inflammasome in liver disease. J. Dig. Dis. 2020, 21, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Wu, Y.; Zhao, Z.; Wu, B.; Sun, K.; Wang, H.; Qin, L.; Bai, F.; Leng, Y.; Tang, W. A new mechanism of obeticholic acid on NASH treatment by inhibiting NLRP3 inflammasome activation in macrophage. Metabolism 2021, 120, 154797. [Google Scholar] [CrossRef]
- Ruan, S.; Han, C.; Sheng, Y.; Wang, J.; Zhou, X.; Guan, Q.; Li, W.; Zhang, C.; Yang, Y. Antcin A alleviates pyroptosis and inflammatory response in Kupffercells of non-alcoholic fatty liver disease by targeting NLRP3. Int. Immunopharmacol. 2021, 100, 108126. [Google Scholar] [CrossRef]
- Hwangbo, H.; Kim, M.Y.; Ji, S.Y.; Kim, S.Y.; Lee, H.; Kim, G.Y.; Park, C.; Keum, Y.S.; Hong, S.H.; Cheong, J.; et al. Auranofin attenuates non-alcoholic fatty liver disease by suppressing lipid accumulation and nlrp3 inflammasome-mediated hepatic inflammation in vivo and in vitro. Antioxidants 2020, 9, 1040. [Google Scholar] [CrossRef]
- Liu, J.; Wang, T.; He, K.; Xu, M.; Gong, J.P. Cardiolipin inhibitor ameliorates the non-alcoholic steatohepatitis through suppressing IMLRP3 inflammasome activation. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 8158–8167. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Cao, G.; Min, X.; Wang, T.; Sun, S.; Du, X.; Zhang, W. Cathepsin B inhibition ameliorates the non-alcoholic steatohepatitis through suppressing caspase-1 activation. J. Physiol. Biochem. 2018, 74, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Sui, Y.H.; Luo, W.J.; Xu, Q.Y.; Hua, J. Dietary saturated fatty acid and polyunsaturated fatty acid oppositely affect hepatic NOD-like receptor protein 3 inflammasome through regulating nuclear factor-kappa B activation. World J. Gastroenterol. 2016, 22, 2533–2544. [Google Scholar] [CrossRef]
- Yu, Y.; Chen, D.; Zhao, Y.; Zhu, J.; Dong, X. Melatonin ameliorates hepatic steatosis by inhibiting NLRP3 inflammasome in db/db mice. Int. J. Immunopathol. Pharmacol. 2021, 35, 205873842110368. [Google Scholar] [CrossRef] [PubMed]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.H.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef]
- Negash, A.A.; Ramos, H.J.; Crochet, N.; Lau, D.T.Y.; Doehle, B.; Papic, N.; Delker, D.A.; Jo, J.; Bertoletti, A.; Hagedorn, C.H.; et al. IL-1β production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 2013, 9, e1003330. [Google Scholar] [CrossRef] [PubMed]
- Negash, A.A.; Olson, R.M.; Griffin, S.; Gale, M. Modulation of calcium signaling pathway by hepatitis C virus core protein stimulates NLRP3 inflammasome activation. PLoS Pathog. 2019, 15, e1007593. [Google Scholar] [CrossRef]
- Serti, E.; Werner, J.M.; Chattergoon, M.; Cox, A.L.; Lohmann, V.; Rehermann, B. Monocytes Activate Natural Killer Cells via Inflammasome-Induced Interleukin 18 in Response to Hepatitis C Virus Replication. Gastroenterology 2014, 147, 209–220.e3. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Lan, P.; Hou, X.; Han, Q.; Lu, N.; Li, T.; Jiao, C.; Zhang, J.; Zhang, C.; Tian, Z. HBV inhibits LPS-induced NLRP3 inflammasome activation and IL-1β production via suppressing the NF-κB pathway and ROS production. J. Hepatol. 2017, 66, 693–702. [Google Scholar] [CrossRef]
- Arvanitakis, K.; Koletsa, T.; Mitroulis, I.; Germanidis, G. Tumor-Associated Macrophages in Hepatocellular Carcinoma Pathogenesis, Prognosis and Therapy. Cancers 2022, 14, 226. [Google Scholar] [CrossRef]
- Sun, Y.; Wu, L.; Zhong, Y.; Zhou, K.; Hou, Y.; Wang, Z.; Zhang, Z.; Xie, J.; Wang, C.; Chen, D.; et al. Single-cell landscape of the ecosystem in early-relapse hepatocellular carcinoma. Cell 2021, 184, 404–421.e16. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Zhou, L.; Lv, D.; Zhu, X.; Tang, H. Exosome-mediated communication in the tumor microenvironment contributes to hepatocellular carcinoma development and progression. J. Hematol. Oncol. 2019, 12, 53. [Google Scholar] [CrossRef] [PubMed]
- Thi, H.T.H.; Hong, S. Inflammasome as a Therapeutic Target for Cancer Prevention and Treatment. J. Cancer Prev. 2017, 22, 62–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; He, Y.; Luo, N.; Patel, S.J.; Han, Y.; Gao, R.; Modak, M.; Carotta, S.; Haslinger, C.; Kind, D.; et al. Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell 2019, 179, 829–845.e20. [Google Scholar] [CrossRef]
- Hao, N.B.; Lü, M.H.; Fan, Y.H.; Cao, Y.L.; Zhang, Z.R.; Yang, S.M. Macrophages in tumor microenvironments and the progression of tumors. Clin. Dev. Immunol. 2012, 2012, 948098. [Google Scholar] [CrossRef] [Green Version]
- Arvanitakis, K.; Mitroulis, I.; Germanidis, G. Tumor-associated neutrophils in hepatocellular carcinoma pathogenesis, prognosis, and therapy. Cancers 2021, 13, 2899. [Google Scholar] [CrossRef]
- Peña-Romero, A.C.; Orenes-Piñero, E. Dual Effect of Immune Cells within Tumour Microenvironment: Pro- and Anti-Tumour Effects and Their Triggers. Cancers 2022, 14, 1681. [Google Scholar] [CrossRef]
- Yang, Y.; Ye, Y.C.; Chen, Y.; Zhao, J.L.; Gao, C.C.; Han, H.; Liu, W.C.; Qin, H.Y. Crosstalk between hepatic tumor cells and macrophages via Wnt/β-catenin signaling promotes M2-like macrophage polarization and reinforces tumor malignant behaviors. Cell Death Dis. 2018, 9, 793. [Google Scholar] [CrossRef]
- Zhou, S.L.; Zhou, Z.J.; Hu, Z.Q.; Huang, X.W.; Wang, Z.; Chen, E.B.; Fan, J.; Cao, Y.; Dai, Z.; Zhou, J. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology 2016, 150, 1646–1658.e17. [Google Scholar] [CrossRef] [Green Version]
- Du, D.; Liu, C.; Qin, M.; Zhang, X.; Xi, T.; Yuan, S.; Hao, H.; Xiong, J. Metabolic dysregulation and emerging therapeutical targets for hepatocellular carcinoma. Acta Pharm. Sin. B 2022, 12, 558–580. [Google Scholar] [CrossRef]
- Jiang, Z.-Z.; Peng, Z.-P.; Liu, X.-C.; Guo, H.-F.; Zhou, M.-M.; Jiang, D.; Ning, W.-R.; Huang, Y.-F.; Zheng, L.; Wu, Y. Neutrophil extracellular traps induce tumor metastasis through dual effects on cancer and endothelial cells. Oncoimmunology 2022, 11, 2052418. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, H.; Abe, M.; Yang, Y.; Cui, D.; Seki, T.; Nakamura, M.; Hosaka, K.; Lim, S.; Wu, J.; He, X.; et al. Cancer Lipid Metabolism Confers Antiangiogenic Drug Resistance. Cell Metab. 2018, 28, 104–117.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berndt, N.; Eckstein, J.; Heucke, N.; Gajowski, R.; Stockmann, M.; Meierhofer, D.; Holzhütter, H.-G. Characterization of Lipid and Lipid Droplet Metabolism in Human HCC. Cells 2019, 8, 512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Lin, J.Z.; Yang, X.B.; Sang, X.T. Aberrant lipid metabolism in hepatocellular carcinoma cells as well as immune microenvironment: A review. Cell Prolif. 2020, 53, e12772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moris, D.; Beal, E.W.; Chakedis, J.; Burkhart, R.A.; Schmidt, C.; Dillhoff, M.; Zhang, X.; Theocharis, S.; Pawlik, T.M. Role of exosomes in treatment of hepatocellular carcinoma. Surg. Oncol. 2017, 26, 219–228. [Google Scholar] [CrossRef]
- Gordon-Weeks, A.; Yuzhalin, A.E. Cancer Extracellular Matrix Proteins Regulate Tumour Immunity. Cancers 2020, 12, 3331. [Google Scholar] [CrossRef]
- Gaul, S.; Leszczynska, A.; Alegre, F.; Kaufmann, B.; Johnson, C.D.; Adams, L.A.; Wree, A.; Damm, G.; Seehofer, D.; Calvente, C.J.; et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J. Hepatol. 2021, 74, 156–167. [Google Scholar] [CrossRef]
- Ding, Y.; Yan, Y.; Dong, Y.; Xu, J.; Su, W.; Shi, W.; Zou, Q.; Yang, X. NLRP3 promotes immune escape by regulating immune checkpoints: A pan-cancer analysis. Int. Immunopharmacol. 2022, 104, 108512. [Google Scholar] [CrossRef]
- Lee, H.H.; Kim, D.; Jung, J.; Kang, H.; Cho, H. NLRP3 Deficiency in Hepatocellular Carcinoma Enhances Surveillance of NK-92 through a Modulation of MICA/B. Int. J. Mol. Sci. 2021, 22, 9285. [Google Scholar] [CrossRef]
- Shi, G.; Tang, N.; Qiu, J.; Zhang, D.; Huang, F.; Cheng, Y.; Ding, K.; Li, W.; Zhang, P.; Tan, X. Irisin stimulates cell proliferation and invasion by targeting the PI3K/AKT pathway in human hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2017, 493, 585–591. [Google Scholar] [CrossRef]
- Liu, H.; Wang, M.; Jin, Z.; Sun, D.; Zhu, T.; Liu, X.; Tan, X.; Shi, G. FNDC5 induces M2 macrophage polarization and promotes hepatocellular carcinoma cell growth by affecting the PPARγ/NF-κB/NLRP3 pathway. Biochem. Biophys. Res. Commun. 2021, 582, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Q.; Lou, Y.; Fu, Q.; Chen, Q.; Wei, T.; Yang, J.; Tang, J.; Wang, J.; Chen, Y.; et al. Hypoxia-inducible factor-1α/interleukin-1β signaling enhances hepatoma epithelial-mesenchymal transition through macrophages in a hypoxic-inflammatory microenvironment. Hepatology 2018, 67, 1872–1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Zhang, X.; Zheng, L.; Zhao, H.; Yan, G.; Zhang, Q.; Zhou, Y.; Lei, J.; Zhang, J.; Wang, J.; et al. RIPK3 orchestrates fatty acid metabolism in tumor-associated macrophages and hepatocarcinogenesis. Cancer Immunol. Res. 2020, 8, 710–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, C.E.; Hu, Y.; Zhou, P.; Guo, X.; Gu, C.; Zhang, Y.; Li, A.; Liu, S. Lactate and TGF-β antagonistically regulate inflammasome activation in the tumor microenvironment. J. Cell. Physiol. 2021, 236, 4528–4537. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Da, Q.; Li, Z.; Lin, Q.; Yi, J.; Su, Y.; Yu, G.; Ren, Q.; Liu, X.; Lin, Z.; et al. Inhibition of NEK7 Suppressed Hepatocellular Carcinoma Progression by Mediating Cancer Cell Pyroptosis. Front. Oncol. 2022, 12, 812655. [Google Scholar] [CrossRef]
- Vogel, A.; Martinelli, E.; Cervantes, A.; Chau, I.; Daniele, B.; Llovet, J.M.; Meyer, T.; Nault, J.C.; Neumann, U.; Ricke, J.; et al. Updated treatment recommendations for hepatocellular carcinoma (HCC) from the ESMO Clinical Practice Guidelines. Ann. Oncol. 2021, 32, 801–805. [Google Scholar] [CrossRef]
- Llovet, J.M.; Pinyol, R.; Kelley, R.K.; El-Khoueiry, A.; Reeves, H.L.; Wang, X.W.; Gores, G.J.; Villanueva, A. Molecular pathogenesis and systemic therapies for hepatocellular carcinoma. Nat. Cancer 2022, 3, 386–401. [Google Scholar] [CrossRef]
- Petrowsky, H.; Fritsch, R.; Guckenberger, M.; De Oliveira, M.L.; Dutkowski, P.; Clavien, P.A. Modern therapeutic approaches for the treatment of malignant liver tumours. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 755–772. [Google Scholar] [CrossRef]
- Jaeck, D.; Oussoultzoglou, E.; Rosso, E.; Greget, M.; Weber, J.C.; Bachellier, P. A two-stage hepatectomy procedure combined with portal vein embolization to achieve curative resection for initially unresectable multiple and bilobar colorectal liver metastases. Ann. Surg. 2004, 240, 1037–1051. [Google Scholar] [CrossRef]
- Clavien, P.-A.; Petrowsky, H.; DeOliveira, M.L.; Graf, R. Strategies for safer liver surgery and partial liver transplantation. N. Engl. J. Med. 2007, 356, 1545–1559. [Google Scholar] [CrossRef]
- Schnitzbauer, A.A.; Lang, S.A.; Goessmann, H.; Nadalin, S.; Baumgart, J.; Farkas, S.A.; Fichtner-Feigl, S.; Lorf, T.; Goralcyk, A.; Hörbelt, R.; et al. Right portal vein ligation combined with in situ splitting induces rapid left lateral liver lobe hypertrophy enabling 2-staged extended right hepatic resection in small-for-size settings. Ann. Surg. 2012, 255, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Baili, E.; Tsilimigras, D.I.; Filippou, D.; Ioannidis, A.; Bakopoulos, A.; Machairas, N.; Papalampros, A.; Petrou, A.; Schizas, D.; Moris, D. Associating liver partition and portal vein ligation for staged hepatectomy in patients with primary liver malignancies: A systematic review of the literature. J. Balk. Union Oncol. JBUON 2019, 24, 1371–1381. [Google Scholar]
- Wakabayashi, G.; Cherqui, D.; Geller, D.A.; Buell, J.F.; Kaneko, H.; Han, H.S.; Asbun, H.; O’Rourke, N.; Tanabe, M.; Koffron, A.J.; et al. Recommendations for laparoscopic liver resection: A report from the second international consensus conference held in Morioka. Ann. Surg. 2015, 261, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, H.; Sun, M.; He, T.; Liu, Y.; Yang, X.; Shi, X.; Liu, X. Alpinumisoflavone suppresses hepatocellular carcinoma cell growth and metastasis via NLRP3 inflammasome-mediated pyroptosis. Pharmacol. Rep. 2020, 72, 1370–1382. [Google Scholar] [CrossRef]
- Feng, M.; He, S.; Huang, S.; Lin, J.; Yang, H.; Wang, J.; Pang, J. Inhibitory effect of Biejiajian pills against diethylnitrosamine-induced hepatocarcinogenesis in rats. J. South. Med. Univ. 2020, 40, 1148–1154. [Google Scholar] [CrossRef]
- Fan, S.; Wang, Y.; Lu, J.; Zheng, Y.; Wu, D.; Li, M.; Hu, B.; Zhang, Z.; Cheng, W.; Shan, Q. Luteoloside Suppresses Proliferation and Metastasis of Hepatocellular Carcinoma Cells by Inhibition of NLRP3 Inflammasome. PLoS ONE 2014, 9, e89961. [Google Scholar] [CrossRef]
- Shen, Z.; Zhou, H.; Li, A.; Wu, T.; Ji, X.; Guo, L.; Zhu, X.; Zhang, D.; He, X. Metformin inhibits hepatocellular carcinoma development by inducing apoptosis and pyroptosis through regulating FOXO3. Aging 2021, 13, 22120–22133. [Google Scholar] [CrossRef]
- Yabuta, S.; Shidoji, Y. TLR4-mediated pyroptosis in human hepatoma-derived HuH-7 cells induced by a branched-chain polyunsaturated fatty acid, geranylgeranoic acid. Biosci. Rep. 2020, 40, BSR20194118. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Wang, H.; Mao, C.; Sun, M.; Dominah, G.; Chen, L.; Zhuang, Z. Fatty acid oxidation contributes to IL-1β secretion in M2 macrophages and promotes macrophage-mediated tumor cell migration. Mol. Immunol. 2018, 94, 27–35. [Google Scholar] [CrossRef]
- Wei, Q.; Guo, P.; Mu, K.; Zhang, Y.; Zhao, W.; Huai, W.; Qiu, Y.; Li, T.; Ma, X.; Liu, Y.; et al. Estrogen suppresses hepatocellular carcinoma cells through ERβ-mediated upregulation of the NLRP3 inflammasome. Lab. Investig. 2015, 95, 804–816. [Google Scholar] [CrossRef]
- Wei, Q.; Zhu, R.; Zhu, J.; Zhao, R.; Li, M. E2-induced activation of the NLRP3 inflammasome triggers pyroptosis and inhibits autophagy in HCC cells. Oncol. Res. 2019, 27, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wei, T.; Chen, Y.; Yang, L.; Wu, X. Downregulation of irak1 prevents the malignant behavior of hepatocellular carcinoma cells by blocking activation of the mapks/nlrp3/il-1β pathway. OncoTargets Ther. 2020, 13, 12787–12796. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Wei, B.; Lan, T.; Xiao, Y.; Quan, X.; Chen, J.; Zhao, C.; Gao, J. Low NLRP3 expression predicts a better prognosis of colorectal cancer. Biosci. Rep. 2021, 41, BSR20210280. [Google Scholar] [CrossRef] [PubMed]
- Saponaro, C.; Scarpi, E.; Sonnessa, M.; Cioffi, A.; Buccino, F.; Giotta, F.; Pastena, M.I.; Zito, F.A.; Mangia, A. Prognostic Value of NLRP3 Inflammasome and TLR4 Expression in Breast Cancer Patients. Front. Oncol. 2021, 11, 705331. [Google Scholar] [CrossRef]
- Wang, Q.; Lyu, J.; Zhang, W.; Shi, F.; Ren, Y.; Mao, Q.; Liu, Y.; Li, Y.; Wang, S. Immunological and clinical immunotherapy implications of NLRP3 mutations in melanoma. Aging 2021, 13, 24271–24289. [Google Scholar] [CrossRef]
- Wang, J.; Huang, Z.; Lu, H.; Zhang, R.; Feng, Q.; He, A. A Pyroptosis-Related Gene Signature to Predict Patients’ Prognosis and Immune Landscape in Liver Hepatocellular Carcinoma. Comput. Math. Methods Med. 2022, 2022, 1258480. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef] [Green Version]
- Wlodarska, M.; Thaiss, C.A.; Nowarski, R.; Henao-Mejia, J.; Zhang, J.P.; Brown, E.M.; Frankel, G.; Levy, M.; Katz, M.N.; Philbrick, W.M.; et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 2014, 156, 1045–1059. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Drug/Therapeutic Target | Study/Year/Reference | Study Subjects | Pathway | Outcomes |
|---|---|---|---|---|
| Obeticholic acid | Huang S. (2021) [38] | BMDM cells, hepatocytes/DIO + CCl4 mice | Inhibition of NLRP3 inflammasome activation in macrophages | Reduction in steatosis, fibrosis and immune infiltration |
| Inhibition of lipid-induced NLRP3 inflammasome activation in hepatocytes | ||||
| Antcin A | Ruan S. (2021) [39] | KC cells/NAFLD mice | Inhibition of NLRP3 inflammasome activation in vitro/in vitro | Inhibition of immune infiltration |
| Auranofin | Hwangbo H. (2020) [40] | High-fat diet (HFD) NAFLD model | Inhibition of NLRP3 inflammasome, NOX4 and PPARγ activation | Inhibition of immune infiltration |
| Cardiolipin inhibitors (shRNA-CLS1) | Liu J. (2019) [41] | KC cells/methionine choline-deficient (MCD) diet mice | Inhibition of NLRP3 inflammasome activation in vitro/in vitro | Improvement in liver biochemistry |
| Cathepsin B inhibition | Tang Y. (2018) [42] | KC cells/MCD diet NASH mice model | Inhibition of NLRP3 inflammasome activation | Inhibition of immune infiltration and steatosis |
| Polyunsaturated fatty acid (PUFA) | Sui Y. (2016) [43] | HFD NASH mice model | Inhibition of NLRP3 inflammasome activation in vitro and in vivo | |
| Melatonin | Yu Y. (2021) [44] | db/m mice, db/db mice | Improvement in mitochondrial membrane potential (MMP) | Reduction in steatosis, fibrosis and immune infiltration |
| Inhibition in NLRP3 inflammasome activation |
| Drug/Therapeutic Target | Study/Year/Reference | Study Subjects | Pathway/Mechanism | Outcomes |
|---|---|---|---|---|
| Alpinumisoflavone (AIF) | Zhang Y. (2020) [84] | SMMC 7721, Huh7 cells | NLRP3-mediated pyroptosis | Reduction of tumor growth and metastatic potential |
| NEK7 inhibition | Yan Z. (2022) [75] | MHCC97L, HepG2 cell/mice | NLRP3-mediated pyroptosis | Reduction of tumor growth and metastatic potential |
| Promotion of cancer cell-stromal communication | ||||
| Biejiajian pills (BJJ) | Feng M. (2020) [85] | Diethyl nitrosamine-mediated hepatocarcinogenesis in SD rats | Dose-dependent reduction in NLRP3 activation | Reduction of tumor growth |
| Luteoloside | Fan S. (2014) [86] | Hep3B, SNU-449, Huh-7, MHCC- LM3 and MHCC97-H cell lines/BALB/c-nu/nu male mice | Downregulation of NLRP3 activation | Reduction of tumor growth and metastatic potential in vitro and in vivo |
| Metformin | Shen Z. (2021) [87] | BALB/c nude male mice | FOXO3-dependent induction of the NLRP3 inflammasome and autophagy | Reduction of tumor growth |
| Geranylgeranoic acid (GGA) | Yabuta S. (2020) [88] | HuH-7 cells | TLR4-induced ROS generation activating both non-canonical and canonical phases of pyroptosis | Reduction of tumor growth |
| NLRP3 siRNA or CPT1A blockage or N-acetyl cysteine (NAC) or etomoxir | Zhang Q. (2018) [89] | HepG2, Hep3B cells | Reduction in NLRP3 activation by FAO-mediated ROS | Reduction of HCC metastatic potential |
| 17β-estradiol (E2) | Wei Q. (2015) [90] | BEL7402, SMMC7721 and HepG2 cells | ERβ/MAPK/ERK-mediated activation of NLRP3 inflammasome | Reduction of tumor growth |
| 17β-estradiol (E2) | Wei Q. (2019) [91] | HepG2 cells | Autophagy reduction through E2/ERβ/AMPK/mTOR-induced NLRP3 activation | Reduction of tumor growth |
| IRAK1 blockage | Chen W. (2020) [92] | Huh7, Hep3B cells | Downregulation of NLRP3 activation through ERK/JNK pathway | Reduction of tumor growth |
| PPARγ inhibitors or FNDC5 blockage | Liu H. (2021) [71] | HepG2, SMCC7721 cells overexpressing FNDC5 | Activation of the NF-κB/NLRP3 pathway | M1 TAM polarization |
| NLRP3 blockage | Lee H. (2021) [69] | HCC SK-Hep1 Luc, NK-92 cells | Upregulation of MICA/B on the HCC cells induced by NK activation through NKG2D receptor | Reduction in tumor growth and metastasis |
| RIPK3 mimic or FAO blockage | Wu L. (2020) [73] | Human HCC tissues | Activation of the ROS–Caspase1–PPAR pathway reversed M2 programming | Reduction in tumor growth |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papadakos, S.P.; Dedes, N.; Kouroumalis, E.; Theocharis, S. The Role of the NLRP3 Inflammasome in HCC Carcinogenesis and Treatment: Harnessing Innate Immunity. Cancers 2022, 14, 3150. https://doi.org/10.3390/cancers14133150
Papadakos SP, Dedes N, Kouroumalis E, Theocharis S. The Role of the NLRP3 Inflammasome in HCC Carcinogenesis and Treatment: Harnessing Innate Immunity. Cancers. 2022; 14(13):3150. https://doi.org/10.3390/cancers14133150
Chicago/Turabian StylePapadakos, Stavros P., Nikolaos Dedes, Elias Kouroumalis, and Stamatios Theocharis. 2022. "The Role of the NLRP3 Inflammasome in HCC Carcinogenesis and Treatment: Harnessing Innate Immunity" Cancers 14, no. 13: 3150. https://doi.org/10.3390/cancers14133150
APA StylePapadakos, S. P., Dedes, N., Kouroumalis, E., & Theocharis, S. (2022). The Role of the NLRP3 Inflammasome in HCC Carcinogenesis and Treatment: Harnessing Innate Immunity. Cancers, 14(13), 3150. https://doi.org/10.3390/cancers14133150