
LncRNA ZNF582-AS1 Expression and Methylation in Breast Cancer and Its Biological and Clinical Implications

 ,
, 
 ,
,
Abstract
Simple Summary
Abstract
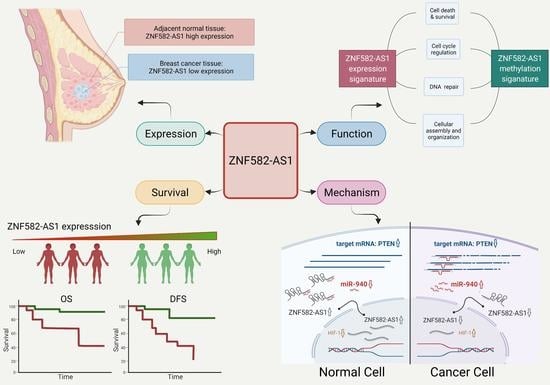
1. Introduction
2. Materials and Methods
2.1. Breast Cancer Patients and Tumor Samples in Our Study
2.2. RNA Extraction and ZNF582-AS1 Measurement
2.3. The Cancer Genome Atlas (TCGA) Data
2.4. Gene Expression Omnibus (GEO) Data
2.5. Meta-Analysis of ZNF582-AS1 Expression in Association with Survival
2.6. In Silico Prediction of ZNF582-AS1 Function and Regulation
2.7. Statistical Analysis
3. Results
3.1. ZNF582-AS1 Expression in Breast Cancer
3.2. ZNF582-AS1 Expression and Patient Characteristics
3.3. ZNF582-AS1 Expression and Breast Cancer Survival
3.4. Meta-Analysis of ZNF582-AS1 in Association with Survival
3.5. DNA Methylation and ZNF582-AS1 Expression
3.6. Bioinformatic Interrogation of ZNF582-AS1 Expression and Methylation Signatures
3.7. ZNF582-AS1 and hsa-miR-940
3.8. Regulation of ZNF582-AS1 Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S. Initial impact of the sequencing of the human genome. Nature 2011, 470, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Kapranov, P.; Willingham, A.T.; Gingeras, T.R. Genome-wide transcription and the implications for genomic organization. Nat. Rev. Genet. 2007, 8, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.; Chai, P.; Yu, J.; Xing, Y.; Wang, S.; Fan, J.; Ge, S.; Wang, Y.; Jia, R.; Fan, X. LncRNA CANT1 suppresses retinoblastoma progression by repellinghistone methyltransferase in PI3Kgamma promoter. Cell Death Dis. 2020, 11, 306. [Google Scholar] [CrossRef]
- Bhan, A.; Soleimani, M.; Mandal, S.S. Long Noncoding RNA and Cancer: A New Paradigm. Cancer Res. 2017, 77, 3965–3981. [Google Scholar] [CrossRef]
- Li, J.; Han, L.; Roebuck, P.; Diao, L.; Liu, L.; Yuan, Y.; Weinstein, J.N.; Liang, H. TANRIC: An Interactive Open Platform to Explore the Function of lncRNAs in Cancer. Cancer Res. 2015, 75, 3728–3737. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Jensen, M.A.; Ferretti, V.; Grossman, R.L.; Staudt, L.M. The NCI Genomic Data Commons as an engine for precision medicine. Blood 2017, 130, 453–459. [Google Scholar] [CrossRef]
- Diez-Villanueva, A.; Mallona, I.; Peinado, M.A. Wanderer, an interactive viewer to explore DNA methylation and gene expression data in human cancer. Epigenetics Chromatin 2015, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Pawitan, Y.; Bjohle, J.; Amler, L.; Borg, A.L.; Egyhazi, S.; Hall, P.; Han, X.; Holmberg, L.; Huang, F.; Klaar, S.; et al. Gene expression profiling spares early breast cancer patients from adjuvant therapy: Derived and validated in two population-based cohorts. Breast Cancer Res. 2005, 7, R953–R964. [Google Scholar] [CrossRef] [PubMed]
- Haibe-Kains, B.; Desmedt, C.; Di Leo, A.; Azambuja, E.; Larsimont, D.; Selleslags, J.; Delaloge, S.; Duhem, C.; Kains, J.P.; Carly, B.; et al. Genome-wide gene expression profiling to predict resistance to anthracyclines in breast cancer patients. Genom. Data 2013, 1, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zou, L.; Li, Q.; Haibe-Kains, B.; Tian, R.; Li, Y.; Desmedt, C.; Sotiriou, C.; Szallasi, Z.; Iglehart, J.D.; et al. Amplification of LAPTM4B and YWHAZ contributes to chemotherapy resistance and recurrence of breast cancer. Nat. Med. 2010, 16, 214–218. [Google Scholar] [CrossRef]
- Kao, K.J.; Chang, K.M.; Hsu, H.C.; Huang, A.T. Correlation of microarray-based breast cancer molecular subtypes and clinical outcomes: Implications for treatment optimization. BMC Cancer 2011, 11, 143. [Google Scholar] [CrossRef]
- Sabatier, R.; Finetti, P.; Adelaide, J.; Guille, A.; Borg, J.P.; Chaffanet, M.; Lane, L.; Birnbaum, D.; Bertucci, F. Down-regulation of ECRG4, a candidate tumor suppressor gene, in human breast cancer. PLoS ONE 2011, 6, e27656. [Google Scholar] [CrossRef]
- Clarke, C.; Madden, S.F.; Doolan, P.; Aherne, S.T.; Joyce, H.; O’Driscoll, L.; Gallagher, W.M.; Hennessy, B.T.; Moriarty, M.; Crown, J.; et al. Correlating transcriptional networks to breast cancer survival: A large-scale coexpression analysis. Carcinogenesis 2013, 34, 2300–2308. [Google Scholar] [CrossRef]
- Ivshina, A.V.; George, J.; Senko, O.; Mow, B.; Putti, T.C.; Smeds, J.; Lindahl, T.; Pawitan, Y.; Hall, P.; Nordgren, H.; et al. Genetic reclassification of histologic grade delineates new clinical subtypes of breast cancer. Cancer Res. 2006, 66, 10292–10301. [Google Scholar] [CrossRef]
- Metzger-Filho, O.; Michiels, S.; Bertucci, F.; Catteau, A.; Salgado, R.; Galant, C.; Fumagalli, D.; Singhal, S.K.; Desmedt, C.; Ignatiadis, M.; et al. Genomic grade adds prognostic value in invasive lobular carcinoma. Ann. Oncol. 2013, 24, 377–384. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2012, 41, D991–D995. [Google Scholar] [CrossRef]
- Kramer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.R.; Liu, S.; Sun, W.J.; Zheng, L.L.; Zhou, H.; Yang, J.H.; Qu, L.H. ChIPBase v2.0: Decoding transcriptional regulatory networks of non-coding RNAs and protein-coding genes from ChIP-seq data. Nucleic Acids Res. 2017, 45, D43–D50. [Google Scholar] [CrossRef] [PubMed]
- Paraskevopoulou, M.D.; Vlachos, I.S.; Karagkouni, D.; Georgakilas, G.; Kanellos, I.; Vergoulis, T.; Zagganas, K.; Tsanakas, P.; Floros, E.; Dalamagas, T.; et al. DIANA-LncBase v2: Indexing microRNA targets on non-coding transcripts. Nucleic Acids Res. 2016, 44, D231–D238. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Kumegawa, K.; Maruyama, R.; Yamamoto, E.; Ashida, M.; Kitajima, H.; Tsuyada, A.; Niinuma, T.; Kai, M.; Yamano, H.O.; Sugai, T.; et al. A genomic screen for long noncoding RNA genes epigenetically silenced by aberrant DNA methylation in colorectal cancer. Sci. Rep. 2016, 6, 26699. [Google Scholar] [CrossRef]
- Cheng, G.; Liu, D.; Liang, H.; Yang, H.; Chen, K.; Zhang, X. A cluster of long non-coding RNAs exhibit diagnostic and prognostic values in renal cell carcinoma. Aging 2019, 11, 9597–9615. [Google Scholar] [CrossRef]
- Yuan, L.Y.; Qin, X.; Li, L.; Zhou, J.; Zhou, M.; Li, X.; Xu, Y.; Wang, X.J.; Xing, H. The transcriptome profiles and methylation status revealed the potential cancer-related lncRNAs in patients with cervical cancer. J. Cell. Physiol. 2019, 234, 9756–9763. [Google Scholar] [CrossRef]
- Yang, W.; Zhang, K.; Li, L.; Xu, Y.; Ma, K.; Xie, H.; Zhou, J.; Cai, L.; Gong, Y.; Gong, K. Downregulation of lncRNA ZNF582-AS1 due to DNA hypermethylation promotes clear cell renal cell carcinoma growth and metastasis by regulating the N(6)-methyladenosine modification of MT-RNR1. J. Exp. Clin. Cancer Res. 2021, 40, 92. [Google Scholar] [CrossRef]
- Cheng, S.J.; Chang, C.F.; Lee, J.J.; Chen, H.M.; Wang, H.J.; Liou, Y.L.; Yen, C.; Chiang, C.P. Hypermethylated ZNF582 and PAX1 are effective biomarkers for detection of oral dysplasia and oral cancer. Oral. Oncol. 2016, 62, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Juan, Y.C.; Su, Y.F.; Zhang, W.B.; Yu, Y.; Yang, H.Y.; Yu, G.Y.; Peng, X. Hypermethylated PAX1 and ZNF582 genes in the tissue sample are associated with aggressive progression of oral squamous cell carcinoma. J. Oral. Pathol. Med. 2020, 49, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Park, S.L.; Li, Y.; Sheng, X.; Hom, V.; Xia, L.; Zhao, K.; Pooler, L.; Setiawan, V.W.; Lim, U.; Monroe, K.R.; et al. Genome-Wide Association Study of Liver Fat: The Multiethnic Cohort Adiposity Phenotype Study. Hepatol. Commun. 2020, 4, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, G.; Tang, J.; Zhuang, W.; Wang, L.P.; Liou, Y.L.; Liu, Y.Z.; Zhou, H.H.; Zhu, Y.S. DNA Methylation Status of PAX1 and ZNF582 in Esophageal Squamous Cell Carcinoma. Int. J. Env. Res. Public Health 2017, 14, 216. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Liou, Y.L.; Wan, Z.R.; Tang, J.; Zhou, Y.; Zhuang, W.; Wang, G. Aberrant DNA methylation of PAX1, SOX1 and ZNF582 genes as potential biomarkers for esophageal squamous cell carcinoma. Biomed. Pharmacother. 2019, 120, 109488. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.Y.; Zhang, X.; Chu, T.; Zhu, S.; Deng, Y.; Zhou, Y.; Wang, Y.; Zhao, X.; Liu, L.; Fang, C.; et al. High methylation of ZNF582 in cervical adenocarcinoma affects radiosensitivity and prognosis. Ann. Transl. Med. 2019, 7, 328. [Google Scholar] [CrossRef]
- Harty, P.S.; Sieglinger, B.; Heymsfield, S.B.; Shepherd, J.A.; Bruner, D.; Stratton, M.T.; Tinsley, G.M. Novel body fat estimation using machine learning and 3-dimensional optical imaging. Eur. J. Clin. Nutr. 2020, 74, 842–845. [Google Scholar] [CrossRef]
- Mitri, Z.; Constantine, T.; O’Regan, R. The HER2 Receptor in Breast Cancer: Pathophysiology, Clinical Use, and New Advances in Therapy. Chemother. Res. Pract. 2012, 2012, 743193. [Google Scholar] [CrossRef]
- Park, S.Y.; Boushey, C.J.; Wilkens, L.R.; Haiman, C.A.; Le Marchand, L. High-Quality Diets Associate With Reduced Risk of Colorectal Cancer: Analyses of Diet Quality Indexes in the Multiethnic Cohort. Gastroenterology 2017, 153, 386–394.e2. [Google Scholar] [CrossRef]
- Salomon, D.S.; Normanno, N.; Ciardiello, F.; Brandt, R.; Shoyab, M.; Todaro, G.J. The role of amphiregulin in breast cancer. Breast Cancer Res. Treat. 1995, 33, 103–114. [Google Scholar] [CrossRef]
- O’Regan, R.M.; Nahta, R. Targeting forkhead box M1 transcription factor in breast cancer. Biochem. Pharmacol. 2018, 154, 407–413. [Google Scholar] [CrossRef]
- Myatt, S.S.; Lam, E.W. The emerging roles of forkhead box (Fox) proteins in cancer. Nat. Rev. Cancer 2007, 7, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Koo, C.Y.; Muir, K.W.; Lam, E.W. FOXM1: From cancer initiation to progression and treatment. Biochim. Biophys. Acta 2012, 1819, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Francis, R.E.; Myatt, S.S.; Krol, J.; Hartman, J.; Peck, B.; McGovern, U.B.; Wang, J.; Guest, S.K.; Filipovic, A.; Gojis, O.; et al. FoxM1 is a downstream target and marker of HER2 overexpression in breast cancer. Int. J. Oncol. 2009, 35, 57–68. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, Y.Y.; Fu, S.; Wang, X.P.; Wang, H.Y.; Zeng, M.S.; Shao, J.Y. Down-regulation of c9orf86 in human breast cancer cells inhibits cell proliferation, invasion and tumor growth and correlates with survival of breast cancer patients. PLoS ONE 2013, 8, e71764. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, J.; Wang, W. Construction and investigation of breast-cancer-specific ceRNA network based on the mRNA and miRNA expression data. IET Syst. Biol. 2014, 8, 96–103. [Google Scholar] [CrossRef]
- Paci, P.; Colombo, T.; Farina, L. Computational analysis identifies a sponge interaction network between long non-coding RNAs and messenger RNAs in human breast cancer. BMC Syst. Biol. 2014, 8, 83. [Google Scholar] [CrossRef]
- Zhang, H.; Peng, J.; Lai, J.; Liu, H.; Zhang, Z.; Li, X.; Liang, B.; Chen, X.; Zou, B.; Lin, S.; et al. MiR-940 promotes malignant progression of breast cancer by regulating FOXO3. Biosci. Rep. 2020, 40, BSR20201337. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Maggiolini, M.; Musti, A.M. Crosstalk between Notch, HIF-1alpha and GPER in Breast Cancer EMT. Int. J. Mol. Sci. 2018, 19, 2011. [Google Scholar] [CrossRef]
- Das, J.K.; Felty, Q.; Poppiti, R.; Jackson, R.M.; Roy, D. Nuclear Respiratory Factor 1 Acting as an Oncoprotein Drives Estrogen-Induced Breast Carcinogenesis. Cells 2018, 7, 234. [Google Scholar] [CrossRef]
- Tharakaraman, K.; Bodenreider, O.; Landsman, D.; Spouge, J.L.; Marino-Ramirez, L. The biological function of some human transcription factor binding motifs varies with position relative to the transcription start site. Nucleic Acids Res. 2008, 36, 2777–2786. [Google Scholar] [CrossRef]
- Tabach, Y.; Brosh, R.; Buganim, Y.; Reiner, A.; Zuk, O.; Yitzhaky, A.; Koudritsky, M.; Rotter, V.; Domany, E. Wide-scale analysis of human functional transcription factor binding reveals a strong bias towards the transcription start site. PLoS ONE 2007, 2, e807. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, T.W.; Wang, J.; Collins, P.J.; Partridge, E.C.; Aldred, S.F.; Trinklein, N.D.; Myers, R.M.; Weng, Z. Functional analysis of transcription factor binding sites in human promoters. Genome Biol. 2012, 13, R50. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ZNF582-AS1 Expression | |||||
|---|---|---|---|---|---|
| Variables | Total (n = 361) No. (%) | Low No. (%) | Mid No. (%) | High No. (%) | p-Value |
| Age | |||||
| Age < 58.14 | 181 (50.14) | 61 (33.70) | 59 (32.60) | 61 (33.70) | 0.965 |
| Age ≥ 58.14 | 180 (49.86) | 60 (33.33) | 61 (33.89) | 59 (32.78) | |
| Disease stage | |||||
| Stage I | 114 (33.33) | 28 (24.56) | 39 (34.21) | 47 (41.23) | 0.055 |
| Stage II | 174 (50.88) | 59 (33.91) | 59 (33.91) | 56 (32.18) | |
| Stage III and IV | 54 (15.79) | 27 (50.00) | 16 (29.63) | 11 (20.37) | |
| Tumor grade | |||||
| Grade 1 | 40 (11.33) | 8 (20.00) | 11 (27.50) | 21 (52.50) | 5.86 × 10−5 |
| Grade 2 | 135 (38.24) | 30 (22.22) | 47 (34.81) | 58 (42.96) | |
| Grade 3 | 178 (50.42) | 80 (44.94) | 58 (32.58) | 40 (22.47) | |
| ER status | |||||
| Positive | 245 (68.63) | 68 (27.76) | 85 (34.69) | 92 (37.55) | 0.003 |
| Negative | 112 (31.37) | 53 (47.32) | 32 (28.57) | 27 (24.11) | |
| PR status | |||||
| Positive | 212 (59.38) | 60 (28.30) | 73 (34.43) | 79 (37.26) | 0.053 |
| Negative | 145 (40.62) | 61 (42.07) | 44 (30.34) | 40 (27.59) | |
| ZNF582-AS1 Expression | Relapse | Death | ||||
|---|---|---|---|---|---|---|
| HR | 95%CI | p-Value | HR | 95%CI | p-Value | |
| Univariate analysis | ||||||
| Low | 1 | 1 | ||||
| Mid | 0.70 | 0.43–1.13 | 0.143 | 0.88 | 0.51–1.51 | 0.646 |
| High | 0.34 | 0.19–0.61 | <0.001 | 0.36 | 0.18–0.73 | 0.005 |
| Multivariate analysis * | ||||||
| Low | 1 | 1 | ||||
| Mid | 0.84 | 0.49–1.42 | 0.507 | 1.15 | 0.62–2.14 | 0.646 |
| High | 0.42 | 0.21–0.61 | 0.012 | 0.71 | 0.33–1.52 | 0.373 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Katsaros, D.; Biglia, N.; Fu, Y.; Benedetto, C.; Loo, L.; Wang, Z.; Yu, H. LncRNA ZNF582-AS1 Expression and Methylation in Breast Cancer and Its Biological and Clinical Implications. Cancers 2022, 14, 2788. https://doi.org/10.3390/cancers14112788
Wang J, Katsaros D, Biglia N, Fu Y, Benedetto C, Loo L, Wang Z, Yu H. LncRNA ZNF582-AS1 Expression and Methylation in Breast Cancer and Its Biological and Clinical Implications. Cancers. 2022; 14(11):2788. https://doi.org/10.3390/cancers14112788
Chicago/Turabian StyleWang, Junlong, Dionyssios Katsaros, Nicoletta Biglia, Yuanyuan Fu, Chiara Benedetto, Lenora Loo, Zhanwei Wang, and Herbert Yu. 2022. "LncRNA ZNF582-AS1 Expression and Methylation in Breast Cancer and Its Biological and Clinical Implications" Cancers 14, no. 11: 2788. https://doi.org/10.3390/cancers14112788
APA StyleWang, J., Katsaros, D., Biglia, N., Fu, Y., Benedetto, C., Loo, L., Wang, Z., & Yu, H. (2022). LncRNA ZNF582-AS1 Expression and Methylation in Breast Cancer and Its Biological and Clinical Implications. Cancers, 14(11), 2788. https://doi.org/10.3390/cancers14112788