
Crosstalk of Epigenetic and Metabolic Signaling Underpinning Glioblastoma Pathogenesis

 ,
,  ,
,  and
and 
Abstract
Simple Summary
Abstract
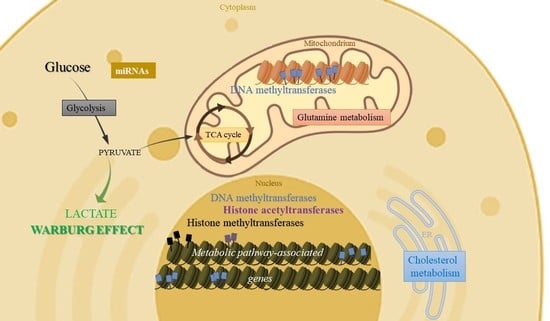
1. Introduction
2. Metabolic Pathways Regulated by Epigenetic Mechanisms in Glioblastomas
2.1. Epigenetic Regulation of Glycolysis
2.1.1. Effects of DNA Methylation in Glycolysis
2.1.2. Effects of Histone Methyltransferases and Demethylases in Glycolysis
2.1.3. Effects of Histone Acetyltransferases and Deacetylases in Glycolysis
2.1.4. Noncoding RNA Effects in Glycolysis
2.2. Epigenetic Regulation of Pentose Phosphate Pathway
2.3. Epigenetic Regulation of Gluconeogenesis
2.4. Epigenetic Regulation of TCA Cycle
2.4.1. DNA Methylation Effects in the TCA Cycle
2.4.2. Histone Methylation Effects in the TCA Cycle
2.5. Epigenetic Regulation of Oxidative Phosphorylation (OXPHOS)
2.5.1. DNA Methylation Effects in OXPHOS
2.5.2. Histone Methylation Effects in OXPHOS
2.5.3. Noncoding RNAs Effects in OXPHOS
2.6. Epigenetic Regulation of Lipid Metabolism
microRNA Effects in Lipid Metabolism
2.7. Epigenetic Regulation of Glutamine Metabolism
3. Therapeutic Targeting Options
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Stetson, L.; Virk, S.M.; Barnholtz-Sloan, J.S. Epidemiology of gliomas. Cancer Treat. Res. 2015, 163, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro. Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Kanderi, T.; Gupta, V. Glioblastoma Multiforme; StatPearls Publishing: Treasure Island, UK, 2021. [Google Scholar]
- Ostrom, Q.T.; Gittleman, H.; Xu, J.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2009–2013. Neuro. Oncol. 2016, 18, v1–v75. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.-A.; Jones, D.T.W.; Konermann, C.; Pfaff, E.; Tönjes, M.; Sill, M.; Bender, S.; et al. Hotspot Mutations in H3F3A and IDH1 Define Distinct Epigenetic and Biological Subgroups of Glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef]
- Ohgaki, H.; Kleihues, P. The Definition of Primary and Secondary Glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef]
- Ho, P.-C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.-C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef]
- Zhu, X.; Xuan, Z.; Chen, J.; Li, Z.; Zheng, S.; Song, P. How DNA methylation affects the Warburg effect. Int. J. Biol. Sci. 2020, 16, 2029–2041. [Google Scholar] [CrossRef]
- Gimple, R.C.; Bhargava, S.; Dixit, D.; Rich, J.N. Glioblastoma stem cells: Lessons from the tumor hierarchy in a lethal cancer. Genes Dev. 2019, 33, 591–609. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Rea, S.; Eisenhaber, F.; O’Carroll, D.; Strahl, B.D.; Sun, Z.W.; Schmid, M.; Opravil, S.; Mechtler, K.; Ponting, C.P.; Allis, C.D.; et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 2000, 406, 593–599. [Google Scholar] [CrossRef]
- Feng, Q.; Wang, H.; Ng, H.H.; Erdjument-Bromage, H.; Tempst, P.; Struhl, K.; Zhang, Y. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr. Biol. 2002, 12, 1052–1058. [Google Scholar] [CrossRef]
- Paik, W.K.; Paik, D.C.; Kim, S. Historical review: The field of protein methylation. Trends Biochem. Sci. 2007, 32, 146–152. [Google Scholar] [CrossRef]
- Biggar, K.K.; Li, S.S.-C. Non-histone protein methylation as a regulator of cellular signalling and function. Nat. Rev. Mol. Cell Biol. 2015, 16, 5–17. [Google Scholar] [CrossRef]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Malecki, J.; Aileni, V.K.; Ho, A.Y.Y.; Schwarz, J.; Moen, A.; Sørensen, V.; Nilges, B.S.; Jakobsson, M.E.; Leidel, S.A.; Falnes, P.Ø. The novel lysine specific methyltransferase METTL21B affects mRNA translation through inducible and dynamic methylation of Lys-165 in human eukaryotic elongation factor 1 alpha (eEF1A). Nucleic Acids Res. 2017, 45, 4370–4389. [Google Scholar] [CrossRef]
- Di Blasi, R.; Blyuss, O.; Timms, J.F.; Conole, D.; Ceroni, F.; Whitwell, H.J. Non-Histone Protein Methylation: Biological Significance and Bioengineering Potential. ACS Chem. Biol. 2021, 16, 238–250. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Strickland, M.; Stoll, E.A. Metabolic Reprogramming in Glioma. Front. Cell Dev. Biol. 2017, 5, 43. [Google Scholar] [CrossRef]
- Epstein, T.; Xu, L.; Gillies, R.J.; Gatenby, R.A. Separation of metabolic supply and demand: Aerobic glycolysis as a normal physiological response to fluctuating energetic demands in the membrane. Cancer Metab. 2014, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. Links between metabolism and cancer. Genes Dev. 2012, 26, 877–890. [Google Scholar] [CrossRef] [PubMed]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef]
- Kant, S.; Kesarwani, P.; Prabhu, A.; Graham, S.F.; Buelow, K.L.; Nakano, I.; Chinnaiyan, P. Enhanced fatty acid oxidation provides glioblastoma cells metabolic plasticity to accommodate to its dynamic nutrient microenvironment. Cell Death Dis. 2020, 11, 253. [Google Scholar] [CrossRef]
- Kanwore, K.; Kanwore, K.; Adzika, G.K.; Abiola, A.A.; Guo, X.; Kambey, P.A.; Xia, Y.; Gao, D. Cancer Metabolism: The Role of Immune Cells Epigenetic Alteration in Tumorigenesis, Progression, and Metastasis of Glioma. Front. Immunol. 2022, 13, 831636. [Google Scholar] [CrossRef]
- Cheng, S.-C.; Quintin, J.; Cramer, R.A.; Shepardson, K.M.; Saeed, S.; Kumar, V.; Giamarellos-Bourboulis, E.J.; Martens, J.H.A.; Rao, N.A.; Aghajanirefah, A.; et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 2014, 345, 1250684. [Google Scholar] [CrossRef]
- Lopez-Serra, P.; Marcilla, M.; Villanueva, A.; Ramos-Fernandez, A.; Palau, A.; Leal, L.; Wahi, J.E.; Setien-Baranda, F.; Szczesna, K.; Moutinho, C.; et al. A DERL3-associated defect in the degradation of SLC2A1 mediates the Warburg effect. Nat. Commun. 2014, 5, 3608. [Google Scholar] [CrossRef]
- Ha, T.-K.; Her, N.-G.; Lee, M.-G.; Ryu, B.-K.; Lee, J.-H.; Han, J.; Jeong, S.-I.; Kang, M.-J.; Kim, N.-H.; Kim, H.-J.; et al. Caveolin-1 increases aerobic glycolysis in colorectal cancers by stimulating HMGA1-mediated GLUT3 transcription. Cancer Res. 2012, 72, 4097–4109. [Google Scholar] [CrossRef]
- Dong, Z.; Cui, H. Epigenetic modulation of metabolism in glioblastoma. Semin. Cancer Biol. 2019, 57, 45–51. [Google Scholar] [CrossRef]
- Singh, S.; Narayanan, S.P.; Biswas, K.; Gupta, A.; Ahuja, N.; Yadav, S.; Panday, R.K.; Samaiya, A.; Sharan, S.K.; Shukla, S. Intragenic DNA methylation and BORIS-mediated cancer-specific splicing contribute to the Warburg effect. Proc. Natl. Acad. Sci. USA 2017, 114, 11440–11445. [Google Scholar] [CrossRef]
- Thakur, C.; Chen, F. Connections between metabolism and epigenetics in cancers. Semin. Cancer Biol. 2019, 57, 52–58. [Google Scholar] [CrossRef]
- Wolf, A.; Agnihotri, S.; Munoz, D.; Guha, A. Developmental profile and regulation of the glycolytic enzyme hexokinase 2 in normal brain and glioblastoma multiforme. Neurobiol. Dis. 2011, 44, 84–91. [Google Scholar] [CrossRef]
- Chen, C.; Shi, Y.; Li, Y.; He, Z.-C.; Zhou, K.; Zhang, X.-N.; Yang, K.-D.; Wu, J.-R.; Kung, H.-F.; Ping, Y.-F.; et al. A glycolysis-based ten-gene signature correlates with the clinical outcome, molecular subtype and IDH1 mutation in glioblastoma. J. Genet. Genom. 2017, 44, 519–530. [Google Scholar] [CrossRef]
- Pang, B.; Zheng, X.-R.; Tian, J.-X.; Gao, T.-H.; Gu, G.-Y.; Zhang, R.; Fu, Y.-B.; Pang, Q.; Li, X.-G.; Liu, Q. EZH2 promotes metabolic reprogramming in glioblastomas through epigenetic repression of EAF2-HIF1α signaling. Oncotarget 2016, 7, 45134–45143. [Google Scholar] [CrossRef]
- Bao, L.; Chen, Y.; Lai, H.-T.; Wu, S.-Y.; Wang, J.E.; Hatanpaa, K.J.; Raisanen, J.M.; Fontenot, M.; Lega, B.; Chiang, C.-M.; et al. Methylation of hypoxia-inducible factor (HIF)-1α by G9a/GLP inhibits HIF-1 transcriptional activity and cell migration. Nucleic Acids Res. 2018, 46, 6576–6591. [Google Scholar] [CrossRef]
- Heddleston, J.M.; Wu, Q.; Rivera, M.; Minhas, S.; Lathia, J.D.; Sloan, A.E.; Iliopoulos, O.; Hjelmeland, A.B.; Rich, J.N. Hypoxia-induced mixed-lineage leukemia 1 regulates glioma stem cell tumorigenic potential. Cell Death Differ. 2012, 19, 428–439. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Yang, H.; Zhao, X.; Zhao, L.; Liu, L.; Li, J.; Jia, W.; Liu, J.; Huang, G. PRMT5 competitively binds to CDK4 to promote G1-S transition upon glucose induction in hepatocellular carcinoma. Oncotarget 2016, 7, 72131–72147. [Google Scholar] [CrossRef]
- Tsai, W.-W.; Niessen, S.; Goebel, N.; Yates, J.R., 3rd; Guccione, E.; Montminy, M. PRMT5 modulates the metabolic response to fasting signals. Proc. Natl. Acad. Sci. USA 2013, 110, 8870–8875. [Google Scholar] [CrossRef]
- Hu, J.; Sun, T.; Wang, H.; Chen, Z.; Wang, S.; Yuan, L.; Liu, T.; Li, H.-R.; Wang, P.; Feng, Y.; et al. MiR-215 Is Induced Post-transcriptionally via HIF-Drosha Complex and Mediates Glioma-Initiating Cell Adaptation to Hypoxia by Targeting KDM1B. Cancer Cell 2016, 29, 49–60. [Google Scholar] [CrossRef]
- Zhang, S.; Sun, G.; Wang, Z.; Wan, Y.; Guo, J.; Shi, L. PCAF-mediated Akt1 acetylation enhances the proliferation of human glioblastoma cells. Tumor Biol. 2015, 36, 1455–1462. [Google Scholar] [CrossRef]
- Lv, D.; Jia, F.; Hou, Y.; Sang, Y.; Alvarez, A.A.; Zhang, W.; Gao, W.-Q.; Hu, B.; Cheng, S.-Y.; Ge, J.; et al. Histone Acetyltransferase KAT6A Upregulates PI3K/AKT Signaling through TRIM24 Binding. Cancer Res. 2017, 77, 6190–6201. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Masui, K.; Tanaka, K.; Akhavan, D.; Babic, I.; Gini, B.; Matsutani, T.; Iwanami, A.; Liu, F.; Villa, G.R.; Gu, Y.; et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab. 2013, 18, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Xia, Y.; Cao, Y.; Zheng, Y.; Bu, W.; Zhang, L.; You, M.J.; Koh, M.Y.; Cote, G.; Aldape, K.; et al. EGFR-induced and PKCε monoubiquitylation-dependent NF-κB activation upregulates PKM2 expression and promotes tumorigenesis. Mol. Cell 2012, 48, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Sheikh, T.; Sen, E. SIRT6 regulated nucleosomal occupancy affects Hexokinase 2 expression. Exp. Cell Res. 2017, 357, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Yin, N.; Chhangawala, S.; Xu, K.; Leslie, C.S.; Li, M.O. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 2016, 354, 481–484. [Google Scholar] [CrossRef]
- Liu, P.-S.; Ho, P.-C. Mitochondria: A master regulator in macrophage and T cell immunity. Mitochondrion 2018, 41, 45–50. [Google Scholar] [CrossRef]
- Guan, Y.; Cao, Z.; Du, J.; Liu, T.; Wang, T. Circular RNA circPITX1 knockdown inhibits glycolysis to enhance radiosensitivity of glioma cells by miR-329-3p/NEK2 axis. Cancer Cell Int. 2020, 20, 80. [Google Scholar] [CrossRef]
- Wang, J.; Liu, X.; Yan, C.; Liu, J.; Wang, S.; Hong, Y.; Gu, A.; Zhao, P. LEF1-AS1, a long-noncoding RNA, promotes malignancy in glioblastoma. Onco Targets Ther. 2017, 10, 4251–4260. [Google Scholar] [CrossRef]
- Dai, D.-W.; Lu, Q.; Wang, L.-X.; Zhao, W.-Y.; Cao, Y.-Q.; Li, Y.-N.; Han, G.-S.; Liu, J.-M.; Yue, Z.-J. Decreased miR-106a inhibits glioma cell glucose uptake and proliferation by targeting SLC2A3 in GBM. BMC Cancer 2013, 13, 478. [Google Scholar] [CrossRef]
- Zhao, S.; Liu, H.; Liu, Y.; Wu, J.; Wang, C.; Hou, X.; Chen, X.; Yang, G.; Zhao, L.; Che, H.; et al. miR-143 inhibits glycolysis and depletes stemness of glioblastoma stem-like cells. Cancer Lett. 2013, 333, 253–260. [Google Scholar] [CrossRef]
- Kefas, B.; Comeau, L.; Erdle, N.; Montgomery, E.; Amos, S.; Purow, B. Pyruvate kinase M2 is a target of the tumor-suppressive microRNA-326 and regulates the survival of glioma cells. Neuro. Oncol. 2010, 12, 1102–1112. [Google Scholar] [CrossRef]
- Luan, W.; Wang, Y.; Chen, X.; Shi, Y.; Wang, J.; Zhang, J.; Qian, J.; Li, R.; Tao, T.; Wei, W.; et al. PKM2 promotes glucose metabolism and cell growth in gliomas through a mechanism involving a let-7a/c-Myc/hnRNPA1 feedback loop. Oncotarget 2015, 6, 13006–13018. [Google Scholar] [CrossRef]
- Rao, S.A.M.; Santosh, V.; Somasundaram, K. Genome-wide expression profiling identifies deregulated miRNAs in malignant astrocytoma. Mod. Pathol. 2010, 23, 1404–1417. [Google Scholar] [CrossRef]
- Rao, S.A.M.; Arimappamagan, A.; Pandey, P.; Santosh, V.; Hegde, A.S.; Chandramouli, B.A.; Somasundaram, K. miR-219-5p Inhibits Receptor Tyrosine Kinase Pathway by Targeting EGFR in Glioblastoma. PLoS ONE 2013, 8, e63164. [Google Scholar] [CrossRef]
- Kefas, B.; Godlewski, J.; Comeau, L.; Li, Y.; Abounader, R.; Hawkinson, M.; Lee, J.; Fine, H.; Chiocca, E.A.; Lawler, S.; et al. microRNA-7 inhibits the epidermal growth factor receptor and the Akt pathway and is down-regulated in glioblastoma. Cancer Res. 2008, 68, 3566–3572. [Google Scholar] [CrossRef]
- Papagiannakopoulos, T.; Friedmann-Morvinski, D.; Neveu, P.; Dugas, J.C.; Gill, R.M.; Huillard, E.; Liu, C.; Zong, H.; Rowitch, D.H.; Barres, B.A.; et al. Pro-neural miR-128 is a glioma tumor suppressor that targets mitogenic kinases. Oncogene 2012, 31, 1884–1895. [Google Scholar] [CrossRef]
- Chan, J.A.; Krichevsky, A.M.; Kosik, K.S. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005, 65, 6029–6033. [Google Scholar] [CrossRef]
- Zhou, X.; Ren, Y.; Moore, L.; Mei, M.; You, Y.; Xu, P.; Wang, B.; Wang, G.; Jia, Z.; Pu, P.; et al. Downregulation of miR-21 inhibits EGFR pathway and suppresses the growth of human glioblastoma cells independent of PTEN status. Lab. Investig. 2010, 90, 144–155. [Google Scholar] [CrossRef]
- Masui, K.; Harachi, M.; Cavenee, W.K.; Mischel, P.S.; Shibata, N. Codependency of Metabolism and Epigenetics Drives Cancer Progression: A Review. Acta Histochem. Cytochem. 2020, 53, 1–10. [Google Scholar] [CrossRef]
- Lamb, R.F. Amino acid sensing mechanisms: An Achilles heel in cancer? FEBS J. 2012, 279, 2624–2631. [Google Scholar] [CrossRef]
- Masui, K.; Shibata, N.; Cavenee, W.K.; Mischel, P.S. mTORC2 activity in brain cancer: Extracellular nutrients are required to maintain oncogenic signaling. Bioessays 2016, 38, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Xia, Y.; Hawke, D.; Li, X.; Liang, J.; Xing, D.; Aldape, K.; Hunter, T.; Alfred Yung, W.K.; Lu, Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 2012, 150, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Xia, Y.; Ji, H.; Zheng, Y.; Liang, J.; Huang, W.; Gao, X.; Aldape, K.; Lu, Z. Nuclear PKM2 regulates β-catenin transactivation upon EGFR activation. Nature 2011, 480, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z. Nonmetabolic functions of pyruvate kinase isoform M2 in controlling cell cycle progression and tumorigenesis. Chin. J. Cancer 2012, 31, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Fu, X.-L.; Wang, J.-J.; Guan, R.; Sun, Y.; Tony To, S.-S. Inhibition of glycolytic metabolism in glioblastoma cells by Pt3glc combinated with PI3K inhibitor via SIRT3-mediated mitochondrial and PI3K/Akt-MAPK pathway. J. Cell. Physiol. 2019, 234, 5888–5903. [Google Scholar] [CrossRef]
- Rodríguez-García, A.; Samsó, P.; Fontova, P.; Simon-Molas, H.; Manzano, A.; Castaño, E.; Rosa, J.L.; Martinez-Outshoorn, U.; Ventura, F.; Navarro-Sabaté, À.; et al. TGF-β1 targets Smad, p38 MAPK, and PI3K/Akt signaling pathways to induce PFKFB3 gene expression and glycolysis in glioblastoma cells. FEBS J. 2017, 284, 3437–3454. [Google Scholar] [CrossRef]
- Liu, Z.; Jiang, Z.; Huang, J.; Huang, S.; Li, Y.; Yu, S.; Yu, S.; Liu, X. miR-7 inhibits glioblastoma growth by simultaneously interfering with the PI3K/ATK and Raf/MEK/ERK pathways. Int. J. Oncol. 2014, 44, 1571–1580. [Google Scholar] [CrossRef]
- Cai, J.; Zhao, J.; Zhang, N.; Xu, X.; Li, R.; Yi, Y.; Fang, L.; Zhang, L.; Li, M.; Wu, J.; et al. MicroRNA-542-3p Suppresses Tumor Cell Invasion via Targeting AKT Pathway in Human Astrocytoma*. J. Biol. Chem. 2015, 290, 24678–24688. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, X.; Lian, H.; Liu, J.; Zhou, B.; Han, S.; Peng, B.; Yin, J.; Liu, W.; He, X. MicroRNA-503 acts as a tumor suppressor in glioblastoma for multiple antitumor effects by targeting IGF-1R. Oncol. Rep. 2014, 31, 1445–1452. [Google Scholar] [CrossRef]
- Guo, P.; Nie, Q.; Lan, J.; Ge, J.; Qiu, Y.; Mao, Q. C-Myc negatively controls the tumor suppressor PTEN by upregulating miR-26a in glioblastoma multiforme cells. Biochem. Biophys. Res. Commun. 2013, 441, 186–190. [Google Scholar] [CrossRef]
- Xia, X.; Li, Y.; Wang, W.; Tang, F.; Tan, J.; Sun, L.; Li, Q.; Sun, L.; Tang, B.; He, S. MicroRNA-1908 functions as a glioblastoma oncogene by suppressing PTEN tumor suppressor pathway. Mol. Cancer 2015, 14, 154. [Google Scholar] [CrossRef]
- Li, X.-T.; Wang, H.-Z.; Wu, Z.-W.; Yang, T.-Q.; Zhao, Z.-H.; Chen, G.-L.; Xie, X.-S.; Li, B.; Wei, Y.-X.; Huang, Y.-L.; et al. miR-494-3p Regulates Cellular Proliferation, Invasion, Migration, and Apoptosis by PTEN/AKT Signaling in Human Glioblastoma Cells. Cell. Mol. Neurobiol. 2015, 35, 679–687. [Google Scholar] [CrossRef]
- Liu, S.; Sun, J.; Lan, Q. TGF-β-induced miR10a/b expression promotes human glioma cell migration by targeting PTEN. Mol. Med. Rep. 2013, 8, 1741–1746. [Google Scholar] [CrossRef]
- Godlewski, J.; Nowicki, M.O.; Bronisz, A.; Nuovo, G.; Palatini, J.; De Lay, M.; Van Brocklyn, J.; Ostrowski, M.C.; Chiocca, E.A.; Lawler, S.E. MicroRNA-451 regulates LKB1/AMPK signaling and allows adaptation to metabolic stress in glioma cells. Mol. Cell 2010, 37, 620–632. [Google Scholar] [CrossRef]
- Shen, L.; Sun, C.; Li, Y.; Li, X.; Sun, T.; Liu, C.; Zhou, Y.; Du, Z. MicroRNA-199a-3p suppresses glioma cell proliferation by regulating the AKT/mTOR signaling pathway. Tumor Biol. 2015, 36, 6929–6938. [Google Scholar] [CrossRef]
- Rathod, S.S.; Rani, S.B.; Khan, M.; Muzumdar, D.; Shiras, A. Tumor suppressive miRNA-34a suppresses cell proliferation and tumor growth of glioma stem cells by targeting Akt and Wnt signaling pathways. FEBS Open Bio 2014, 4, 485–495. [Google Scholar] [CrossRef]
- Michlewski, G.; Cáceres, J.F. Antagonistic role of hnRNP A1 and KSRP in the regulation of let-7a biogenesis. Nat. Struct. Mol. Biol. 2010, 17, 1011–1018. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, J.; Manley, J.L. Turning on a fuel switch of cancer: hnRNP proteins regulate alternative splicing of pyruvate kinase mRNA. Cancer Res. 2010, 70, 8977–8980. [Google Scholar] [CrossRef]
- Babic, I.; Anderson, E.S.; Tanaka, K.; Guo, D.; Masui, K.; Li, B.; Zhu, S.; Gu, Y.; Villa, G.R.; Akhavan, D.; et al. EGFR mutation-induced alternative splicing of Max contributes to growth of glycolytic tumors in brain cancer. Cell Metab. 2013, 17, 1000–1008. [Google Scholar] [CrossRef]
- David, C.J.; Chen, M.; Assanah, M.; Canoll, P.; Manley, J.L. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 2010, 463, 364–368. [Google Scholar] [CrossRef]
- Chen, X.; Gao, Y.; Li, D.; Cao, Y.; Hao, B. LncRNA-TP53TG1 Participated in the Stress Response Under Glucose Deprivation in Glioma. J. Cell. Biochem. 2017, 118, 4897–4904. [Google Scholar] [CrossRef]
- Miao, F.-A.; Chu, K.; Chen, H.-R.; Zhang, M.; Shi, P.-C.; Bai, J.; You, Y.-P. Increased DKC1 expression in glioma and its significance in tumor cell proliferation, migration and invasion. Investig. New Drugs 2019, 37, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Yu, Y.-T. RNA pseudouridylation: New insights into an old modification. Trends Biochem. Sci. 2013, 38, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ma, S.; Yi, C. Pseudouridine: The fifth RNA nucleotide with renewed interests. Curr. Opin. Chem. Biol. 2016, 33, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Jayachandran, A.; Lo, P.-H.; Chueh, A.C.; Prithviraj, P.; Molania, R.; Davalos-Salas, M.; Anaka, M.; Walkiewicz, M.; Cebon, J.; Behren, A. Transketolase-like 1 ectopic expression is associated with DNA hypomethylation and induces the Warburg effect in melanoma cells. BMC Cancer 2016, 16, 134. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Liu, Y.; Glazer, C.A.; Shao, C.; Bhan, S.; Demokan, S.; Zhao, M.; Rudek, M.A.; Ha, P.K.; Califano, J.A. TKTL1 is activated by promoter hypomethylation and contributes to head and neck squamous cell carcinoma carcinogenesis through increased aerobic glycolysis and HIF1alpha stabilization. Clin. Cancer Res. 2010, 16, 857–866. [Google Scholar] [CrossRef]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1–Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef]
- Sekhar, K.R.; Yan, X.X.; Freeman, M.L. Nrf2 degradation by the ubiquitin proteasome pathway is inhibited by KIAA0132, the human homolog to INrf2. Oncogene 2002, 21, 6829–6834. [Google Scholar] [CrossRef]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J. Biol. Chem. 2004, 279, 31556–31567. [Google Scholar] [CrossRef]
- Muscarella, L.A.; Barbano, R.; D’Angelo, V.; Copetti, M.; Coco, M.; Balsamo, T.; la Torre, A.; Notarangelo, A.; Troiano, M.; Parisi, S.; et al. Regulation of KEAP1 expression by promoter methylation in malignant gliomas and association with patient’s outcome. Epigenetics 2011, 6, 317–325. [Google Scholar] [CrossRef]
- Muscarella, L.A.; Parrella, P.; D’Alessandro, V.; la Torre, A.; Barbano, R.; Fontana, A.; Tancredi, A.; Guarnieri, V.; Balsamo, T.; Coco, M.; et al. Frequent epigenetics inactivation of KEAP1 gene in non-small cell lung cancer. Epigenetics 2011, 6, 710–719. [Google Scholar] [CrossRef]
- Dong, C.; Yuan, T.; Wu, Y.; Wang, Y.; Fan, T.W.M.; Miriyala, S.; Lin, Y.; Yao, J.; Shi, J.; Kang, T.; et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 2013, 23, 316–331. [Google Scholar] [CrossRef]
- Li, B.; Qiu, B.; Lee, D.S.M.; Walton, Z.E.; Ochocki, J.D.; Mathew, L.K.; Mancuso, A.; Gade, T.P.F.; Keith, B.; Nissim, I.; et al. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature 2014, 513, 251–255. [Google Scholar] [CrossRef]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Yang, W.; Lu, Z. Regulation and function of pyruvate kinase M2 in cancer. Cancer Lett. 2013, 339, 153–158. [Google Scholar] [CrossRef]
- Mazurek, S.; Boschek, C.B.; Hugo, F.; Eigenbrodt, E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin. Cancer Biol. 2005, 15, 300–308. [Google Scholar] [CrossRef]
- Marín-Hernández, A.; Rodríguez-Enríquez, S.; Vital-González, P.A.; Flores-Rodríguez, F.L.; Macías-Silva, M.; Sosa-Garrocho, M.; Moreno-Sánchez, R. Determining and understanding the control of glycolysis in fast-growth tumor cells. Flux control by an over-expressed but strongly product-inhibited hexokinase. FEBS J. 2006, 273, 1975–1988. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Giatromanolaki, A.; Sivridis, E.; Gatter, K.C.; Harris, A.L. Pyruvate dehydrogenase and pyruvate dehydrogenase kinase expression in non-small cell lung cancer and tumor-associated stroma. Neoplasia 2005, 7, 1–6. [Google Scholar] [CrossRef]
- Leclerc, D.; Lévesque, N.; Cao, Y.; Deng, L.; Wu, Q.; Powell, J.; Sapienza, C.; Rozen, R. Genes with aberrant expression in murine preneoplastic intestine show epigenetic and expression changes in normal mucosa of colon cancer patients. Cancer Prev. Res. 2013, 6, 1171–1181. [Google Scholar] [CrossRef]
- Sun, X.; Johnson, J.; St. John, J.C. Global DNA methylation synergistically regulates the nuclear and mitochondrial genomes in glioblastoma cells. Nucleic Acids Res. 2018, 46, 5977–5995. [Google Scholar] [CrossRef]
- Böttcher, M.; Renner, K.; Berger, R.; Mentz, K.; Thomas, S.; Cardenas-Conejo, Z.E.; Dettmer, K.; Oefner, P.J.; Mackensen, A.; Kreutz, M.; et al. D-2-hydroxyglutarate interferes with HIF-1α stability skewing T-cell metabolism towards oxidative phosphorylation and impairing Th17 polarization. Oncoimmunology 2018, 7, e1445454. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef]
- Isaacs, J.S.; Jung, Y.J.; Mole, D.R.; Lee, S.; Torres-Cabala, C.; Chung, Y.-L.; Merino, M.; Trepel, J.; Zbar, B.; Toro, J.; et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: Novel role of fumarate in regulation of HIF stability. Cancer Cell 2005, 8, 143–153. [Google Scholar] [CrossRef]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.; Sweha, S.R.; Pratt, D.; Tamrazi, B.; Panwalkar, P.; Banda, A.; Bayliss, J.; Hawes, D.; Yang, F.; Lee, H.-J.; et al. Integrated Metabolic and Epigenomic Reprograming by H3K27M Mutations in Diffuse Intrinsic Pontine Gliomas. Cancer Cell 2020, 38, 334–349.e9. [Google Scholar] [CrossRef] [PubMed]
- Piunti, A.; Hashizume, R.; Morgan, M.A.; Bartom, E.T.; Horbinski, C.M.; Marshall, S.A.; Rendleman, E.J.; Ma, Q.; Takahashi, Y.-H.; Woodfin, A.R.; et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat. Med. 2017, 23, 493–500. [Google Scholar] [CrossRef]
- Stafford, J.M.; Lee, C.-H.; Voigt, P.; Descostes, N.; Saldaña-Meyer, R.; Yu, J.-R.; Leroy, G.; Oksuz, O.; Chapman, J.R.; Suarez, F.; et al. Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Sci. Adv. 2018, 4, eaau5935. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Johnson, J.; Gough, D.J.; Donoghue, J.; Cagnone, G.L.M.; Vaghjiani, V.; Brown, K.A.; Johns, T.G.; St John, J.C. Mitochondrial DNA copy number is regulated by DNA methylation and demethylation of POLGA in stem and cancer cells and their differentiated progeny. Cell Death Dis. 2015, 6, e1664. [Google Scholar] [CrossRef]
- Nakamura, Y.; Arakawa, H. Discovery of Mieap-regulated mitochondrial quality control as a new function of tumor suppressor p53. Cancer Sci. 2017, 108, 809–817. [Google Scholar] [CrossRef]
- Tsuneki, M.; Nakamura, Y.; Kinjo, T.; Nakanishi, R.; Arakawa, H. Mieap suppresses murine intestinal tumor via its mitochondrial quality control. Sci. Rep. 2015, 5, 12472. [Google Scholar] [CrossRef]
- Ciechanover, A. Proteolysis: From the lysosome to ubiquitin and the proteasome. Nat. Rev. Mol. Cell Biol. 2005, 6, 79–87. [Google Scholar] [CrossRef]
- Xu, G.; Li, J.Y. ATP5A1 and ATP5B are highly expressed in glioblastoma tumor cells and endothelial cells of microvascular proliferation. J. Neurooncol. 2016, 126, 405–413. [Google Scholar] [CrossRef]
- Ru, P.; Hu, P.; Geng, F.; Mo, X.; Cheng, C.; Yoo, J.Y.; Cheng, X.; Wu, X.; Guo, J.Y.; Nakano, I.; et al. Feedback Loop Regulation of SCAP/SREBP-1 by miR-29 Modulates EGFR Signaling-Driven Glioblastoma Growth. Cell Rep. 2016, 16, 1527–1535. [Google Scholar] [CrossRef]
- Ru, P.; Guo, D. microRNA-29 mediates a novel negative feedback loop to regulate SCAP/SREBP-1 and lipid metabolism. RNA Dis. 2017, 4, e1525. [Google Scholar] [CrossRef][Green Version]
- Liu, Z.; Wang, J.; Li, Y.; Fan, J.; Chen, L.; Xu, R. MicroRNA-153 regulates glutamine metabolism in glioblastoma through targeting glutaminase. Tumor Biol. 2017, 39, 1010428317691429. [Google Scholar] [CrossRef]
- Cheng, C.; Ru, P.; Geng, F.; Liu, J.; Yoo, J.Y.; Wu, X.; Cheng, X.; Euthine, V.; Hu, P.; Guo, J.Y.; et al. Glucose-Mediated N-glycosylation of SCAP Is Essential for SREBP-1 Activation and Tumor Growth. Cancer Cell 2015, 28, 569–581. [Google Scholar] [CrossRef]
- Obara-Michlewska, M.; Szeliga, M. Targeting Glutamine Addiction in Gliomas. Cancers 2020, 12, 310. [Google Scholar] [CrossRef]
- Virtuoso, A.; Giovannoni, R.; De Luca, C.; Gargano, F.; Cerasuolo, M.; Maggio, N.; Lavitrano, M.; Papa, M. The Glioblastoma Microenvironment: Morphology, Metabolism, and Molecular Signature of Glial Dynamics to Discover Metabolic Rewiring Sequence. Int. J. Mol. Sci. 2021, 22, 3301. [Google Scholar] [CrossRef]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase-2 bound to mitochondria: Cancer’s stygian link to the “Warburg Effect” and a pivotal target for effective therapy. Semin. Cancer Biol. 2009, 19, 17–24. [Google Scholar] [CrossRef]
- Zhang, D.; Li, J.; Wang, F.; Hu, J.; Wang, S.; Sun, Y. 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014, 355, 176–183. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, L.; Chai, W.; Zhao, T.; Jin, X.; Guo, X.; Han, L.; Yuan, C. Dichloroacetic acid upregulates apoptosis of ovarian cancer cells by regulating mitochondrial function. Onco. Targets. Ther. 2019, 12, 1729–1739. [Google Scholar] [CrossRef]
- Pulliam, N.; Fang, F.; Ozes, A.R.; Tang, J.; Adewuyi, A.; Keer, H.; Lyons, J.; Baylin, S.B.; Matei, D.; Nakshatri, H.; et al. An Effective Epigenetic-PARP Inhibitor Combination Therapy for Breast and Ovarian Cancers Independent of BRCA Mutations. Clin. Cancer Res. 2018, 24, 3163–3175. [Google Scholar] [CrossRef]
- Zhou, S.; Li, Y.; Huang, F.; Zhang, B.; Yi, T.; Li, Z.; Luo, H.; He, X.; Zhong, Q.; Bian, C.; et al. Live-attenuated measles virus vaccine confers cell contact loss and apoptosis of ovarian cancer cells via ROS-induced silencing of E-cadherin by methylation. Cancer Lett. 2012, 318, 14–25. [Google Scholar] [CrossRef]
- Das, D.S.; Ray, A.; Das, A.; Song, Y.; Tian, Z.; Oronsky, B.; Richardson, P.; Scicinski, J.; Chauhan, D.; Anderson, K.C. A novel hypoxia-selective epigenetic agent RRx-001 triggers apoptosis and overcomes drug resistance in multiple myeloma cells. Leukemia 2016, 30, 2187–2197. [Google Scholar] [CrossRef]
- Egler, V.; Korur, S.; Failly, M.; Boulay, J.-L.; Imber, R.; Lino, M.M.; Merlo, A. Histone deacetylase inhibition and blockade of the glycolytic pathway synergistically induce glioblastoma cell death. Clin. Cancer Res. 2008, 14, 3132–3140. [Google Scholar] [CrossRef]
- Cuperlovic-Culf, M.; Touaibia, M.; St-Coeur, P.-D.; Poitras, J.; Morin, P.; Culf, A.S. Metabolic Effects of Known and Novel HDAC and SIRT Inhibitors in Glioblastomas Independently or Combined with Temozolomide. Metabolites 2014, 4, 807–830. [Google Scholar] [CrossRef]
- Appelskog, I.B.; Ammerpohl, O.; Svechnikova, I.G.; Lui, W.-O.; Almqvist, P.M.; Ekström, T.J. Histone deacetylase inhibitor 4-phenylbutyrate suppresses GAPDH mRNA expression in glioma cells. Int. J. Oncol. 2004, 24, 1419–1425. [Google Scholar]
- Konstantinopoulos, P.A.; Vandoros, G.P.; Papavassiliou, A.G. FK228 (depsipeptide): A HDAC inhibitor with pleiotropic antitumor activities. Cancer Chemother. Pharmacol. 2006, 58, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Dong, L.; Bao, S.; Wang, M.; Yun, Y.; Zhu, R. FK228 augmented temozolomide sensitivity in human glioma cells by blocking PI3K/AKT/mTOR signal pathways. Biomed. Pharmacother. 2016, 84, 462–469. [Google Scholar] [CrossRef]
- Hennika, T.; Hu, G.; Olaciregui, N.G.; Barton, K.L.; Ehteda, A.; Chitranjan, A.; Chang, C.; Gifford, A.J.; Tsoli, M.; Ziegler, D.S.; et al. Pre-Clinical Study of Panobinostat in Xenograft and Genetically Engineered Murine Diffuse Intrinsic Pontine Glioma Models. PLoS ONE 2017, 12, e0169485. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Tang, Y.; Truffaux, N.; Berlow, N.E.; Liu, L.; Debily, M.-A.; Quist, M.J.; Davis, L.E.; Huang, E.C.; Woo, P.J.; et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat. Med. 2015, 21, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Souweidane, M.M.; Kramer, K.; Pandit-Taskar, N.; Zhou, Z.; Haque, S.; Zanzonico, P.; Carrasquillo, J.A.; Lyashchenko, S.K.; Thakur, S.B.; Donzelli, M.; et al. Convection-enhanced delivery for diffuse intrinsic pontine glioma: A single-centre, dose-escalation, phase 1 trial. Lancet Oncol. 2018, 19, 1040–1050. [Google Scholar] [CrossRef]
- Katagi, H.; Louis, N.; Unruh, D.; Sasaki, T.; He, X.; Zhang, A.; Ma, Q.; Piunti, A.; Shimazu, Y.; Lamano, J.B.; et al. Radiosensitization by Histone H3 Demethylase Inhibition in Diffuse Intrinsic Pontine Glioma. Clin. Cancer Res. 2019, 25, 5572–5583. [Google Scholar] [CrossRef]
- Nagaraja, S.; Vitanza, N.A.; Woo, P.J.; Taylor, K.R.; Liu, F.; Zhang, L.; Li, M.; Meng, W.; Ponnuswami, A.; Sun, W.; et al. Transcriptional Dependencies in Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 31, 635–652.e6. [Google Scholar] [CrossRef]
- Krug, B.; De Jay, N.; Harutyunyan, A.S.; Deshmukh, S.; Marchione, D.M.; Guilhamon, P.; Bertrand, K.C.; Mikael, L.G.; McConechy, M.K.; Chen, C.C.L.; et al. Pervasive H3K27 Acetylation Leads to ERV Expression and a Therapeutic Vulnerability in H3K27M Gliomas. Cancer Cell 2019, 35, 782–797.e8. [Google Scholar] [CrossRef]
- Karimi, M.M.; Goyal, P.; Maksakova, I.A.; Bilenky, M.; Leung, D.; Tang, J.X.; Shinkai, Y.; Mager, D.L.; Jones, S.; Hirst, M.; et al. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell 2011, 8, 676–687. [Google Scholar] [CrossRef]
- Christman, J.K. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene 2002, 21, 5483–5495. [Google Scholar] [CrossRef]
- Jones, P.A.; Ohtani, H.; Chakravarthy, A.; De Carvalho, D.D. Epigenetic therapy in immune-oncology. Nat. Rev. Cancer 2019, 19, 151–161. [Google Scholar] [CrossRef]
- Mendez, F.M.; Núñez, F.J.; Garcia-Fabiani, M.B.; Haase, S.; Carney, S.; Gauss, J.C.; Becher, O.J.; Lowenstein, P.R.; Castro, M.G. Epigenetic reprogramming and chromatin accessibility in pediatric diffuse intrinsic pontine gliomas: A neural developmental disease. Neuro. Oncol. 2020, 22, 195–206. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme/ Molecule | Classification | Effect on Metabolism | Key Metabolic Genes/ Enzymes/Pathways Involved | Reference |
|---|---|---|---|---|
| G9 | HMT | Inhibits glycolysis | HIF-1, SLC6A3, PTGS1, NDNF, Linc01132 | [37] |
| MLL1 | HMT | Inhibits glycolysis, increases OXPHOS | Gluts, HKs, LHDA, PFK | [38,39] |
| EZH2 | HMT | Favors glycolysis | EAF2 | [36] |
| LSD1/HDM1A | HDM | Inhibits glycolysis | p53 | [31] |
| miR-215/KDM1B | microRNA and HDM | Favor glycolysis | HIF2α, Ndrg1, ADM, NDUFA4L2, Glut1, Glut3 | [42] |
| PCAF/KAT2B | HAT | Favors glycolysis | Akt1 | [43] |
| KAT6A | HAT | Favors glycolysis | PI3K/Akt, Glut1, PFK1, HK1/2/3 | [44] |
| HDAC 4/5/7 | HDAC | Favors glycolysis | c-Myc, ENO1, Glut1, LDH-A, SHMT | [46] |
| SIRT6 | HDAC | Inhibits glycolysis | HK2 | [48] |
| miR-143 | microRNA | Inhibits glycolysis | HK2 | [54] |
| miR-let-7a | microRNA | Favors glycolysis | c-Myc, PKM2 | [55,56] |
| miR-29 | microRNA | Decreases lipid synthesis | SCAP, SREBP-1 | [117,118] |
| miR-153 | microRNA | Decreases glutamine metabolism | glutaminases | [119] |
| Drug Name | Mechanism of Action | Type of Study | Reference |
|---|---|---|---|
| Glycolysis inhibitors | |||
| 3-Bromopyruvate (3-BP) | Inhibits glycolysis by targeting HK-2 and the mitochondrial ATP synthasome | Preclinical studies in Various cancers | [123] |
| 2-deoxy-D-glucose (2-DG) | Glucose analog that depletes tumor cell energy, eliciting antitumor effects | Phase 1 clinical trials in GB | [124] |
| Dichloroacetic acid (DCA) | PDK1 inhibitor that minimizes the Warburg effect and shifts tumor cells towards OXPHOS | Preclinical trials in ovarian cancer | [125] |
| DNA methylation inhibitors | |||
| 5-azacitidine | Gets incorporated into the DNA and its residues inhibit DNA methylation | Phase II clinical trial in GB | [141] |
| ROS manipulators | |||
| Decitabine | Upregulates ROS production, increases sensitivity to PARP Inhibitors | Preclinical studies in Ovarian and breast cancer | [126] |
| Live-attenuated measles vaccine | Upregulates ROS, causing DNA methylation and E-cadherin silencing, leading to intercellular loss of contact and cancer cell apoptosis | Preclinical studies in Breast cancer | [127] |
| RRx-001 | Increases ROS and nitrogen production, inhibits DNA methylation, leading to apoptosis | Phase I clinical trial in gliomas | [128] |
| Deacetylase inhibitors | |||
| Nicotinamide | Sirtuin (class III NAD+-dependent HDAC) inhibitor that can affect mitochondrial metabolism, glycolysis, and fatty acid synthesis in GB | Phase I/II clinical trial | [130] |
| Vorinostat (SAHA) | Pan-HDAC inhibitor that can affect mitochondrial metabolism, glycolysis, and fatty acid synthesis in GB | Phase I clinical trial | [130] |
| Tubastatin A | Selective HDAC inhibitor that can affect mitochondrial metabolism, glycolysis, and fatty acid synthesis in GB | Preclinical studies in GB | [130] |
| 4-phenylbutyrate | Selective HDAC inhibitor that suppresses GAPDH mRNA expression, decreasing energy consumption and cell proliferation | Preclinical studies in gliomas | [131] |
| Panobinostat | Pan-HDAC inhibitor, more effective when combined with low dose 5-azacytidine in derepressing ERVs and activating the immune system leading to glioma cell death | Phase II clinical trial | [135] |
| α-KG- producing enzyme inhibitors | |||
| JHU-083 and WT-DH1i13 | Glutamine antagonist and WT-IDH1 antagonist prolonged survival in diffuse midline gliomas with H3K27M | Preclinical studies in diffuse intrinsic pontine gliomas | [109] |
| Lysine demethylase inhibitor | |||
| GSKJ4 | JMJD3 lysine specific demethylase inhibitor increases H3K27me2 and H3K27me3 levels in glioma cells harboring the H3K27M, decreasing cell viability and colony formation | Preclinical studies in diffuse intrinsic pontine gliomas | [137] |
| BET bromodomain and extra- terminal domain inhibitor | |||
| JQ1 | BET bromodomain and extra-terminal domain inhibitor leading to a more differentiated cellular phenotype and decreased proliferation | Preclinical studies in diffuse intrinsic pontine gliomas | [110] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markouli, M.; Strepkos, D.; Papavassiliou, K.A.; Papavassiliou, A.G.; Piperi, C. Crosstalk of Epigenetic and Metabolic Signaling Underpinning Glioblastoma Pathogenesis. Cancers 2022, 14, 2655. https://doi.org/10.3390/cancers14112655
Markouli M, Strepkos D, Papavassiliou KA, Papavassiliou AG, Piperi C. Crosstalk of Epigenetic and Metabolic Signaling Underpinning Glioblastoma Pathogenesis. Cancers. 2022; 14(11):2655. https://doi.org/10.3390/cancers14112655
Chicago/Turabian StyleMarkouli, Mariam, Dimitrios Strepkos, Kostas A. Papavassiliou, Athanasios G. Papavassiliou, and Christina Piperi. 2022. "Crosstalk of Epigenetic and Metabolic Signaling Underpinning Glioblastoma Pathogenesis" Cancers 14, no. 11: 2655. https://doi.org/10.3390/cancers14112655
APA StyleMarkouli, M., Strepkos, D., Papavassiliou, K. A., Papavassiliou, A. G., & Piperi, C. (2022). Crosstalk of Epigenetic and Metabolic Signaling Underpinning Glioblastoma Pathogenesis. Cancers, 14(11), 2655. https://doi.org/10.3390/cancers14112655