
Targeting Estrogens and Various Estrogen-Related Receptors against Non-Small Cell Lung Cancers: A Perspective


Abstract
Simple Summary
Abstract
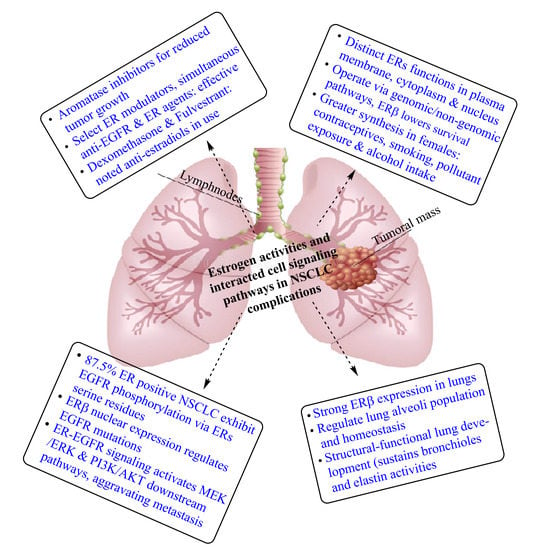
1. Introduction
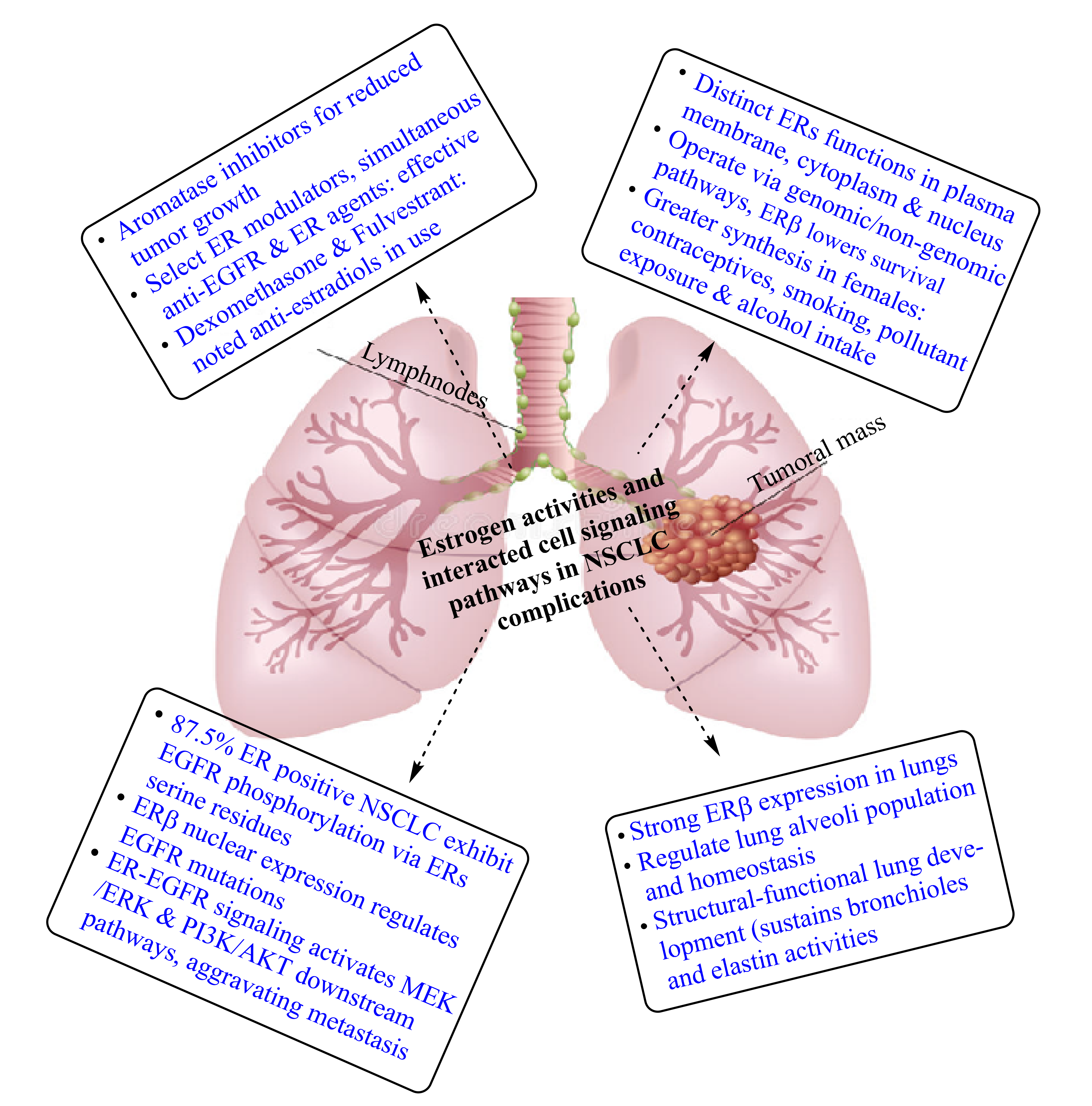
2. Exposure of Mammalian Lungs to Endogenous and Exogenous Estrogens Including Synthetic Estrogens, Phytoestrogens and Xenoestrogens
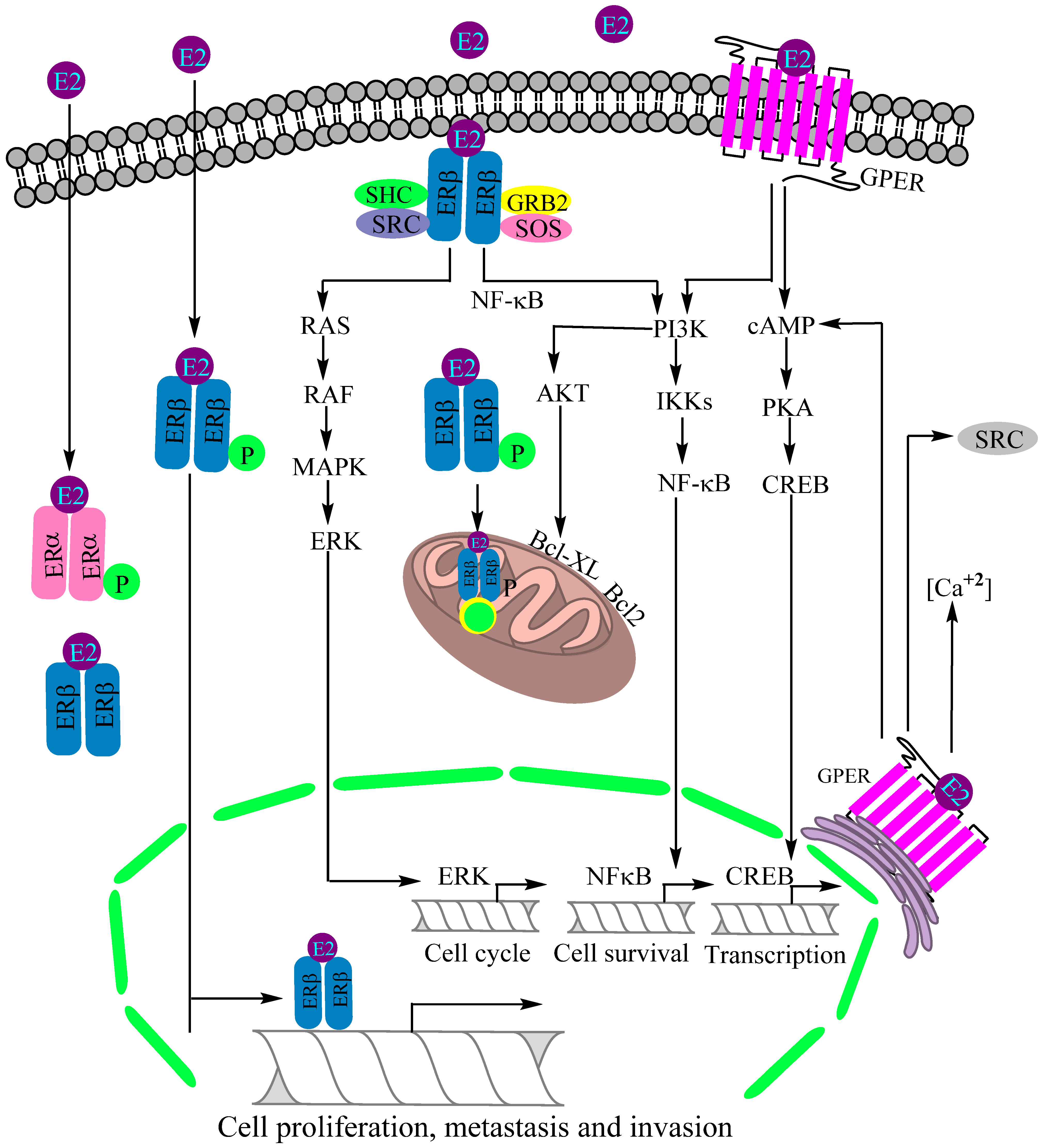
3. Expression of Estrogen Receptors by Mammalian Lungs and Their Physiological Roles
4. Role of Estrogen Receptors in Non-Small Cell Lung Cancer Complication
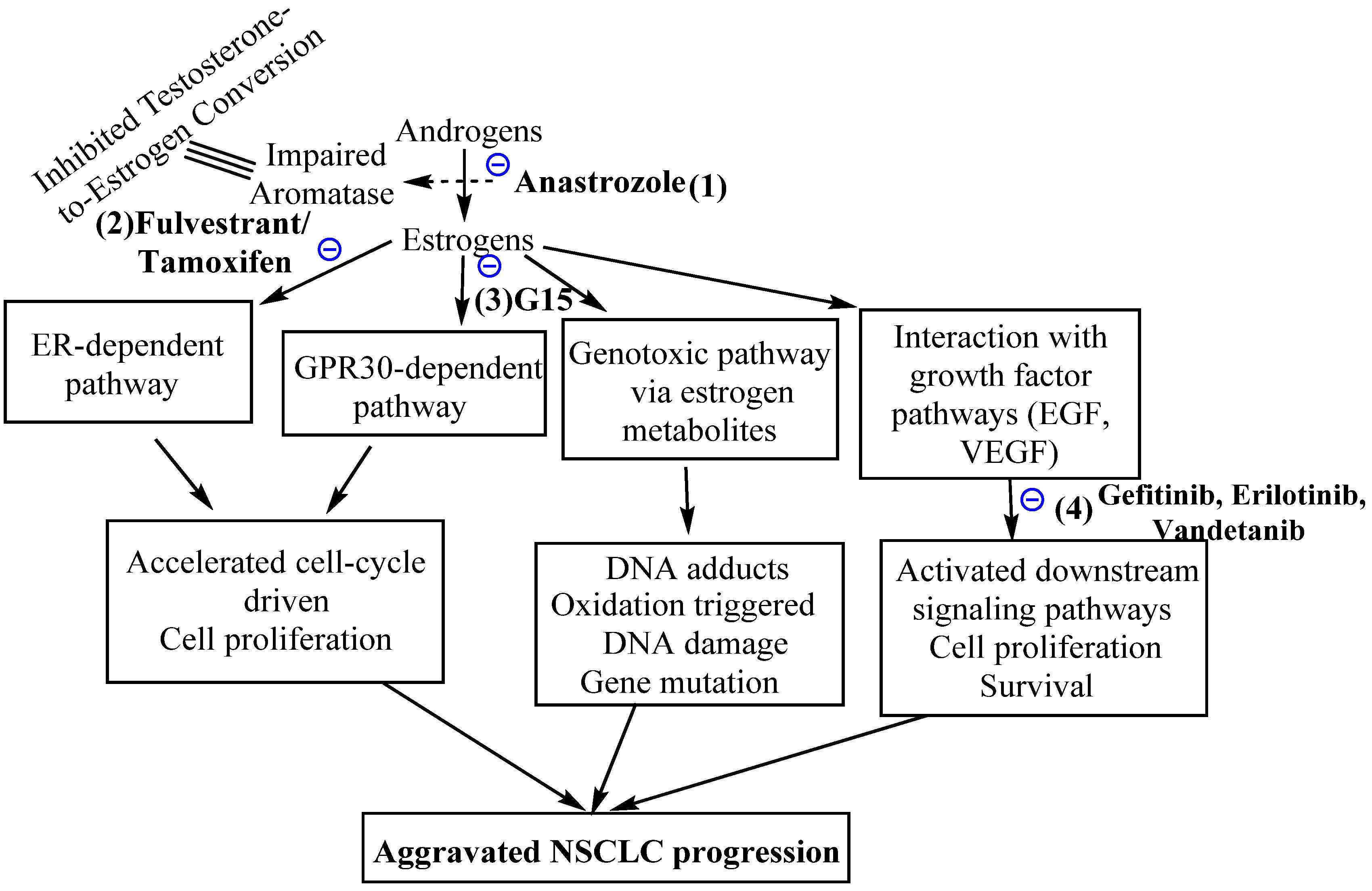
5. Efficacy of Anti-Estrogen and Anti-Estrogen Receptor Molecules against Non-Small Cell Lung Cancers
6. G-Protein-Coupled Estrogen Receptors and Non-Small Cell Lung Cancers
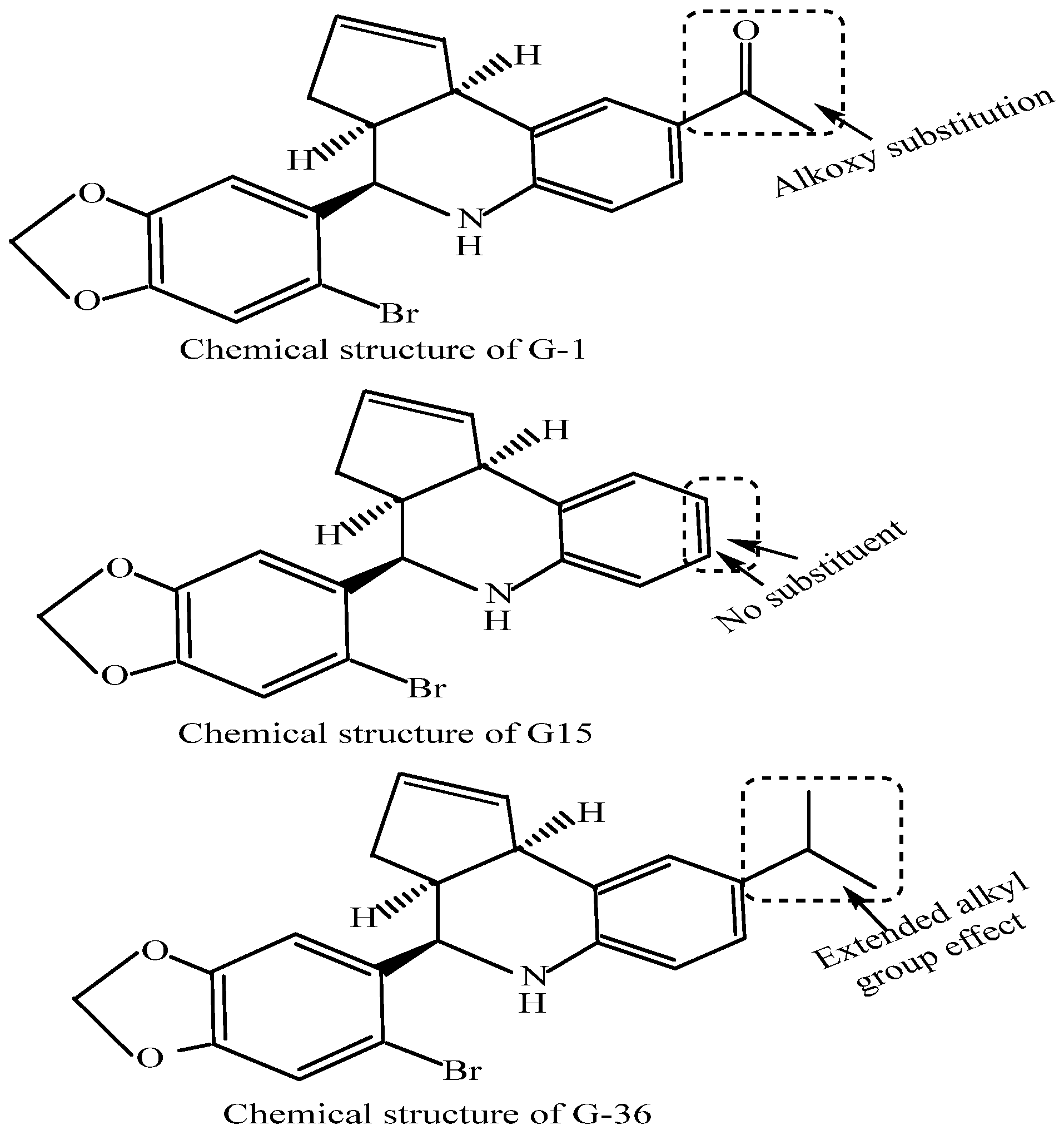
7. The Agonists and Antagonists of G-Protein-Coupled Estrogen Receptors and Potential Non-Small Cell Lung Cancer Therapy
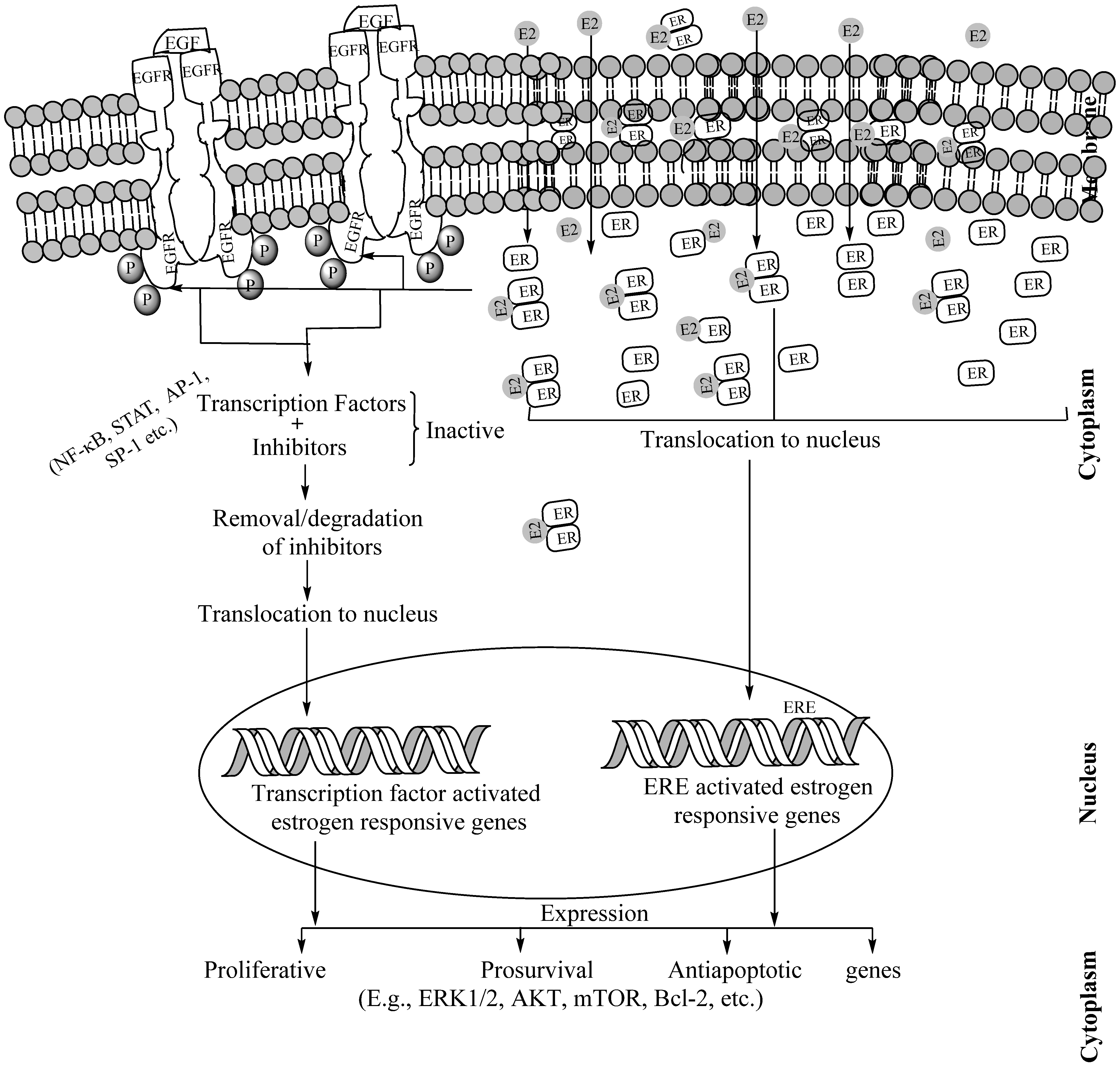
8. Interactions of Estrogen Receptors and Epidermal Growth Factor Receptors for Aggravated Tumorigenesis in Non-Small Cell Lung Cancers
9. Potentials of Dual Targeting of Estrogen Receptors and Epidermal Growth Factor Receptors against Non-Small Cell Lung Cancers
10. Estrogen-Related Receptors and Non-Small Cell Lung Cancer
11. Progress of Clinical Trials on Estrogen-Targeting-Driven Non-Small Cell Lung Cancer Treatment
12. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Koos, R.D. Minireview: Putting physiology back into estrogens’ mechanism of action. Endocrinology 2011, 152, 4481–4488. [Google Scholar] [CrossRef]
- Wierman, M.E. Sex steroid effects at target tissues: Mechanisms of action. Adv. Physiol. Educ. 2007, 31, 26–33. [Google Scholar] [CrossRef]
- Nilsson, S.; Mäkelä, S.; Treuter, E.; Tujague, M.; Thomsen, J.; Andersson, G.; Enmark, E.; Pettersson, K.; Warner, M.; Gustafsson, J.A. Mechanisms of estrogen action. Physiol. Rev. 2001, 81, 1535–1565. [Google Scholar] [CrossRef]
- Nelson, L.R.; Bulun, S.E. Estrogen production and action. J. Am. Acad. Dermatol. 2001, 45, S116–S124. [Google Scholar] [CrossRef]
- Cui, J.; Shen, Y.; Li, R. Estrogen synthesis and signaling pathways during ageing: From periphery to brain. Trends Mol. Med. 2013, 19, 197–209. [Google Scholar] [CrossRef]
- Stocco, C. Tissue physiology and pathology of aromatase. Steroids 2012, 77, 27–35. [Google Scholar] [CrossRef]
- Barakat, R.; Oakley, O.; Kim, H.; Jin, J.; Ko, C.J. Extra-gonadal sites of estrogen biosynthesis and function. BMV Rep. 2016, 49, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Carey, M.A.; Card, J.W.; Voltz, J.W.; Germolec, D.R.; Korach, K.S.; Zeldin, D.C. The impact of sex and sex hormones on lung physiology and disease: Lessons from animal studies. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L272–L278. [Google Scholar] [CrossRef]
- Brandenberger, A.W.; Tee, M.K.; Lee, J.Y.; Chao, V.; Jaffe, R.B. Tissue distribution of estrogen receptors alpha (ER-alpha) and beta (ER-beta) mRNA in the mid-gestational human fetus. J. Clin. Endocrinol. Metab. 1997, 82, 3509–3512. [Google Scholar] [CrossRef][Green Version]
- Ginsburg, E.S.; Gao, X.; Shea, B.F.; Barbieri, R.L. Half-life of estradiol in postmenopausal women. Gynecol. Obstet. Investig. 1998, 45, 45–48. [Google Scholar] [CrossRef]
- Bolton, J.L.; Thatcher, G.R. Potential mechanisms of estrogen quinone carcinogenesis. Chem. Res. Toxicol. 2008, 21, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Goa, K.L.; Warne, G.T.; Easthope, S.E. Transdermal ethinylestradiol/ norelgestromin: A review of its use in hormonal contraception. Treat Endocrinol. 2003, 2, 191–206. [Google Scholar] [CrossRef] [PubMed]
- Shulman, L.P. The state of hormonal contraception today: Benefits and risks of hormonal contraceptives: Combined estrogen and progestin contraceptives. Am. J. Obstet. Gynecol. 2011, 205, S9–S13. [Google Scholar] [CrossRef]
- Cagnacci, A.; Venier, M. The controversial history of hormone replacement therapy. Medicina 2019, 55, 602. [Google Scholar] [CrossRef] [PubMed]
- Lobo, R.A. Hormone-replacement therapy: Current thinking. Nat. Rev. Endocrinol. 2017, 13, 220–231. [Google Scholar] [CrossRef]
- Wang, T.T.; Sathyamoorthy, N.; Phang, J. Molecular effects of genestein on estrogen receptor mediated pathways. Carcinogenesis 1996, 17, 271–275. [Google Scholar] [CrossRef]
- Fucic, A.; Gamulin, M.; Ferencic, Z.; Rokotov, D.S.; Katic, J.; Bartonova, A.; Lovasic, I.B.; Merlo, D.F. Lung cancer and environmental chemical exposure: A review of our current state of knowledge with reference to the role of hormones and hormone receptors as an increased risk factor for developing lung cancer in man. Toxicol. Pathol. 2010, 38, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Bredhult, C.; Backlin, B.M.; Olovsson, M. Effects of some endocrine disruptors on the proliferation and viability of human endometrial endothelial cells in vitro. Reprod. Toxicol. 2007, 23, 550–559. [Google Scholar] [CrossRef]
- Zhou, Y.; Yau, C.; Gray, J.W.; Chew, K.; Dairkee, S.H.; Moore, D.H.; Eppenberger, U.; Eppenberger-Castori, S.; Benz, C.C. Enhanced NF-κB and AP-1 transcriptional activity associated with antiestrogen resistant breast cancer. BMC Cancer 2007, 7, 59. [Google Scholar] [CrossRef]
- Patisaul, H.B.; Jefferson, W. The pros and cons of phytoestrogens. Front. Neuroendocrinol. 2010, 31, 400–419. [Google Scholar] [CrossRef]
- Niikawa, H.; Suzuki, T.; Miki, Y.; Suzuki, S.; Nagasaki, S.; Akahira, J.; Honma, S.; Evans, D.B.; Hayashi, S.; Kondo, T.; et al. Intratumoral estrogens and estrogen receptors in human non-small cell lung carcinoma. Clin. Cancer Res. 2008, 14, 4417–4426. [Google Scholar] [CrossRef] [PubMed]
- Liehr, J.G.; Wan-Fen, F.; Sirbasku, D.A.; Ari-Ulubelen, A. Carcinogenicity of catechol estrogens in Syrian hamsters. J. Steroid Biochem. 1986, 24, 353–356. [Google Scholar] [CrossRef]
- Li, J.J.; Li, S.A. Estrogen carcinogenesis in Syrian hamster tissues: Role of metabolism. Fed. Proc. 1987, 46, 1858–1863. [Google Scholar] [PubMed]
- Cavalieri, E.; Chakravarti, D.; Guttenplan, J.; Hart, E.; Ingle, J.; Jankowiak, R.; Muti, P.; Rogan, E.; Russo, J.; Santen, R.; et al. Catechol estrogen quinones as initiators of breast and other human cancers: Implications for biomarkers of susceptibility and cancer prevention. Biochim. Biophys. Acta Rev. Cancer 2006, 1766, 63–78. [Google Scholar] [CrossRef]
- Su, J.-M.; Lin, P.; Wang, C.-K.; Chang, H. Overexpression of cytochrome P450 1B1 in advanced non-small cell lung cancer: A potential therapeutic target. Anticancer Res. 2009, 29, 509–515. [Google Scholar] [PubMed]
- Márquez-Garbán, D.C.; Chen, H.W.; Fishbein, M.C.; Goodglick, L.; Pietras, R.J. Estrogen receptor signaling pathways in human non-small cell lung cancer. Steroids 2007, 72, 135–143. [Google Scholar] [CrossRef]
- Baik, C.S.; Eaton, K.D. Estrogen signaling in lung cancer: An opportunity for novel therapy. Cancers 2012, 4, 969–988. [Google Scholar] [CrossRef]
- Hsu, L.H.; Liu, K.J.; Tsai, M.F.; Wu, C.R.; Feng, A.C.; Chu, N.M.; Kao, S.H. Estrogen adversely affects the prognosis of patients with lung adenocarcinoma. Cancer Sci. 2015, 106, 51–59. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer (IARC). GLOBOCAN 2012: Estimated Cancer Incidence, Mortality and Prevalence Worldwide; International Agency for Research on Cancer, World Health Organization: Geneva, Switzerland, 2013; ISBN 13-978-92-832-2447-1. [Google Scholar]
- Malvezzi, M.; Carioli, G.; Bertuccio, P.; Boffetta, P.; Levi, F.; La Vecchia, C.; Negri, E. European cancer mortality predictions for the year 2017, with focus on lung cancer. Ann. Oncol. 2017, 28, 1117–1123. [Google Scholar] [CrossRef]
- Jemal, A.; Ward, E.M.; Johnson, C.J.; Cronin, K.A.; Ma, J.; Ryerson, B.; Mariotto, A.; Lake, A.J.; Wilson, R.; Sherman, R.L.; et al. Annual report to the nation on the status of cancer, 1975–2014, featuring survival. J. Natl. Cancer Inst. 2017, 109, djx030. [Google Scholar] [CrossRef]
- Hammes, S.R.; Levin, E.R. Impact of estrogens in males and androgens in females. J. Clin. Investig. 2019, 129, 1818–18269. [Google Scholar] [CrossRef] [PubMed]
- Baiu, I.; Titan, A.L.; Martin, L.W.; Wolf, A.; Backhus, L. The role of gender in non-small cell lung cancer: A narrative review. J. Thorac. Dis. 2021, 13, 3816–3826. [Google Scholar] [CrossRef]
- Ragavan, M.V.; Patel, M.I. Understanding sex disparities in lung cancer incidence: Are women more at risk? Lung Cancer Manag. 2020, 9, LMT34. [Google Scholar] [CrossRef] [PubMed]
- Taioli, E.; Wynder, E.L. Re: Endocrine factors and adenocarcinoma of the lung in women. J. Natl. Cancer Inst. 1994, 86, 869–870. [Google Scholar] [CrossRef] [PubMed]
- Blackman, J.A.; Coogan, P.F.; Rosenberg, L.; Strom, B.L.; Zauber, A.G.; Palmer, J.R.; Langenberg, P.; Shapiro, S. Estrogen replacement therapy and risk of lung cancer. Pharmacoepidemiol. Drug Saf. 2002, 11, 561–567. [Google Scholar] [CrossRef]
- Schabath, M.B.; Wu, X.; Vassilopoulou-Sellin, R.; Vaporciyan, A.A.; Spitz, M.R. Hormone replacement therapy and lung cancer risk: A case-control analysis. Clin. Cancer Res. 2004, 10, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, C.; Spencer, F.H.; Deka, A.; Vaporciyan, A.A.; Spitz, M.R. Postmenopausal hormone therapy and lung cancer risk in the cancer prevention study II nutrition cohort. Cancer Epidemiol. Biomark. Prev. 2008, 17, 655–660. [Google Scholar] [CrossRef]
- Slatore, C.G.; Chien, J.W.; Au, D.H.; Satia, J.A.; White, E. Lung cancer and hormone replacement therapy: Association in the vitamins and lifestyle study. J. Clin. Oncol. 2010, 28, 1540–1546. [Google Scholar] [CrossRef]
- Ganti, A.K.; Sahmoun, A.E.; Panwalkar, A.W.; Tendulkar, K.K.; Potti, A. Hormone replacement therapy is associated with decreased survival in women with lung cancer. J. Clin. Oncol. 2006, 24, 59–63. [Google Scholar] [CrossRef]
- Chlebowski, R.T.; Anderson, G.L.; Gass, M.; Lane, D.S.; Aragaki, A.K.; Kuller, L.H.; Manson, J.E.; Stefanick, M.L.; Ockene, J.; Sarto, G.E.; et al. Estrogen plus progestin and breast cancer incidence and mortality in postmenopausal women. JAMA 2010, 304, 1684–1692. [Google Scholar] [CrossRef]
- Kligerman, S.; White, C. Epidemiology of lung cancer in women: Risk factors, survival, and screening. Am. J. Roentgenol. 2011, 196, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, M.; Watanabe, M.; Yoshida, M.; Sugaya, K.; Endo, Y.; Miyajima, N.; Abe, M.; Sugano, S.; Nakae, D. Enhancement of lung carcinogenesis initiated with 4-(N-hydroxymethylnitrosamino)-1-(3-pyridyl)-1-butanone by Ogg1 gene deficiency in female, but not male, mice. J. Toxicol. Sci. 2009, 34, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Hammoud, Z.; Tan, B.; Badve, S.; Bigsby, R.M. Estrogen promotes tumor progression in a genetically defined mouse model of lung adenocarcinoma. Endocr. Relat. Cancer 2008, 15, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Yaşar, P.; Ayaz, G.; User, S.D.; Güpür, G.; Muyan, M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod. Med. Biol. 2017, 16, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Cheskis, B.J.; Grege, J.G.; Nagpal, S.; Freedman, L.P. Signaling by estrogens. J. Cell. Physiol. 2007, 321, 610–617. [Google Scholar] [CrossRef]
- Couse, J.F.; Korach, K.S. Estrogen receptor null mice: What have we learned and where will they lead us? Endocr. Rev. 1999, 20, 358–417. [Google Scholar] [CrossRef]
- Kumar, S.; Lata, K.; Mukhopadhyay, S.; Mukherjee, T.K. Role of estrogen receptors in pro-oxidative and anti-oxidative actions of estrogens: A perspective. Biochim. Biophys. Acta 2010, 1800, 1127–1135. [Google Scholar] [CrossRef]
- Stabile, L.P.; Davis, A.L.; Gubish, C.T.; Hopkins, T.M.; Luketich, J.D.; Christie, N.; Finkelstein, S.; Siegfried, J.M. Human non-small cell lung tumors and cells derived from normal lung express both estrogen receptor alpha and beta and show biological responses to estrogen. Cancer Res. 2002, 62, 2141–2150. [Google Scholar] [PubMed]
- Ivanova, M.M.; Mazhawidza, W.; Dougherty, S.M.; Minna, J.D.; Klinge, C.M. Activity and intracellular location of estrogen receptors [alpha] and [beta] in human bronchial epithelial cells. Mol. Cell Endocrinol. 2009, 205, 12–21. [Google Scholar] [CrossRef]
- Hong, D.G.; Park, J.Y.; Chong, G.O.; Lee, Y.H.; Lee, H.J.; Shinn, J.U.; Lee, Y.S.; Seong, W.J. Transmembrane G protein-coupled receptor 30 gene polymorphisms and uterine adenomyosis in Korean women. Gynecol. Endocrinol. 2019, 35, 498–501. [Google Scholar] [CrossRef]
- Filardo, E.J.; Quinn, J.A.; Graeber, C.T. Evidence Supporting a Role for GPR30, an Orphan Member of the G-Protein-Coupled Receptor Superfamily, in Rapid Estrogen Signaling. The Identities of Membrane Steroid Receptors; Springer: Boston, MA, USA, 2003. [Google Scholar] [CrossRef]
- Liu, C.; Liao, Y.; Fan, S.; Fu, X.; Xiong, J.; Zhou, S.; Zou, M.; Wang, J. G-protein-coupled estrogen receptor antagonist G-15 decreases estrogen-induced development of non-small cell lung cancer. Oncol. Res. 2019, 27, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Jala, V.R.; Radde, B.N.; Haribabu, B.; Klinge, C.M. Enhanced expression of G-protein coupled estrogen receptor (GPER/GPR30) in lung cancer. BMC Cancer 2012, 12, 624. [Google Scholar] [CrossRef] [PubMed]
- Li-Han, H.; Nei-Min, C.; Shu-Huei, K. Estrogen, estrogen receptor and lung cancer. Int. J. Mol. Sci. 2017, 18, 1713. [Google Scholar] [CrossRef]
- Jacenik, D.; Cygankiewicz, A.I.; Krajewska, W.M. The G protein-coupled estrogen receptor as a modulator of neoplastic transformation. Mol. Cell. Endocrinol. 2016, 429, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Huang, Y.; Wu, C.; Wei, D.; Shi, Y. Activation of G-Protein-Coupled Estrogen Receptor Inhibits the Migration of Human Non-small Cell Lung Cancer Cells via IKK-β/NF-κB Signals. DNA Cell Biol. 2016, 35, 434–442. [Google Scholar] [CrossRef]
- Shen, Y.; Li, C.; Zhou, L.; Huang, J.-A. G protein-coupled oestrogen receptor promotes cell growth of non-small cell lung cancer cells via YAP1/QKI/circNOTCH1/m6A methylated NOTCH1 signalling. J. Cell Mol. Med. 2021, 25, 284–296. [Google Scholar] [CrossRef]
- Prabhakar, C.N. Epidermal growth factor receptor in non-small cell lung cancer. Transl. Lung Cancer Res. 2015, 4, 110–118. [Google Scholar] [CrossRef]
- Pietras, R.J.; Marquez, D.C.; Chen, H.W.; Tsai, E.; Weinberg, O.; Fishbein, M. Estrogen and growth factor receptor interactions in human breast and non-small cell lung cancer cells. Steroids 2005, 70, 372–381. [Google Scholar] [CrossRef]
- Bethune, G.; Bethune, D.; Ridgway, N.; Xu, Z. Epidermal growth factor receptor (EGFR) in lung cancer: An overview and update. J. Thorac. Dis. 2010, 2, 48–51. [Google Scholar]
- Lata, K.; Mukherjee, T.K. Knockdown of receptor for advanced glycation end products attenuate 17α-ethinyl-estradiol dependent proliferation and survival of MCF-7 breast cancer cells. Biochim. Biophys. Acta 2014, 1840, 1083–1091. [Google Scholar] [CrossRef]
- Riggins, R.B.; Lan, J.P.; Zhu, Y.; Klimach, U.; Zwart, A.; Cavalli, L.R.; Haddad, B.R.; Chen, L.; Gong, T.; Xuan, J.; et al. ERR gamma mediates tamoxifen resistance in novel models of invasive lobular breast cancer. Cancer Res. 2008, 68, 8908–8917. [Google Scholar] [CrossRef]
- Giguere, V.; Yang, N.; Segui, P.; Evans, R.M. Identification of a new class of steroid hormone receptors. Nature 1988, 331, 91–94. [Google Scholar] [CrossRef]
- Mukherjee, T.K.; Malik, P.; Hoidal, J.R. The emerging role of estrogen related receptor α in complications of non-small cell lung cancers. Oncol. Lett. 2021, 21, 258. [Google Scholar] [CrossRef]
- Li, P.; Wang, J.; Wu, D.; Ren, X.; Wu, W.; Zuo, R.; Zeng, Q.; Wang, B.; He, X.; Yuan, J.; et al. ERRα is an aggressive factor in lung adenocarcinoma indicating poor prognostic outcomes. Cancer Manag. Res. 2019, 11, 8111–8123. [Google Scholar] [CrossRef] [PubMed]
- Stabile, L.P.; Lyker, J.S.; Gubish, C.T.; Zhang, W.; Grandis, J.R.; Siegfried, J.M. Combined targeting of the estrogen receptor and the epidermal growth factor receptor in non-small cell lung cancer shows enhanced anti-proliferative effects. Cancer Res. 2005, 65, 1459–1470. [Google Scholar] [CrossRef] [PubMed]
- Dowsett, M.; Nicholson, R.I.; Pietras, R.J. Biological characteristics of the pure antiestrogen fulvestrant: Overcoming endocrine resistance. Breast Cancer Res. Treat. 2005, 93, S11–S18. [Google Scholar] [CrossRef]
- Dubey, S.; Siegfried, J.M.; Traynor, A.M. Non-small-cell lung cancer and breast carcinoma: Chemotherapy and beyond. Lancet Oncol. 2006, 7, 416–424. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Barton, M. Signaling, physiological functions and clinical relevance of the G protein-coupled estrogen receptor GPER. Prostaglandins Other Lipid Mediat. 2009, 89, 89–97. [Google Scholar] [CrossRef]
- Study Evaluating the Addition of Fulvestrant to Erlotinib in Stage IIIB/IV Non-Small Cell Lung Cancer. Available online: https://clinicaltrials.gov/Bct2/show/NCT00592007?term=estrogen&rslt=With&cond=Lung+Cancer&draw=2&rank=1 (accessed on 20 December 2020).
- Fulvestrant and Anastrozole as Consolidation Therapy in Postmenopausal Women with Advanced Non-Small Cell Lung Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT00932152?term=estrogen&rslt=With&cond=Lung+Cancer&draw=2&rank=2 (accessed on 20 December 2020).
- Alisertib in Adults with Nonhematological Malignancies, Followed by Alisertib in Lung, Breast, Head and Neck or Gastroesophageal Malignancies. Available online: https://clinicaltrials.gov/ct2/show/NCT01045421?term=estrogen&rslt=With&cond=Lung+Cancer&draw=2&rank=5 (accessed on 20 December 2020).
- Molehin, D.; Rasha, F.; Rahman, R.L.; Pruitt, K. Regulation of aromatase in cancer. Mol. Cell Biochem. 2021, 476, 2449–2464. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Nakajima, M.; Yokoi, T. Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Lett. 2005, 227, 115–124. [Google Scholar] [CrossRef]
- Hachey, D.L.; Dawling, S.; Roodi, N.; Parl, F.F. Sequential action of phase i and ii enzymes cytochrome P450 1B1 and glutathione s-transferase P1 in mammary estrogen metabolism. Cancer Res. 2003, 63, 8492–8499. [Google Scholar]
- Miao, S.; Yang, F.; Wang, Y.; Shao, C.; Zava, D.T.; Ding, Q.; Shi, Y.E. 4-Hydroxy estrogen metabolite, causing genomic instability by attenuating the function of spindle-assembly checkpoint, can serve as a biomarker for breast cancer. Am. J. Transl. Res. 2019, 11, 4992–5007. [Google Scholar] [PubMed]
- Peng, J.; Meireles, S.I.; Xu, X.; Smith, W.E.; Slifker, M.J.; Riel, S.L.; Zhai, S.; Zhang, G.; Ma, X.; Kurzer, M.S.; et al. Estrogen metabolism in the human lung: Impact of tumorigenesis, smoke, sex and race/ethnicity. Oncotarget 2017, 8, 106778–106789. [Google Scholar] [CrossRef] [PubMed]
- Siegfried, J.M.; Stabile, L.P. Estrongenic steroid hormones in lung cancer. Semin. Oncol. 2014, 41, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Siegfried, J.M.; Gubish, C.T.; Rothstein, M.E.; Henry, C.; Stabile, L.P. Combining the multitargeted tyrosine kinase inhibitor vandetanib with the antiestrogen fulvestrant enhances its antitumor effect in non-small cell lung cancer. J. Thorac. Oncol. 2012, 7, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Mayer, J.A.; Mazumdar, A.; Fertuck, K.; Kim, H.; Brown, M.; Powel, H. Brown, Estrogen induces c-myc gene expression via an upstream enhancer activated by the estrogen receptor and the AP-1 transcription factor. Mol. Endocrinol. 2011, 25, 1527–1538. [Google Scholar] [CrossRef]
- Bergh, J.C.S. Gene Amplification in human lung cancer. Am. Rev. Respir. Dis. 1990, 142, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Alkis, N.; Arpaci, E.; Aslan, U.Y. Targeted therapies in non-small cell lung cancer: Review. Turk. Klin. Arch. Lung 2010, 11, 20–25. [Google Scholar]
- Cline, M.J.; Battifora, H. Abnormalities of protooncogenes in non-small cell lung cancer. Cancer 1987, 60, 2669–2674. [Google Scholar] [CrossRef]
- Nelson, A.L.; Kaunitz, A.M.; Kroll, R.; Simon, J.A.; Poindexter, A.N.; Castaño, P.M.; Ackerman, R.T.; Flood, L.; Chiodo, J.A., 3rd; Garner, E.I.; et al. Efficacy, safety, and tolerability of a levonorgestrel/ethinyl estradiol transdermal delivery system: Phase 3 clinical trial results. Contraception 2021, 103, 137–143. [Google Scholar] [CrossRef]
- Bhavnani, B.R.; Stanczyk, F.Z. Pharmacology of conjugated equine estrogens: Efficacy, safety and mechanism of action. J. Steroid Biochem. Mol. Biol. 2014, 142, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Hilakivi-Clarke, L.; de Assis, S.; Warri, A. Exposures to synthetic estrogens at different times during the life, and their effect on breast cancer risk. J. Mammary Gland Biol. Neoplasia 2013, 18, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.S.; Chen, H.Q.; Chen, Y.S.; Qiu, K.F.; Zheng, X.B.; Li, G.C.; Yang, H.D.; Wen, C.J. Bisphenol A stimulates human lung cancer cell migration via upregulation of matrix metalloproteinases by GPER/EGFR/ERK1/2 signal pathway. Biomed. Pharmacother. 2014, 68, 1037–1043. [Google Scholar] [CrossRef]
- Li, J.; Ji, Z.; Luo, X.; Li, Y.; Yuan, P.; Long, J.; Shen, N.; Lu, Q.; Zeng, Q.; Zhong, R.; et al. Urinary bisphenol A andits interaction with ESR1 genetic polymorphism associated with non-small cell lung cancer: Findings from a case-control study in Chinese population. Chemosphere 2020, 254, 126835. [Google Scholar] [CrossRef]
- Darbre, P.D. Environmental oestrogens, cosmetics and breast cancer. Best Pract. Res. Clin. Endocrinol. Metab. 2006, 20, 121–143. [Google Scholar] [CrossRef]
- Barbieri, R.L.; Gochberg, J.; Ryan, K.J. Nicotine, cotinine and anabasine inhibit aromatase in human trophoblast in vitro. J. Clin. Investig. 1986, 77, 1727–1733. [Google Scholar] [CrossRef]
- Wang, S.L.; Chang, Y.C.; Chao, H.R.; Li, C.M.; Li, L.A.; Lin, L.Y.; Papke, O. Body burdens of polychlorinated dibenzo-p-dioxins, dibenzofurans and biphenyls and their relations to estrogen metabolism in pregnant women. Environ. Health Perspect. 2006, 114, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.; Grantha, E.; Liu, B.; Macapagai, R.; Willingham, E.; Baskin, L.S. In utero exposure to benzophenone-2 causes hypospadias through an estrogen receptor dependent mechanisms. J. Urol. 2007, 178, 1637–1642. [Google Scholar] [CrossRef]
- Birnbaum, L.S.; Fenton, S. Cancer and developmental exposure to endocrine disruptors. Environ. Health Perspect. 2003, 111, 389–394. [Google Scholar] [CrossRef]
- Andersen, H.R.; Bonefeld-Jorgensen, E.C.; Nielsen, F.; Jarfeldt, K.; Jayatissa, M.N.; Vinggaard, A.M. Estrogen effects in vitro and in vivo of the fungicide fenarimol. Toxicol. Lett. 2006, 163, 142–152. [Google Scholar] [CrossRef]
- Shen, J.; Liu, J.; Xie, Y.; Diwan, B.A.; Waalkes, M.P. Fetal onset of aberrant gene expression relevant to pulmonary carcinogenesis in lung adenocarcinoma development induced by in utero arsenic exposure. Toxicol. Sci. 2007, 95, 313–320. [Google Scholar] [CrossRef]
- Watson, W.H.; Yager, Y.D. Arsenic: Extension of its endocrine disruption potential to interference with estrogen receptor-mediated signaling. Toxicol. Sci. 2007, 98, 1–4. [Google Scholar] [CrossRef]
- Coutelle, C.; Hohn, B.; Benesova, M.; Oneta, C.M.; Quattrochi, P.; Roth, H.J.; Schmidt-Gayk, H.; Schneeweiss, A.; Bastert, G.; Seitz, H.K. Risk factors in alcohol associated breast cancer: Alcohol dehydrogenase polymorphism and estrogens. Int. J. Oncol. 2004, 25, 1127–1132. [Google Scholar]
- Fan, S.; Meng, Q.; Gao, B.; Grossman, J.; Yadegari, M.; Goldberg, I.D.; Rosen, E.M. Alcohol stimulates estrogen receptor signaling in human breast cancer cell lines. Cancer Res. 2000, 60, 5635–56339. [Google Scholar]
- Matsuo, K.; Ito, H.; Yatabe, Y.; Hiraki, A.; Hirose, K.; Wakai, K. Risk factors differ for non-small-cell lung cancer with and without EFGR mutation: Assessment of smoking and sex by a case-control study in Japanese. Cancer Sci. 2007, 98, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Majidi, M.; Al-Wadel, H.A.; Takahashi, T.; Schuller, H.M. Non-genomic beta estrogen receptor enchance beta1 adrenergic signaling induced by the nicotine-derived carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1- butanone in human small airway epithelial cells. Cancer Res. 2007, 67, 6863–6874. [Google Scholar] [CrossRef]
- Pauly, J.R. Gender differences in tobacco smoking dynamics and the neuropharmacological actions of nicotine. Front Biosci. 2008, 13, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Samet, J.M. Environmental causes of lung cancer: What do we know in 2003? Chest 2004, 125, 80–83. [Google Scholar] [CrossRef]
- Muggi, M.E.; Elbert, J.O.; Robertson, C.; Hurt, R.D. Waking a sleeping ginat: The tobacco industry’s response to the Polonium-210 issue. Am. J. Pub. Health 2008, 98, 1643–1650. [Google Scholar] [CrossRef] [PubMed]
- Fucic, A.; Gamulin, M.; Ferencic, Z.; Katic, J.; von Krauss, M.K.; Bartonova, A.; Merlo, D.F. Environmental exposure to xenoestrogens and oestrogen related cancers: Reproductive system, breast, lung, kidney, pancreas and brain. Environ. Health 2012, 11, S8. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, O.K.; Marquez-Garban, D.C.; Fishbein, M.C.; Goodglick, L.; Garban, H.J.; Dubinett, S.M.; Pietras, R.J. Aromatase inhibitors in human lung cancer therapy. Cancer Res. 2005, 65, 11287–11291. [Google Scholar] [CrossRef] [PubMed]
- Marquez-Garban, D.C.; Chen, H.W.; Goodglick, L.; Fishbein, M.C.; Pietras, R.J. Targeting aromatase and estrogen signaling in human non-small cell lung cancer. Ann. N. Y. Acad. Sci. 2009, 1155, 194–205. [Google Scholar] [CrossRef]
- Skjefstad, K.; Grindstad, T.; Khanehkenari, M.R.; Richardsen, E.; Donnem, T.; Kilvaer, T.; Andersen, S.; Bremnes, R.M.; Busund, L.T.; Al-Saad, S. Prognostic relevance of estrogen receptor alpha, beta and aromatase expression in non-small cell lung cancer. Steroids 2016, 113, 5–13. [Google Scholar] [CrossRef]
- Flouriot, G.; Brand, H.; Denger, S.; Metivier, R.; Kos, M.; Reid, G.; Sonntag-Buck, V.; Gannon, F. Identification of a new isoform of the human estrogen receptor-alpha (hER-alpha) that is encoded by distinct transcripts and that is able to repress hER-alpha activation function 1. EMBO J. 2000, 19, 4688–4700. [Google Scholar] [CrossRef] [PubMed]
- Barraille, P.; Chinestra, P.; Bayard, F.; Faye, J.C. Alternative initiation of translation accounts for a 67/45 kDa dimorphism of the human estrogen receptor ER alpha. Biochem. Biophys. Res. Commun. 1999, 257, 84–88. [Google Scholar] [CrossRef]
- Maaroufi, Y.; Lacroix, M.; Lespagnard, L.; Journé, F.; Larsimont, D.; Leclercq, G. Estrogen receptor of primary breast cancers: Evidence for intracellular proteolysis. Breast Cancer Res. 2000, 2, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, G.G.; Carlsson, B.O.; Grandien, K.A.J.; Enmark, E.; Häggblad, J.; Nilsson, S.; Gustafsson, J.A. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors α and β. Endocrinology 1997, 138, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Mollerup, S.; Jorgensen, K.; Berge, G.; Haugen, A. Expression of estrogen receptors alpha and beta in human lung tissue and cell lines. Lung Cancer 2002, 37, 153–159. [Google Scholar] [CrossRef]
- Ivanova, M.M.; Mazhawidza, W.; Dougherty, S.M.; Klinge, C.M. Sex differences in estrogen receptor sub-cellular location and activity in lung adenocarcinoma cells. Am. J. Respir. Cell Mol. Biol. 2010, 42, 320–330. [Google Scholar] [CrossRef]
- Patrone, C.; Cassel, T.N.; Pettersson, K.; Piao, Y.S.; Cheng, G.; Ciana, P.; Maggi, A.; Warner, M.; Gustafsson, J.A.; Nord, M. Regulation of postnatal lung development and homeostasis by estrogen receptor β. Mol. Cell. Biol. 2003, 23, 8542–8552. [Google Scholar] [CrossRef]
- Morani, A.; Barros, R.P.; Imamov, O.; Kjell, H.; Arner, A.; Warner, M.; Gustafsson, J.A. Lung dysfunction causes systemic hypoxia in estrogen receptor beta knockout (ERβ-/-) mice. Proc. Natl. Acad. Sci. USA 2006, 103, 7165–7169. [Google Scholar] [CrossRef] [PubMed]
- Ciana, P.; DiLuccio, G.; Belcredito, S.; Pollio, G.; Vegeto, E.; Tatangelo, L.; Tiveron, C.; Maggi, A. Engineering of a mouse for the in vivo profiling of estrogen receptor activity. Mol. Endocrinol. 2001, 15, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Lemmen, J.G.; Arends, R.J.; van Boxtel, A.L.; van der Saag, P.T.; van der Burg, B. Tissue- and time-dependent estrogen receptor activation in estrogen reporter mice. J. Mol. Endocrinol. 2004, 32, 689–701. Available online: https://www.jstor.org/stable/3435613 (accessed on 27 October 2021). [CrossRef] [PubMed]
- Kerr, A.; Eliason, J.F.; Wittliff, J.L. Steroid receptor and growth factor receptor expression in human non-small cell lung cancers using cells produced by laser-capture microdissection. Adv. Exp. Med. Biol. 2008, 617, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Lara, V.; Pena-Mirabal, E.; Baez-Saldana, R.; Esparza-Silva, A.L.; Garcia-Zepeda, E.; Cerbon Cervantes, M.A.; Diaz, D.; Fortoul, T.I. Estrogen receptor beta and CXCR4/CXCL12 expression: Differences by sex and hormonal status in lung adenocarcinoma. Arch. Med. Res. 2014, 45, 158–169. [Google Scholar] [CrossRef]
- Hershberger, P.A.; Vasquez, A.C.; Kanterewicz, B.; Land, S.; Siegfried, J.M.; Nichols, M. Regulation of endogenous gene expression in human non-small cell lung cancer cells by estrogen receptor ligands. Cancer Res. 2005, 65, 1598–1605. [Google Scholar] [CrossRef]
- Kawai, H. Estrogen receptors as the novel therapeutic biomarker in non-small cell lung cancer. World J. Clin. Oncol. 2014, 5, 1020–1027. [Google Scholar] [CrossRef]
- Pelekanou, V.; Anastasiou, E.; Bakogeorgou, E.; Notas, G.; Kampa, M.; Garcia-Milian, R.; Lavredaki, K.; Moustou, E.; Chinari, G.; Arapantoni, P.; et al. Estrogen receptor-alpha isoforms are the main estrogen receptors expressed in non-small cell lung carcinoma. Steroids 2019, 142, 65–76. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Z.; Ding, X.; Zhirong, S.; Zhentao, L.; Tongtong, A.; Jianchun, D.; Jia Zhong, J.; Wu, M.; Zhao, J.; et al. ER-beta localization influenced outcomes of EGFR-TKI treatment in NSCLC patients with EGFR mutations. Sci. Rep. 2015, 5, 11392. [Google Scholar] [CrossRef]
- Thomas, P.; Pang, Y.; Filardo, E.J.; Dong, J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 2005, 146, 624–632. [Google Scholar] [CrossRef]
- Gao, X.; Cai, Y.; Zhuo, W.; He, W.; Cao, S.; Xu, R.; Chen, H. Estrogen receptors promote NSCLC progression by modulating the membrane receptor signaling network: A systems biology perspective. J. Transl. Med. 2019, 17, 308. [Google Scholar] [CrossRef]
- Kato, S.; Endoh, H.; Masuhiro, Y.; Kitamoto, T.; Uchiyama, S.; Sasaki, H.; Masushige, S.; Gotoh, Y.; Nishida, E.; Kawashima, H.; et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science 1995, 270, 1491–1494. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.T.; Chang, Y.L.; Shih, J.Y.; Lee, Y.C. The significance of estrogen receptor beta in 301 surgically treated non-small cell lung cancers. J. Thorac. Cardiovasc. Surg. 2005, 130, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.G.; Prysak, G.M.; Murphy, V.; Lonardo, F.; Pass, H.; Schwartz, J.; Brooks, S. Nuclear estrogen receptor beta in lung cancer: Expression and survival differences by sex. Clin. Cancer Res. 2005, 11, 7280–7287. [Google Scholar] [CrossRef]
- Skov, B.G.; Fischer, B.M.; Pappot, H. Oestrogen receptor β over expression in males with non-small cell lung cancer is associated with better survival. Lung Cancer 2008, 59, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Pinton, G.; Manzotti, B.; Balzano, C.; Moro, L. Expression and clinical implications of estrogen receptors in thoracic malignancies: A narrative review. Thorac. Dis. 2021, 13, 1851–1863. [Google Scholar] [CrossRef]
- Rodriguez-Lara, V.; Costa, M.R.A. An overview of lung cancer in women and the impact of estrogen in lung carcinogenesis and lung cancer treatment. Front. Med. 2021, 8, 600121. [Google Scholar] [CrossRef]
- Musial, C.; Zaucha, R.; Kuban-Jankowska, A.; Konieczna, L.; Belka, M.; Gammazza, A.M.; Baczek, T.; Cappello, F.; Wozniak, M.; Gorska-Ponikowska, M. Plausible role of estrogens in pathogenesis, progression and therapy of lung cancer. Int. J. Environ. Res. Public Health 2021, 18, 648. [Google Scholar] [CrossRef]
- Koutras, A.; Giannopoulou, E.; Kritikou, I.; Antonacopoulou, A.; Evans, T.R.; Papavassiliou, A.G.; Kalofonos, H. Antiproliferative effect of exemestane in lung cancer cells. Mol. Cancer 2009, 8, 109. [Google Scholar] [CrossRef]
- Giannopoulou, E.; Siatis, K.E.; Metsiou, D.; Kritikou, I.; Papachristou, D.J.; Kalofonou, M.; Koutras, A.; Athanassiou, G.; Kalofonos, H.P. The inhibition of aromatase alters the mechanical and rheological properties of non-small-cell lung cancer cell lines affecting cell migration. Biochim. Biophys. Acta 2015, 1853, 328–337. [Google Scholar] [CrossRef]
- Tang, H.; Liao, Y.; Zhang, C.; Chen, G.; Xu, L.; Liu, Z.; Fu, S.; Yu, L.; Zhou, S. Fulvestrant-mediated inhibition of estrogen receptor signaling slows lung cancer progression. Oncol. Res. 2014, 22, 13–20. [Google Scholar] [CrossRef]
- Miki, Y.; Abe, K.; Suzuki, S.; Suzuki, T.; Sasano, H. Suppression of estrogen actions in human lung cancer. Mol. Cell Endocrinol. 2011, 340, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.J.; Li, J.; Hao, F.R.; Yuan, Y.; Li, J.Y.; Wei, L.; Tian-Yan, Z. Dexamethasone suppresses the growth of human non-small cell lung cancer via inducing estrogen sulfotransferase and inactivating estrogen. Acta Pharmacol. Sin. 2016, 37, 845–856. [Google Scholar] [CrossRef]
- Bouchardy, C.; Benhamou, S.; Schaffar, R.; Verkooijen, H.M.; Fioretta, G.; Schubert, H.; Vinh-Hung, V.; Soria, J.C.; Vlastos, G.; Rapiti, E. Lung cancer mortality risk among breast cancer patients treated with anti-estrogens. Cancer 2011, 117, 1288–1295. [Google Scholar] [CrossRef]
- Lother, S.A.; Harding, G.A.; Musto, G.; Navaratnam, S.; Pitz, M.W. Antiestrogen use and survival of women with non-small cell lung cancer in Manitoba, Canada. Horm. Cancer 2013, 4, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Lara, V.; Hernandez-Martinez, J.-M.; Arrieta, O. Influence of estrogen in non-small cell lung cancer and its clinical implications. J. Thorac. Dis. 2018, 10, 482–497. [Google Scholar] [CrossRef]
- Smida, T.; Bruno, T.C.; Stabile, L.P. Influence of Estrogen on the NSCLC microenvironment: A comprehensive picture and clinical implications. Front. Oncol. 2020, 10, 137. [Google Scholar] [CrossRef] [PubMed]
- Traynor, A.M.; Schiller, J.H.; Stabile, L.P.; Kolesar, J.M.; Eickhoff, J.C.; Dacic, S.; Hoang, T.; Dubey, S.; Marcotte, S.M.; Siegfried, J.M. Pilot study of gefitinib and fulvestrant in the treatment of post-menopausal women with advanced non-small cell lung cancer. Lung Cancer 2009, 64, 51–59. [Google Scholar] [CrossRef]
- Garon, E.B.; Sadeghi, S.F.F.; Kabbinavar, K.L.; Reckamp, D.C.; Marquez-Garban, L.P.; Stabile, L.; Goodglick, S.M.; Dubinett, J.M.; Siegfried, R.; Pietras, J. Interim safety analysis of a phase II study of erlotinib (E) alone or combined with fulvestrant (F) in previously treated patients with advanced non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2008, 26, 19091. [Google Scholar] [CrossRef]
- Collins, I.M.; Nicholson, S.A.; O’Byrne, K.J. A lung cancer responding to hormonal therapy. J. Thorac. Oncol. 2010, 5, 749–750. [Google Scholar] [CrossRef]
- Langer, C.J.; O’Byrne, K.J.; Socinski, M.A.; Mikhailov, S.M.; Leśniewski-Kmak, K.; Smakal, M.; Ciuleanu, T.E.; Orlov, S.V.; Dediu, M.; Heigener, D.; et al. Phase III trial comparing paclitaxel poliglumex (CT-2103, PPX) in combination with carboplatin versus standard paclitaxel and carboplatin in the treatment of PS 2 patients with chemotherapy-naïve advanced non-small cell lung cancer. J. Thorac. Oncol. 2008, 3, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Young, P.A.; Márquez-Garbán, D.C.; Noor, Z.S.; Moatamed, N.; Elashoff, D.; Grogan, T.; Romero, T.; Sasano, H.; Saito, R.; Rausch, R.; et al. Investigation of combination treatment with an aromatase inhibitor exemestane and carboplatin-based therapy for postmenopausal women with advanced NSCLC. JTO Clin. Res. Rep. 2021, 2, 100150. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Siegfried, J.M.; Stabile, L.P.; Young, P.A.; Marquez-Garban, D.C.; Park, D.J.; Patel, R.; Hu, E.H.; Sadeghi, S.; Parikh, R.J.; et al. Randomized phase II study of fulvestrant and erlotinib compared with erlotinib alone in patients with advanced or metastatic non-small cell lung cancer. Lung Cancer 2018, 123, 91–98. [Google Scholar] [CrossRef]
- Xu, R.; Shen, H.; Guo, R.; Sun, J.; Gao, W.; Shu, Y. Combine therapy of gefitinib and fulvestrant enhances antitumor effects on NSCLC cell lines with acquired resistance to gefitinib. Biomed. Pharmacother. 2012, 66, 384–389. [Google Scholar] [CrossRef]
- Garon, E.S.; Siegfried, J.M.; Dubinett, S.M.; Elashoff, R.M.; Park, D.J.; Parikh, R.J.; Patel, R.; Hu, E.H.; Reckamp, K.L.; Adams, B.; et al. Results of TORI-L-03, a randomized, multicenter phase II clinical trial of erlotinib (E) or E + fulvestrant (F) in previously treated advanced nonsmall cell lung cancer (NSCLC). In Proceedings of the 104th Annual Meeting of the American, Philadelphia, WA, USA, 6–10 April 2013. [Google Scholar] [CrossRef]
- Carmeci, C.; Thompson, D.A.; Ring, H.Z.; Francke, U.; Weigel, R.J. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics 1997, 45, 607–617. [Google Scholar] [CrossRef]
- Takada, Y.; Kato, C.; Kondo, S.; Korenaga, R.; Ando, J. Cloning of cDNAs encoding G protein-coupled receptor expressed in human endothelial cells exposed to fluid shear stress. Biochem. Biophys. Res. Commun. 1997, 240, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.P.; Mathie, A.; Peters, J.A. Guide to receptors and channels (GRAC), 5th edition. Br. J. Pharmacol. 2011, 164, S1–S324. [Google Scholar] [CrossRef]
- O’Dowd, B.F.; Nguyen, T.; Marchese, A.; Cheng, R.; Lynch, K.R.; Heng, H.H.; Kolakowski, L.F., Jr.; George, S.R. Discovery of three novel G-protein-coupled receptor genes. Genomics 1998, 47, 310–313. [Google Scholar] [CrossRef]
- Filardo, E.J.; Quinn, J.; Pang, Y.; Graeber, C.; Shaw, S.; Dong, J.; Thomas, P. Activation of the novel estrogen receptor G Protein-Coupled Receptor 30 (GPR30) at the plasma membrane. Endocrinology 2007, 148, 3236–3245. [Google Scholar] [CrossRef]
- Govind, A.P.; Thampan, R.V. Membrane associated estrogen receptors and related proteins: Localization at the plasma membrane and the endoplasmic reticulum. Mol. Cell Biochem. 2003, 253, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liao, Y.; Fan, S.; Tang, H.; Jiang, Z.; Zhou, B.; Xiong, J.; Zhou, S.; Zou, M.; Wang, J. G protein-coupled estrogen receptor (GPER) mediates NSCLC progression induced by 17beta-estradiol (E2) and selective agonist G1. Med. Oncol. 2015, 32, 104. [Google Scholar] [CrossRef]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R., Jr. Receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol. Endocrinol. 2000, 14, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.; Filardo, E.J.; Lolait, S.J.; Thomas, P.; Maggiolini, M.; Prossnitz, E.R. Twenty years of the G protein-coupled estrogen receptor GPER: Historical and personal perspectives. J. Steroid Biochem. Mol. Biol. 2018, 176, 4–15. [Google Scholar] [CrossRef]
- Kvingedal, A.M.; Smeland, E.B. A novel putative g-protein-coupled receptor expressed in lung, heart and lymphoid tissue. FEBS Lett. 1997, 407, 59–62. [Google Scholar] [CrossRef]
- Avino, S.; De Marco, P.; Cirillo, F.; Santolla, M.F.; De Francesco, E.M.; Perri, M.G.; Rigiracciolo, D.; Dolce, V.; Belfiore, A.; Maggiolini, M.; et al. Stimulatory actions of IGF-I are mediated by IGF-IR cross-talk with GPER and DDR1 in mesothelioma and lung cancer cells. Oncotarget 2016, 7, 52710–52728. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Barton, M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat. Rev. Endocrinol. 2011, 7, 715–726. [Google Scholar] [CrossRef]
- Cheng, S.B.; Dong, J.; Pang, Y.; LaRocca, J.; Hixon, M.; Thomas, P.; Filardo, E.J. Anatomical location and redistribution of G protein-coupled estrogen receptor-1 during the estrus cycle in mouse kidney and specific binding to estrogens but not aldosterone. Mol. Cell Endocrinol. 2014, 382, 950–959. [Google Scholar] [CrossRef]
- Osborne, C.; Wakeling, A.; Nicholson, R.I. Fulvestrant: An oestrogen receptor antagonist with a novel mechanism of action. Br. J. Cancer 2004, 90, S2–S6. [Google Scholar] [CrossRef]
- Filardo, E.J.; Quinn, J.A.; Frackelton, A.R., Jr.; Bland, K.I. Estrogen action via the G protein-coupled receptor, GPR30: Stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol. Endocrinol. 2002, 16, 70–84. [Google Scholar] [CrossRef]
- Catalano, S.; Giordano, C.; Panza, S.; Chemi, F.; Bonofiglio, D.; Lanzino, M.; Rizza, P.; Romeo, F.; Fuqua, S.A.W.; Maggiolini, M.; et al. Tamoxifen through GPER upregulates aromatase expression: A novel mechanism sustaining tamoxifen-resistant breast cancer cell growth. Breast Cancer Res. Treat. 2014, 146, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Chourasia, T.K.; Pang, Y.; Thomas, P. The catechol estrogen, 2-hydroxyestradiol-17 beta, acts as a G protein-coupled estrogen receptor 1 (GPER/GPR30) antagonist to promote the resumption of meiosis in zebrafish oocytes. Biol. Reprod. 2015, 92, 69. [Google Scholar] [CrossRef] [PubMed]
- Prossnitz, E.R. GPER modulators: Opportunity Nox on the heels of a class Akt. J. Steroid Biochem. Mol. Biol. 2018, 176, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Kajta, M.; Litwa, E.; Rzemieniec, J.; Wnuk, A.; Lason, W.; Zelek-Molik, A.; Nalepa, I.; Grzegorzewska-Hiczwa, M.; Tokarski, K.; Golas, A.; et al. Isomer-nonspecific action of dichlorodiphenyltrichloroethane on aryl hydrocarbon receptor and G-protein-coupled receptor 30 intracellular signaling in apoptotic neuronal cells. Mol. Cell Endocrinol. 2014, 392, 90–105. [Google Scholar] [CrossRef]
- Thomas, P.; Dong, J. Binding and activation of the seven-transmembrane estrogen receptor GPR30 by environmental estrogens: A potential novel mechanism of endocrine disruption. J. Steroid Biochem. Mol. Biol. 2006, 102, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Bologa, C.G.; Revankar, C.M.; Young, S.M.; Edwards, B.S.; Arterburn, J.B.; Kiselyov, A.S.; Parker, M.A.; Tkachenko, S.E.; Savchuck, N.P.; Sklar, L.A.; et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat. Chem. Biol. 2006, 2, 207–212. [Google Scholar] [CrossRef]
- Blasko, E.; Haskell, C.A.; Leung, S.; Gualtieri, G.; Halks-Miller, M.; Mahmoudi, M.; Dennis, M.K.; Prossnitz, E.R.; Karpus, W.J.; Horuk, R. Beneficial role of the GPR30 agonist G-1 in an animal model of multiple sclerosis. J. Neuroimmunol. 2009, 29, 67–77. [Google Scholar] [CrossRef]
- Dennis, M.K.; Burai, R.; Ramesh, C.; Petrie, W.K.; Alcon, S.N.; Nayak, T.K.; Bologa, C.G.; Leitao, A.; Brailoiu, E.; Deliu, E.; et al. In vivo effects of a GPR30 antagonist. Nat. Chem. Biol. 2009, 5, 421. [Google Scholar] [CrossRef]
- Dennis, M.K.; Field, A.S.; Burai, R.; Ramesh, C.; Petrie, W.K.; Bologa, C.G.; Oprea, T.I.; Yamaguchi, Y.; Hayashi, S.I.; Sklar, L.A.; et al. Identification of a GPER/GPR30 antagonist with improved estrogen receptor counter selectivity. J. Steroid Biochem. Mol. Biol. 2011, 127, 358–366. [Google Scholar] [CrossRef]
- Xu, S.; Yu, S.; Dong, D.; Lee, L.T.O. G-Protein coupled estrogen receptor: A potential therapeutic target in cancer. Front. Endocrinol. 2019, 10, 275. [Google Scholar] [CrossRef]
- Rouhimoghadam, M.; Lu, A.S.; Salem, A.K.; Filardo, E.J. Therapeutic perspectives on the modulation of G-Protein coupled estrogen receptor, GPER, Function. Front. Endocrinol. 2020, 11, 591217. [Google Scholar] [CrossRef]
- Levin, E.R. Bidirectional signaling between the estrogen receptor and the epidermal growth factor receptor. Mol. Endocrinol. 2003, 17, 309–317. [Google Scholar] [CrossRef]
- Deng, F.; Li, M.; Shan, W.L.; Qian, L.T.; Meng, S.P.; Zhang, X.L.; Wang, B.L. Correlation between epidermal growth factor receptor mutations and the expression of estrogen receptor-beta in advanced non-small cell lung cancer. Oncol. Lett. 2017, 13, 2359–2365. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Koh, Y.; Ando, M.; Ito, N.; Takeo, S.; Adachi, H.; Tagawa, T.; Kakegawa, S.; Yamashita, M.; Kataoka, K.; et al. Prospective analysis of oncogenic driver mutations and environmental factors: Japan molecular epidemiology for lung cancer study. J. Clin. Oncol. 2016, 34, 2247–2257. [Google Scholar] [CrossRef]
- Pietras, R.J.; Márquez-Garbán, D.C. Membrane-associated estrogen receptor signaling pathways in human cancers. Clin. Cancer Res. 2007, 13, 4672–4676. [Google Scholar] [CrossRef]
- Nose, N.; Sugio, K.; Oyama, T.; Nozoe, T.; Uramoto, H.; Iwata, T.; Onitsuka, T.; Yasumoto, K. Association between estrogen receptor expression and epidermal growth factor receptor mutation in the postoperative prognosis of adenocarcinoma of the lung. J. Clin. Oncol. 2009, 27, 411–417. [Google Scholar] [CrossRef]
- Zhao, X.Z.; Liu, Y.; Zhou, L.J.; Wang, Z.Q.; Wu, Z.H.; Yang, X.Y. Role of estrogen in lung cancer based on the estrogen receptor-epithelial mesenchymal transduction signaling pathways. OncoTargets Ther. 2015, 8, 2849–2863. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Pietras, R.J.; Finn, R.S.; Kamranpour, N.; Pitts, S.; Márquez-Garbán, D.C.; Desai, A.J.; Dering, J.; Hosmer, W.; von Euw, E.M.; et al. Antiestrogen fulvestrant enhances the antiproliferative effects of epidermal growth factor receptor inhibitors in human non-small-cell lung cancer. J. Thorac. Oncol. 2013, 8, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Nuclear Receptors Nomenclature Committee. A unified nomenclature system for the nuclear receptor superfamily. Cell 1999, 97, 161–163. [Google Scholar] [CrossRef]
- Tremblay, A.M.; Giguère, V. The NR3B subgroup: An overview. Nucl. Recept. Signal. 2007, 5, e009. [Google Scholar] [CrossRef]
- Heard, D.J.; Norby, P.L.; Holloway, J.; Vissing, H. Human ERRgamma, a third member of the estrogen receptor-related receptor (ERR) subfamily of orphan nuclear receptors: Tissue-specific isoforms are expressed during development and in the adult. Mol. Endocrinol. 2000, 14, 382–392. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bonnelye, E.; Vanacker, J.M.; Dittmar, T.; Begue, A.; Desbiens, X.; Denhardt, D.T.; Aubin, J.E.; Laudet, V.; Fournier, B. The ERR-1 orphan receptor is a transcriptional activator expressed during bone development. Mol. Endocrinol. 1997, 11, 905–916. [Google Scholar] [CrossRef][Green Version]
- Pettersson, K.; Svensson, K.; Mattsson, R.; Carlsson, B.; Ohlsson, R.; Berkenstam, A. Expression of a novel member of estrogen response element-binding nuclear receptors is restricted to the early stages of chorion formation during mouse embryogenesis. Mech. Dev. 1996, 54, 211–223. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.; Wong, C. Oestrogen-related receptor alpha inverse agonist XCT-790 arrests A549 lung cancer cell population growth by inducing mitochondrial reactive oxygen species production. Cell Prolif. 2010, 43, 103–113. [Google Scholar] [CrossRef]
- Makowiecki, C.; Nolte, A.; Sutaj, B.; Keller, T.; Avci-Adali, M.; Stoll, H.; Schlensak, C.; Wendel, H.P.; Walker, T. New basic approach to treat non-small cell lung cancer based on RNA-interference. Thorac. Cancer 2014, 5, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.W.; Guan, B.Z.; Yin, L.H.; Liu, F.N.; Hu, B.; Zheng, Q.Y.; Li, F.L.; Zhong, Y.X.; Chen, Y. Effects of estrogen-related receptor alpha (ERRα) on proliferation and metastasis of human lung cancer A549 cells. J. Huazhong Univ. Sci. Technolog. Med. Sci. 2014, 34, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Guan, X.; Liang, N.; Li, S. Estrogen-related receptor alpha triggers the proliferation and migration of human non-small cell lung cancer via interleukin-6. Cell Biochem. Funct. 2018, 36, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, M.; Liu, J.; Ni, J.; Jiao, Y.; Bai, C. Up regulation of IL-6 is involved in di (2-ethylhexyl) phthalate (DEHP) induced migration and invasion of non-small cell lung cancer (NSCLC) cells. Biomed. Pharmacother. 2017, 89, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H. Di(2-ethylhexyl) phthalate promotes lung cancer cell line A549 progression via Wnt/β-catenin signaling. J. Toxicol. Sci. 2019, 44, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Busch, B.B.; Stevens, W.C., Jr.; Martin, R.; Ordentlich, P.; Zhou, S.; Sapp, D.W.; Horlick, R.A.; Mohan, R. Identification of a selective inverse agonist for the orphan nuclear receptor estrogen-related receptor alpha. J. Med. Chem. 2004, 47, 5593–5596. [Google Scholar] [CrossRef]
- Willey, P.J.; Murray, I.R.; Qian, J.; Busch, B.B.; Stevens, W.C., Jr.; Martin, R.; Mohan, R.; Zhou, S.; Ordentlich, P.; Wei, P.; et al. Regulation of PPARgamma coactivator 1alpha (PGC-1alpha) signaling by an estrogen-related receptor alpha (ERRalpha) ligand. Proc. Natl. Acad. Sci. USA 2004, 101, 8912–8917. [Google Scholar] [CrossRef] [PubMed]
- Chisamore, M.J.; Cunningham, M.E.; Flores, O.; Wilkinson, H.A.; Chen, J.D. Characterization of a novel small molecule subtype specific estrogen-related receptor alpha antagonist in MCF-7 breast cancer cells. PLoS ONE 2009, 4, e5624. [Google Scholar] [CrossRef]
- Teng, C.T.; Beames, B.; Merrick, B.A.; Martin, N.; Romeo, C.; Jetten, A.M. Development of a stable cell line with an intact PGC-1α/ERRα axis for screening environmental chemicals. Biochem. Biophys. Res. Commun. 2014, 444, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Teng, C.T.; Hsieh, J.H.; Zhao, J.; Huang, R.; Xia, M.; Martin, N.; Gao, X.; Dixon, D.; Auerbach, S.S.; Witt, K.L.; et al. Development of novel cell lines for high-throughput screening to detect estrogen-related receptor alpha modulators. SLAS Discov. 2017, 22, 720–731. [Google Scholar] [CrossRef]
- Wei, W.; Schwaid, A.G.; Wang, X.; Wang, X.; Chen, S.; Chu, Q.; Saghatelian, A.; Wan, Y. Ligand activation of ERRα by cholesterol mediates statin and bisphosphonate effects. Cell Metab. 2016, 23, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Mazieres, J.; Barlesi, F.; Rouquette, I.; Molinier, O.; Besse, B.; Monnet, I.; Audigier-Valette, C.; Toffart, A.-C.; Renault, P.A.; Fraboulet, S.; et al. Randomized phase II trial evaluating treatment with EGFR-TKI associated with antiestrogen in women with non-squamous advanced-stage NSCLC: IFCT-1003 LADIE trial. Clin. Cancer Res. 2020, 26, 3172–3181. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Estrogen | Sources |
|---|---|
| Natural (Endogenous) (Estrone, 17β-estradiol, estriol, estretrol) | Extensively by gonadal organs (ovary/testis), low-level production in various organs due to aromatase generation (e.g., lungs, brain, etc.) |
| Synthetic (Exogenous) (Ethinyl estradiol, diethylstilbestrol, estradiol valerate, estropipate, conjugate esterified estrogen and quinestrol) | Either as constituent of contraceptive pills or hormone response therapy |
| Phytoestrogen (Exogenous) (Genistein, coumestans, quercetin, isoflavones, flavones, lignans, saponins and stilbenes) | Plant foods such as soy beans, tofu, tempeh, soy beverages, linseed (flax), sesame seeds, wheat (as lignans), berries (resveratrol), oats, barley, dried beans, lentils, rice, mung beans, apples, carrots, wheat germ, ricebran, soy linseed bread. |
| Xenoestrogen (Exogenous) (Bisphenol A (BPA), Dichlorodiphenyltrichloroethane (DDT), polychlorinated biphenyl (PCB), heavy metals, phthalates, alkylphenols, epileptic drugs | Plastics (water bottles, disposable cups, plastic wrap, food containers), pesticides (used on non-organic fruits and vegetables), tap water (chlorine and runoff byproducts), chemicals in cosmetics, lotions, shampoos and other body care materials |
| Clinical Trial Registry | Primary Objective of the Trial | Phase of Study, Tumor Stage and Current Status | Findings Published (Ref.) |
|---|---|---|---|
| NCT01556191 | Evaluating an EGFR tyrosine kinase inhibitor (EGFR-TKI), gefitinib and an EGFR-TKI-anti-oestrogen (erlotinib, fulvestrant) combined potency in women with advanced-stage non-squamous lung cancer | Phase I, stage IV lung cancer, completed | Improved outcome [143] |
| NCT00100854 | Evaluation of synergistic fulvestrant delivery with erlotinib for the non-small cell lung cancer (NSCLC) treatment | Phase II, stage IIIB or IV non-small cell lung cancer, completed | Improved outcome [144] |
| NCT02666105 | Evaluation of adding exemestane therapy in postmenopausal women suffering from NSCLC while on treatment with an immune checkpoint antibody (pembrolizumab, atezolizumab or nivolumab) | Phase II, advanced stage NSCLC, ongoing | Improved outcome [145] |
| NCT01664754 | Determining the safety and tolerability of escalating exemestane doses on being co-delivered with pemetrexed (pemetrexed disodium) and carboplatin in postmenopausal womensuffering from NSCLC | Phase I, stage IV non-squamous NSCLC, ongoing | Combination is safe and well-tolerated, response rate correlates with tumor aromatase expression [146] |
| NCT02751385 | Screening the effect of nintedanib on the (ethinylestradiol + levonorgestrel) pharmacokinetics in NSCLC patients | Phase I, all NSCLC patients, completed | No findings published todate |
| NCT00576225 | Screening the effect of paclitaxel poliglumex (CT-103)/carboplatin versus paclitaxel/carboplatin for women NSCLC sufferers | Phase III, sufferers having >25 pg·mL−1 estradiol, completed | CT-103 did not provide superior survival over the paclitaxel-carboplatin for first-line treatment of NSCLC patients, results were comparable for progression-free and overall survival [147] |
| NCT03099174 | Ascertaining a safe dosage of xentuzumab in combination with abemaciclib with or without hormonal therapies in lung and breast cancer | Phase I, no stage distinction, ongoing | Findings not yet published |
| NCT00592007 | Screening the impact of adding fulvestrant to erlotinib in NSCLC patients | Phase II, stage IIIB or IV, concluded | [148] |
| NCT00932152 | Fulvestrant and anastrozole (aromatase inhibitor) as consolidation therapy in postmenopausal women NSCLC sufferers | Phase II, advanced stage NSCLC, concluded | [149] |
| NCT01594398 | Assessing the food effect on entinostat pharmacokinetics in NSCLC sufferers (ENCORE110) | Phase I, no stage distinction, completed | No findings published, study listed from [141] |
| AM2013-4664 | Evaluation of erlotinib antitumor activity in NSCLC on fulvestrant inclusion in the patients received > 1 chemotherapy regimen | Phase II, advanced state NSCLC patients | [150] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maitra, R.; Malik, P.; Mukherjee, T.K. Targeting Estrogens and Various Estrogen-Related Receptors against Non-Small Cell Lung Cancers: A Perspective. Cancers 2022, 14, 80. https://doi.org/10.3390/cancers14010080
Maitra R, Malik P, Mukherjee TK. Targeting Estrogens and Various Estrogen-Related Receptors against Non-Small Cell Lung Cancers: A Perspective. Cancers. 2022; 14(1):80. https://doi.org/10.3390/cancers14010080
Chicago/Turabian StyleMaitra, Radhashree, Parth Malik, and Tapan Kumar Mukherjee. 2022. "Targeting Estrogens and Various Estrogen-Related Receptors against Non-Small Cell Lung Cancers: A Perspective" Cancers 14, no. 1: 80. https://doi.org/10.3390/cancers14010080
APA StyleMaitra, R., Malik, P., & Mukherjee, T. K. (2022). Targeting Estrogens and Various Estrogen-Related Receptors against Non-Small Cell Lung Cancers: A Perspective. Cancers, 14(1), 80. https://doi.org/10.3390/cancers14010080