
Activation of the Unfolded Protein Response (UPR) Is Associated with Cholangiocellular Injury, Fibrosis and Carcinogenesis in an Experimental Model of Fibropolycystic Liver Disease

 , ,
, ,  , , , ,
, , , ,  ,
,  ,
,  , , , , ,
, , , , ,  and
and  add
Show full author list
add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Models
2.2. Immunoblot Analysis
2.3. Histological Evaluation of Samples
2.4. RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction (RT-PCR)
2.5. Statistical Analysis
3. Results
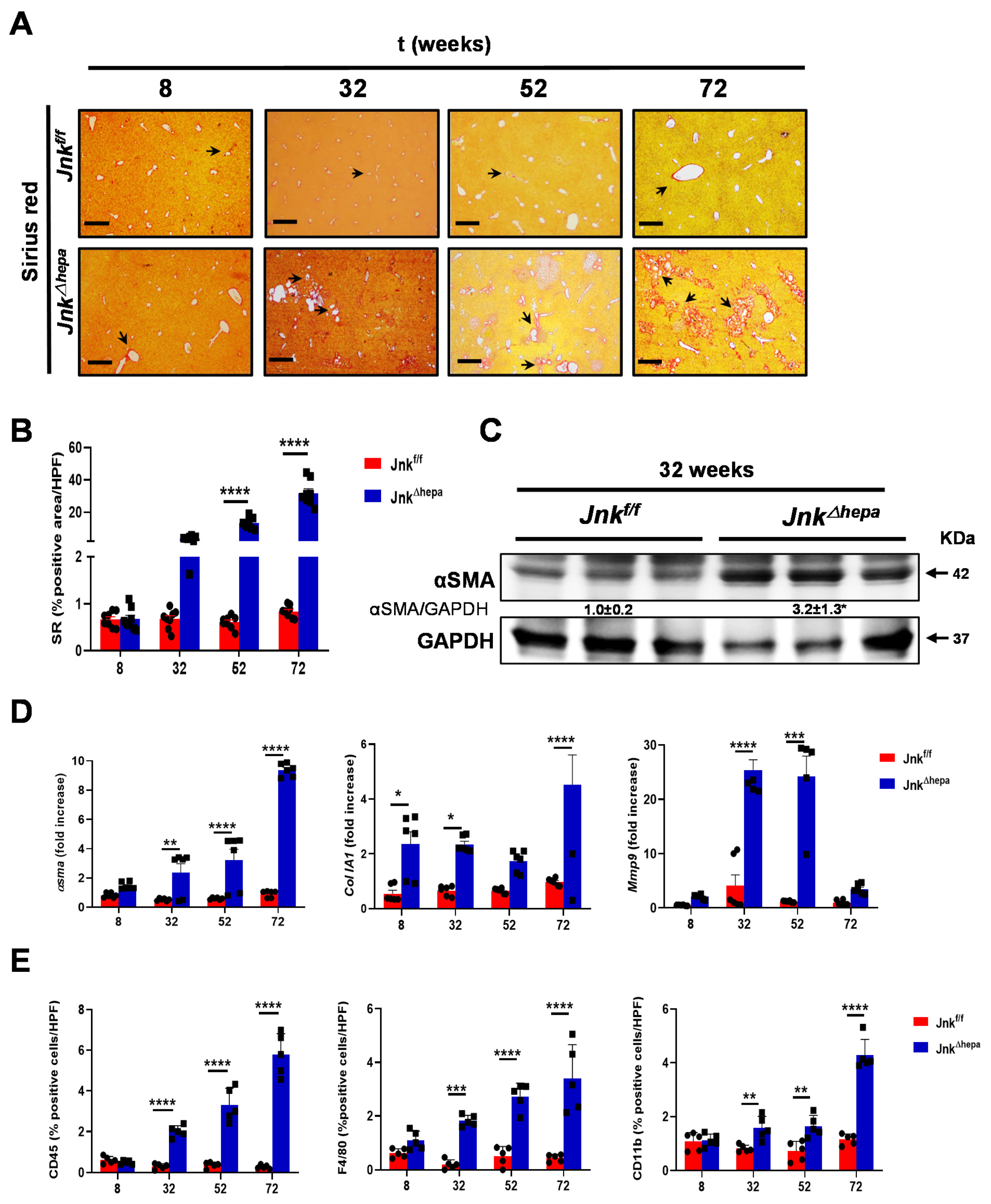
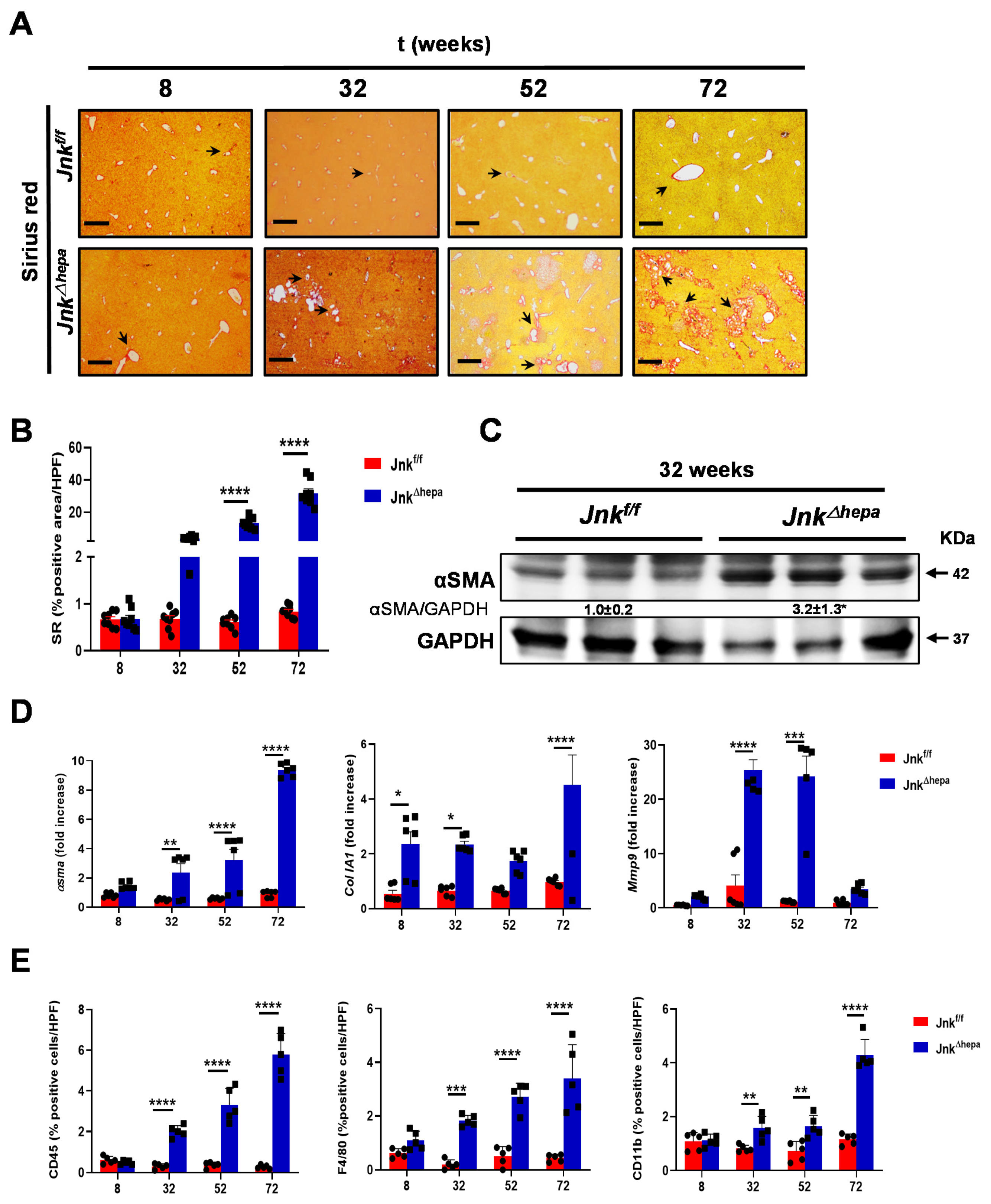
3.1. Hepatocytic Deletion of Jnk1/2 Results in Progressive Fibropolycystic Disease Characterized by Extracellular Matrix Deposition and Inflammation
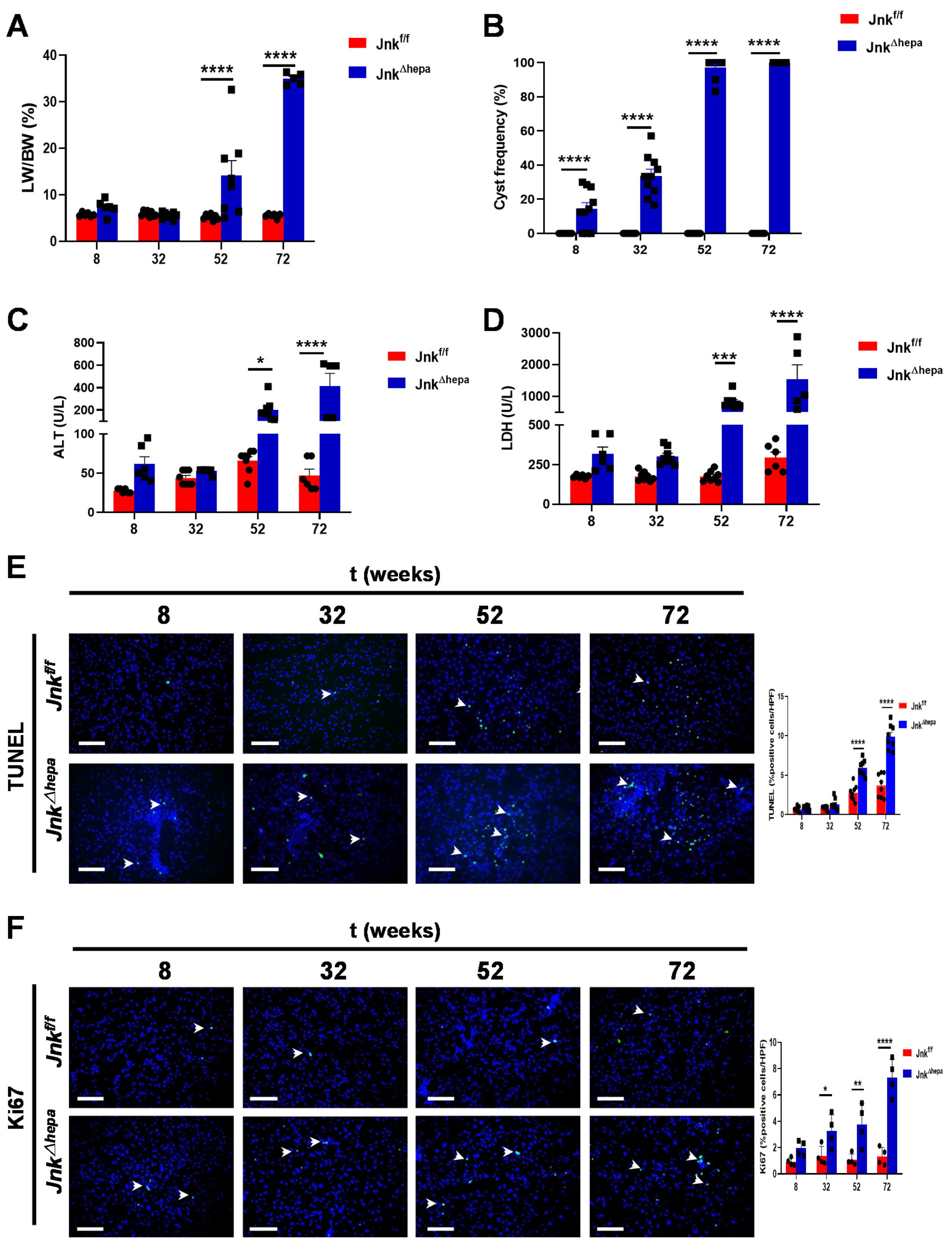
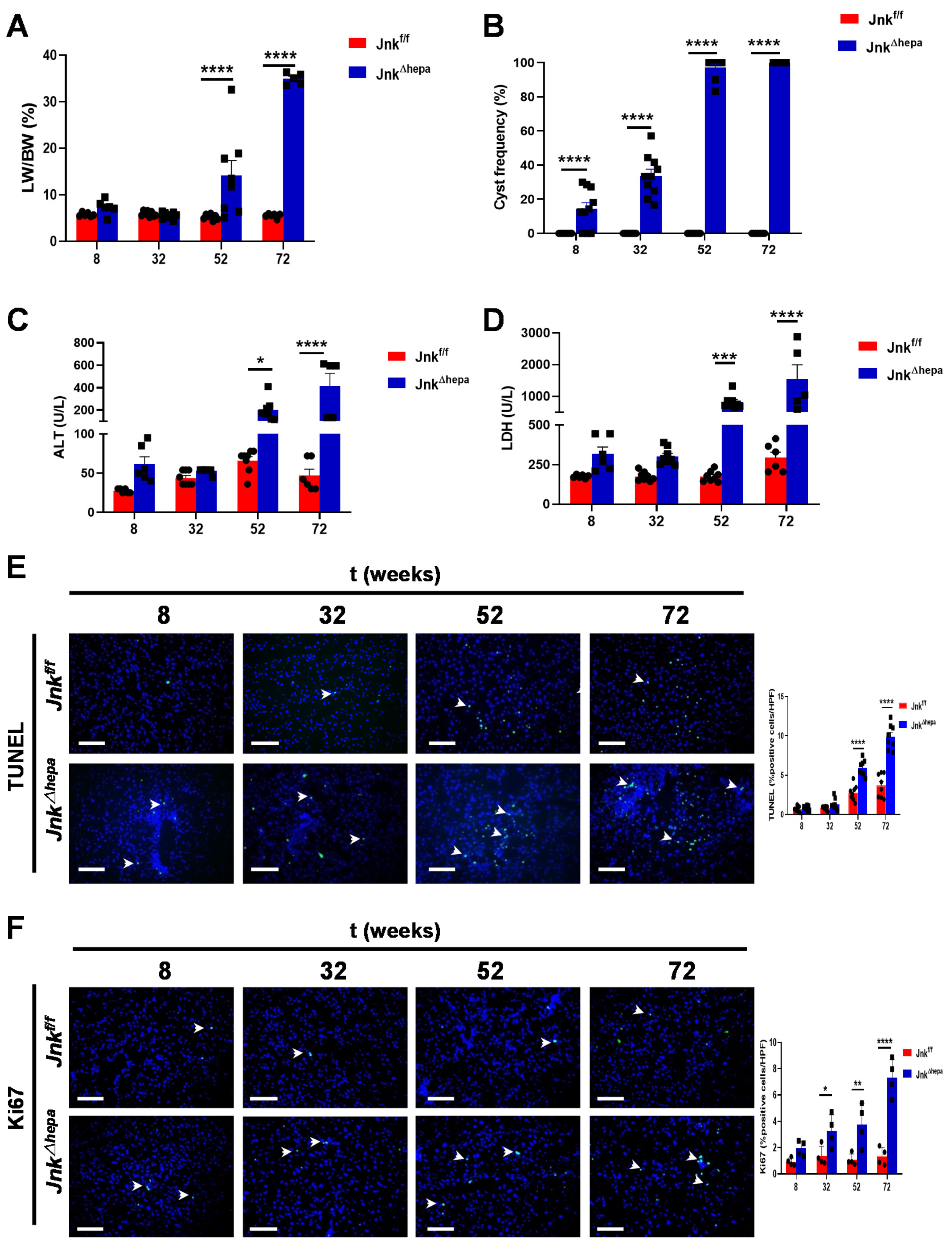
3.2. Hepatocytic Deletion of Jnk1/2 Promotes Hepatomegaly and Liver Damage
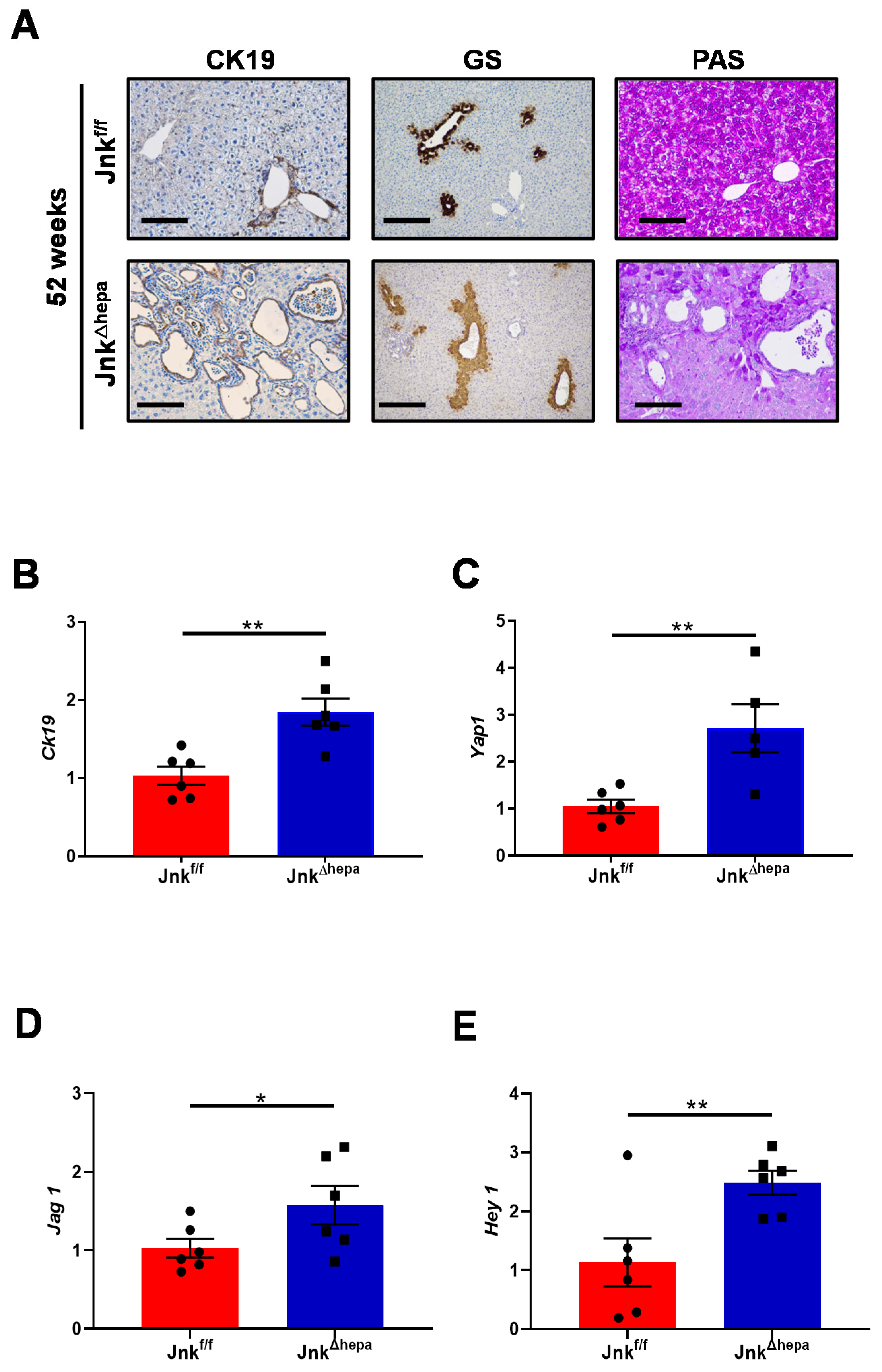
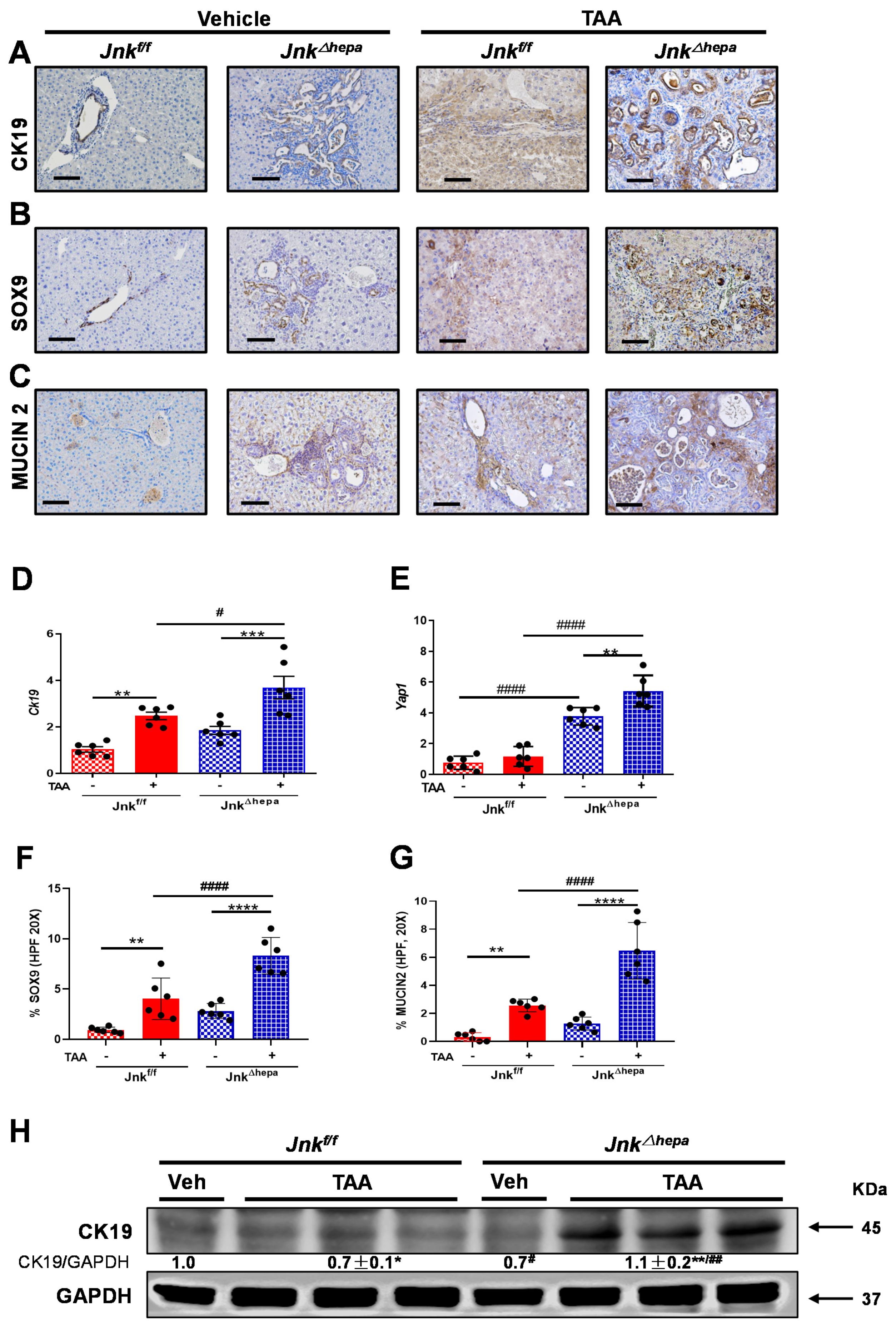
3.3. Hepatocytic Deletion of Jnk1/2 Triggers Cystic Hyperproliferation and Cholangiocyte Malignancy
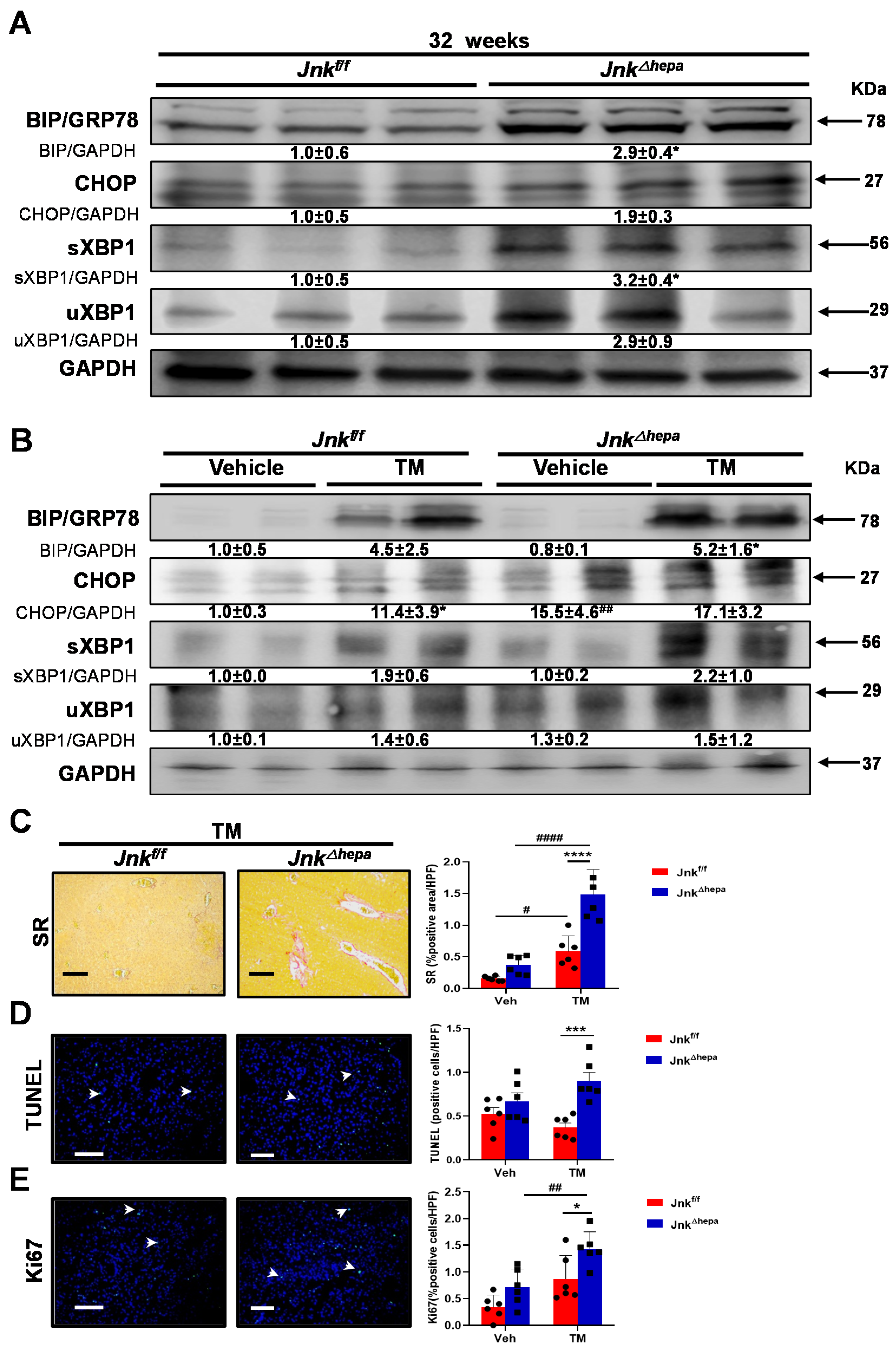
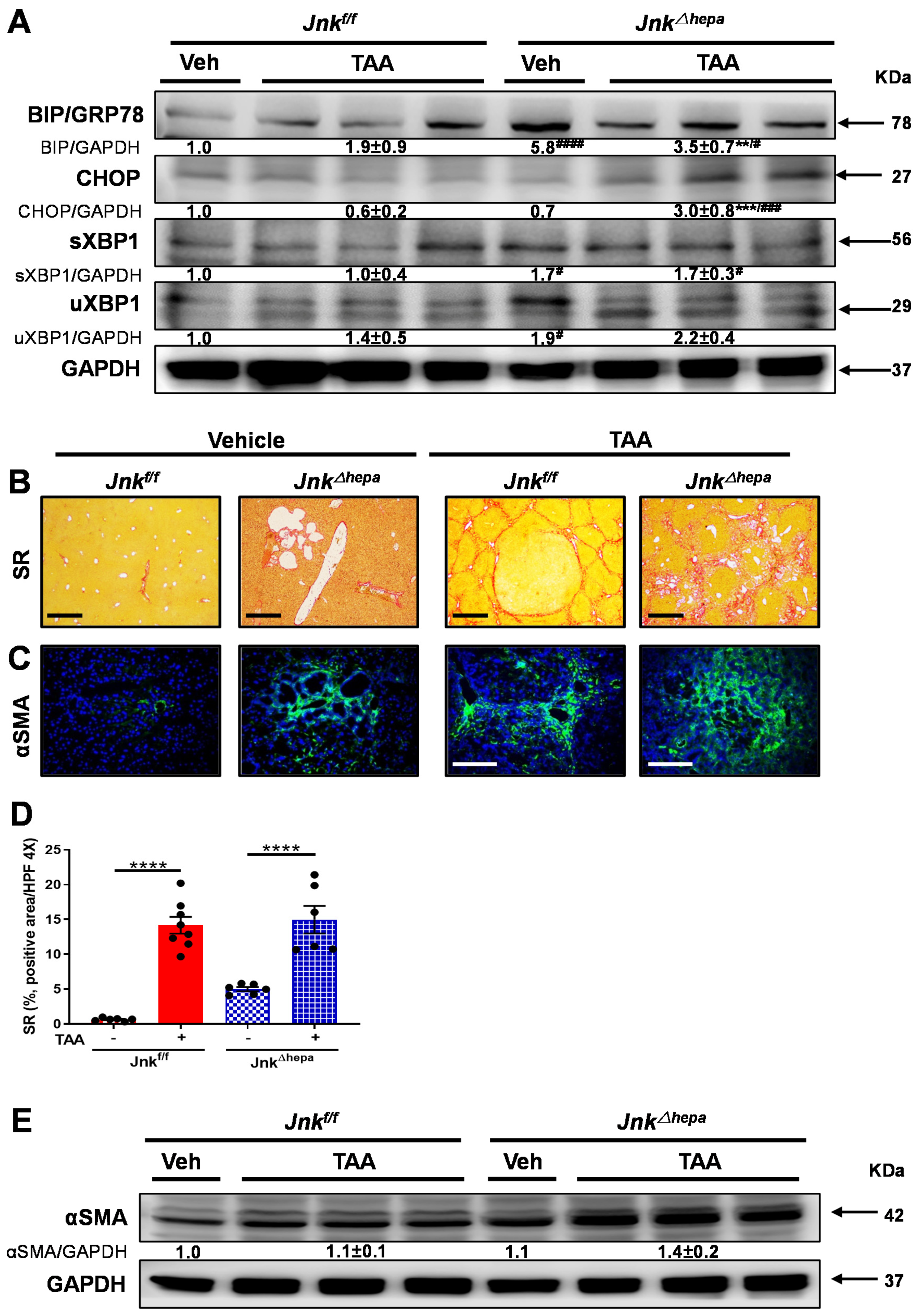
3.4. Activation of the Unfolded Protein Response (UPR) Is Associated with Increased Liver Injury in Jnk∆hepa Mice
3.5. Jnk∆hepa Mice Display an Exacerbated Profibrogenic Response
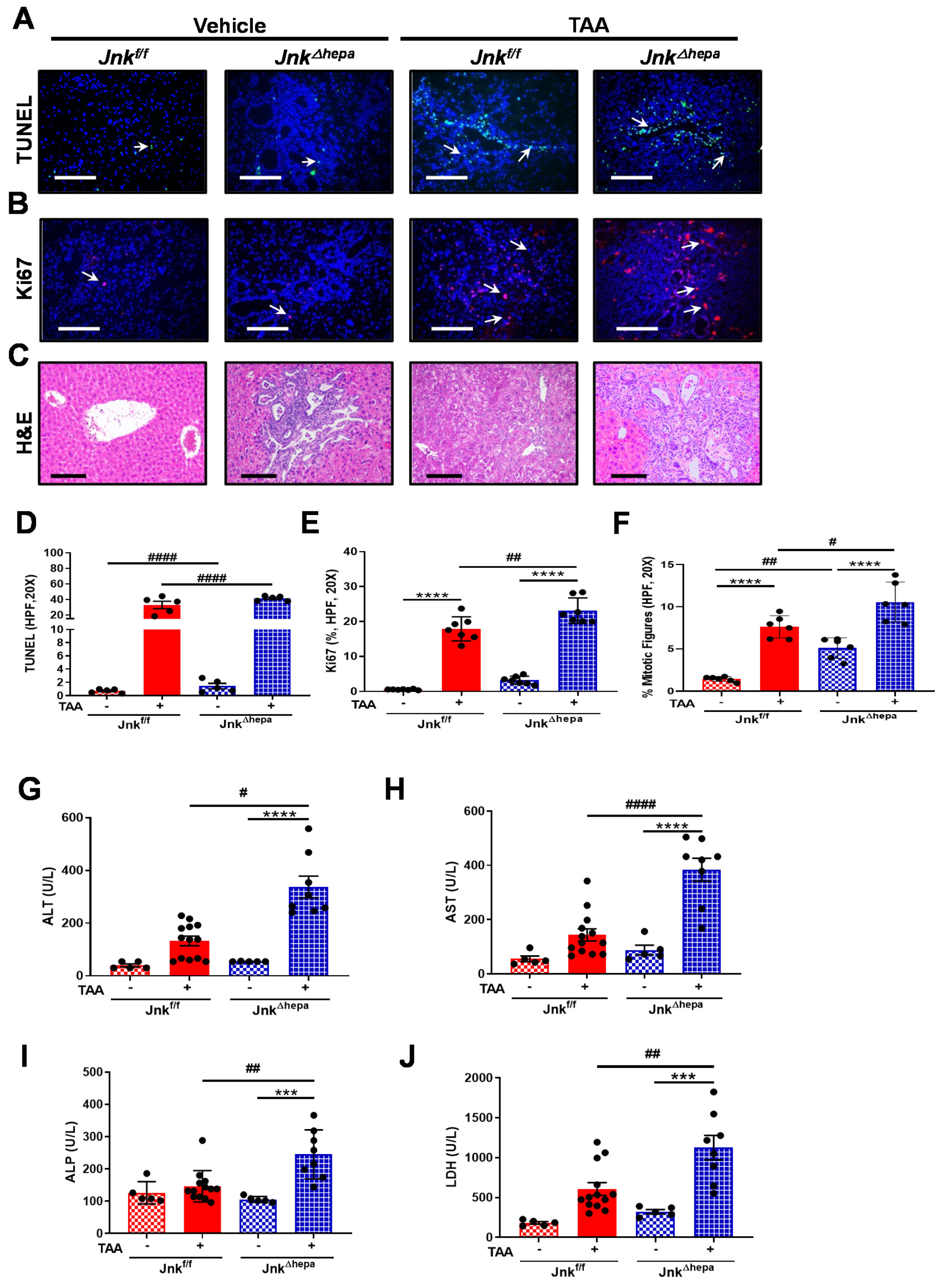
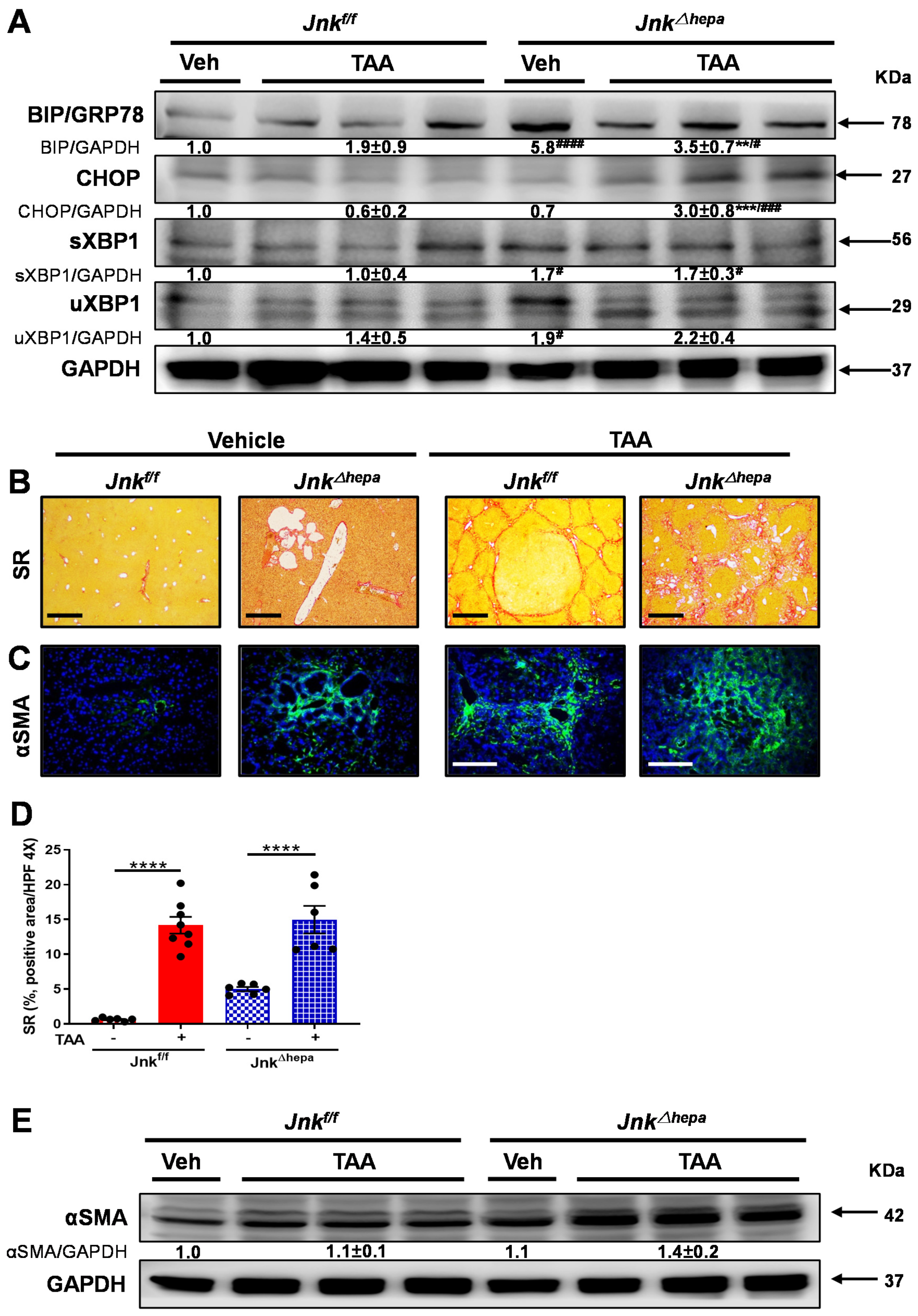
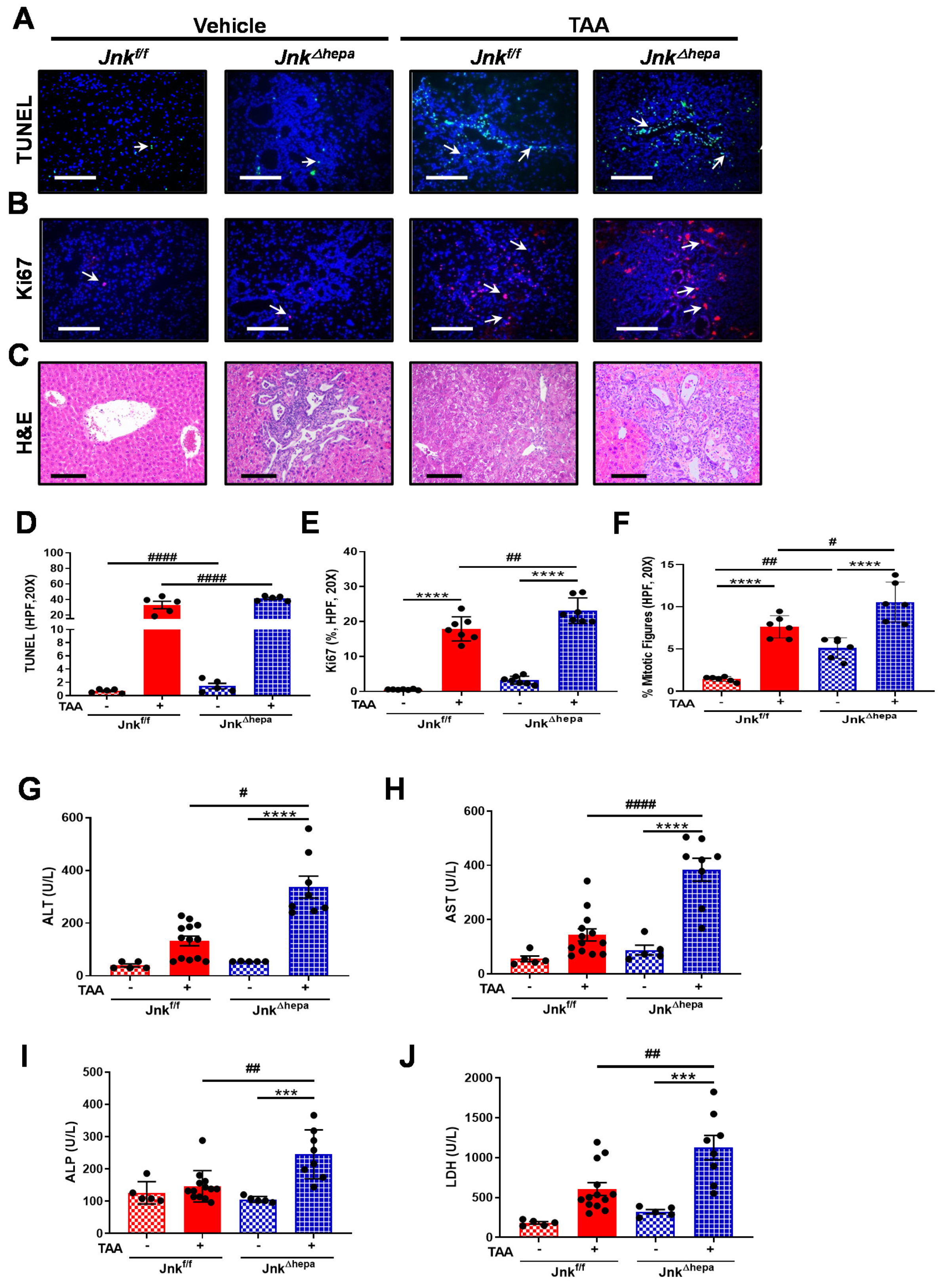
3.6. Jnk∆hepa Mice Display Extensive Hepatocellular Injury in Response to TAA
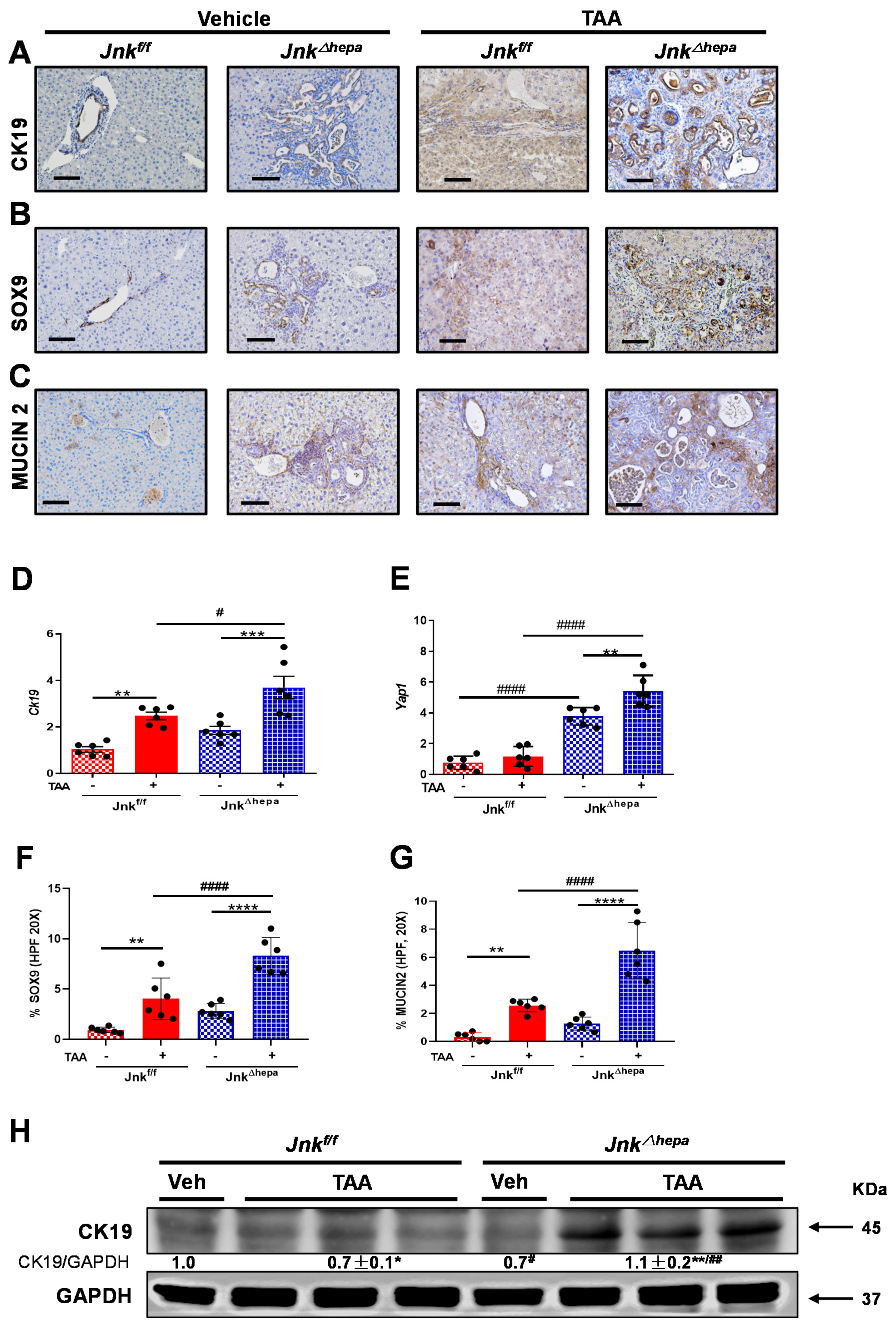
3.7. Chronic TAA Administration Triggers Cellular Atypia and Markers of Cholangiocarcinogenesis in Jnk∆hepa Mice
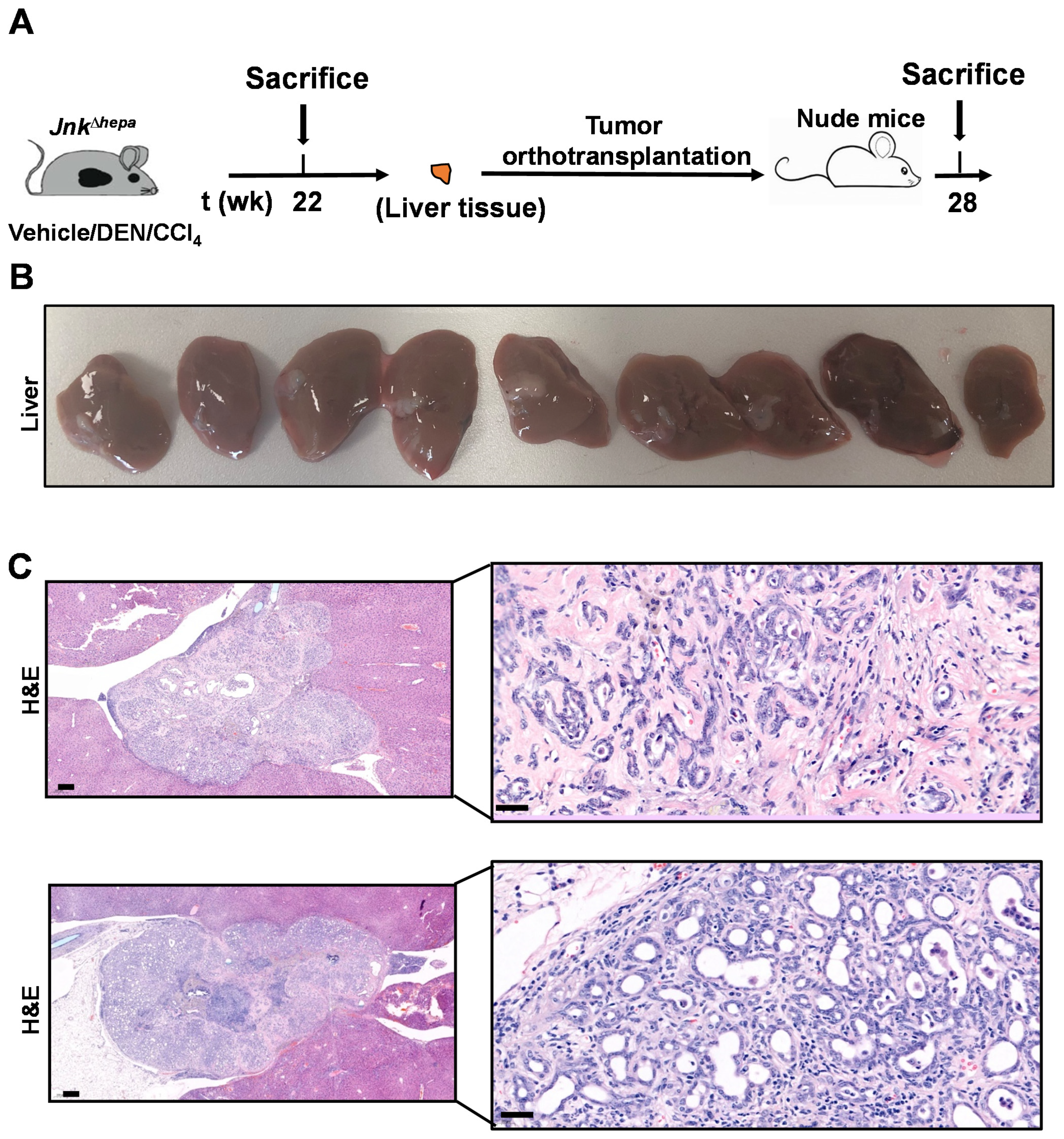
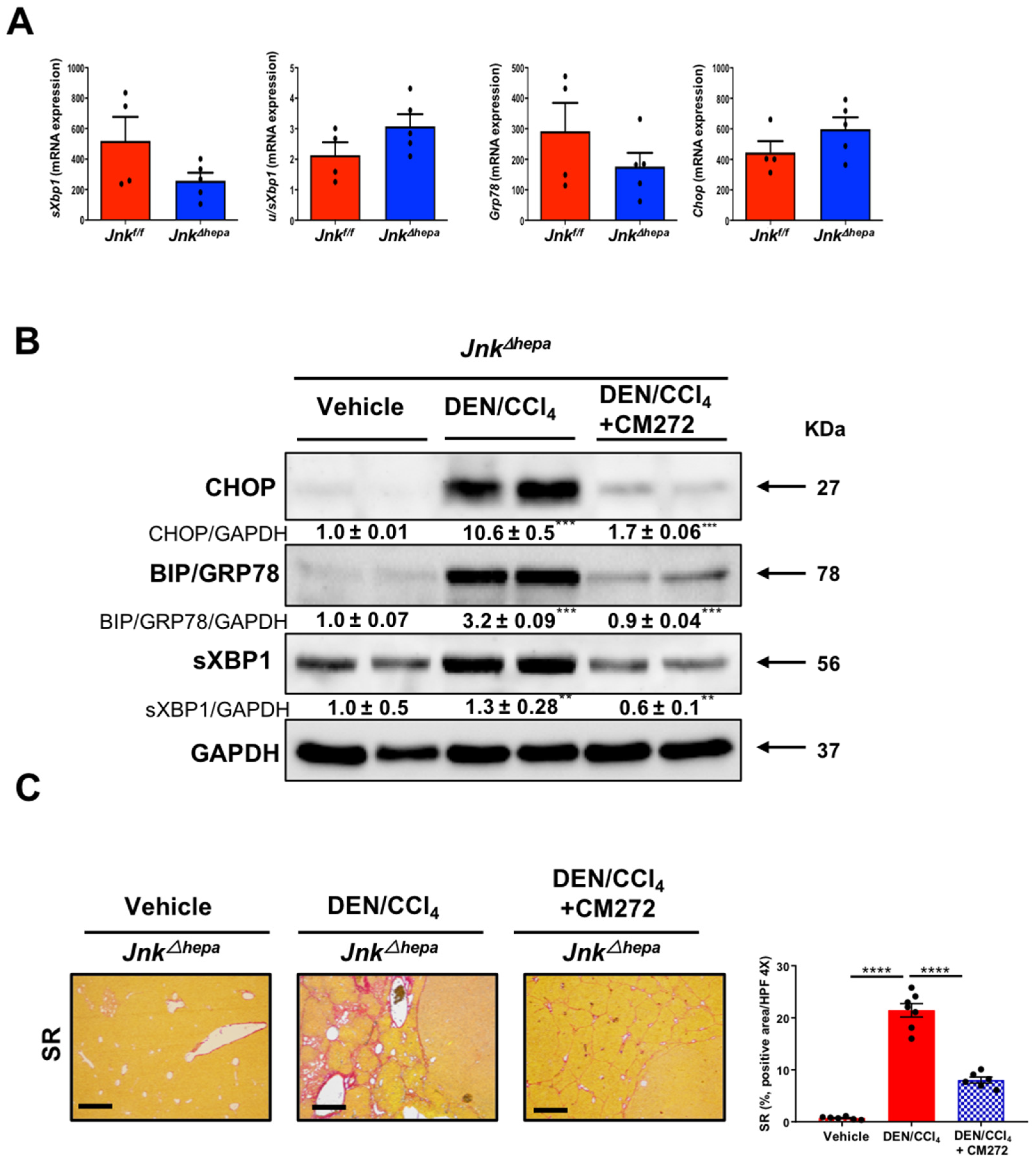
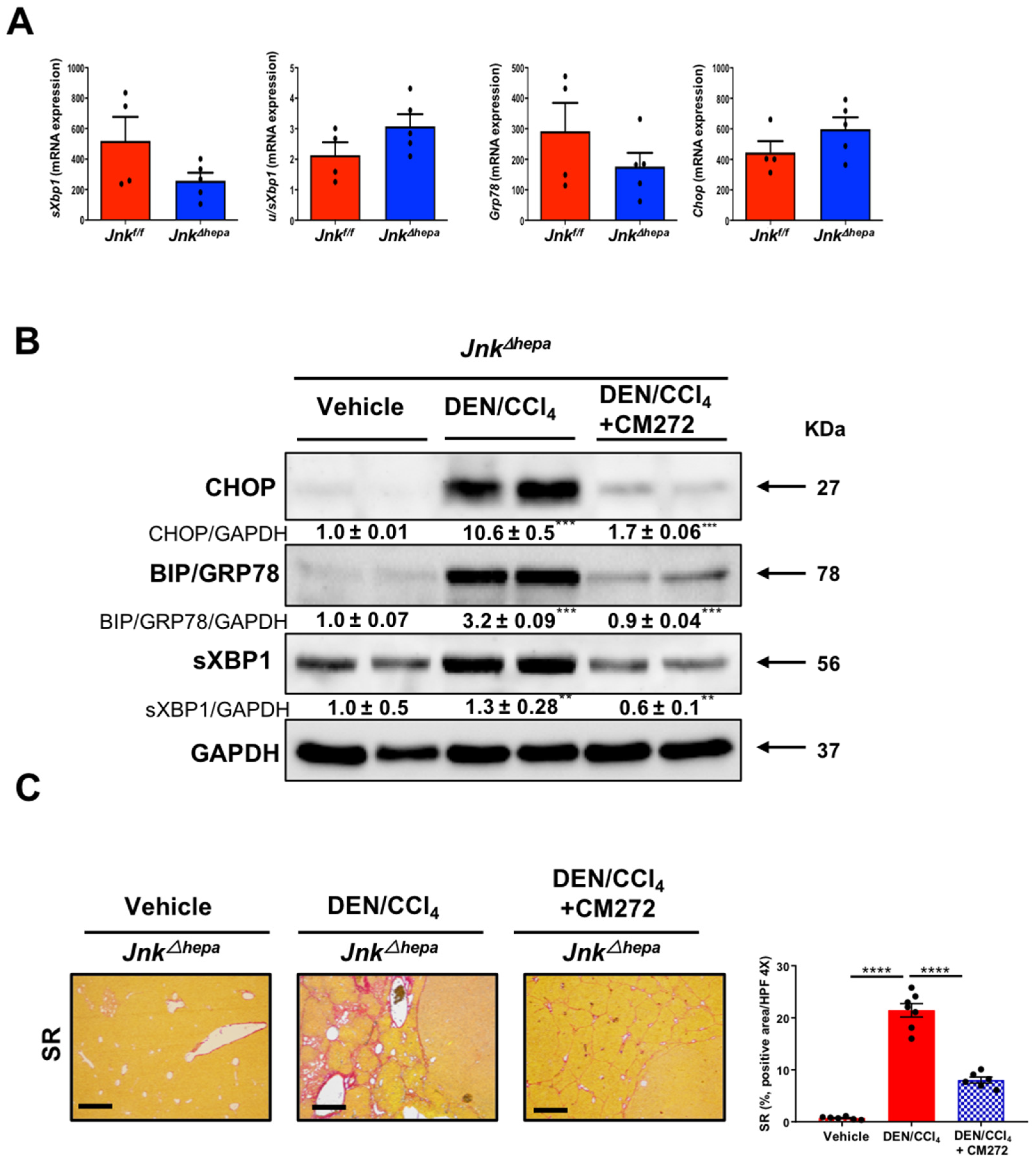
3.8. Hepatotoxin-Challenged Jnk∆hepa Mice Develop Fibrocystic Liver Disease and CCA in Association with a Strong UPR Activation: Therapeutic Potential of an Innovative Epigenetic Inhibitor
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cubero, F.J.; Mohamed, M.R.; Woitok, M.M.; Zhao, G.; Hatting, M.; Nevzorova, Y.A.; Chen, C.; Haybaeck, J.; de Bruin, A.; Avila, M.A.; et al. Loss of c-jun n-terminal kinase 1 and 2 function in liver epithelial cells triggers biliary hyperproliferation resembling cholangiocarcinoma. Hepatol. Commun. 2020, 4, 834–851. [Google Scholar] [CrossRef] [PubMed]
- Sabio, G.; Davis, R.J. Tnf and map kinase signalling pathways. Semin. Immunol. 2014, 26, 237–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seki, E.; Brenner, D.A.; Karin, M. A liver full of jnk: Signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology 2012, 143, 307–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, R.J. Signal transduction by the jnk group of map kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef] [Green Version]
- Cubero, F.J.; Zoubek, M.E.; Hu, W.; Peng, J.; Zhao, G.; Nevzorova, Y.A.; Al Masaoudi, M.; Bechmann, L.P.; Boekschoten, M.V.; Muller, M.; et al. Combined activities of jnk1 and jnk2 in hepatocytes protect against toxic liver injury. Gastroenterology 2016, 150, 968–981. [Google Scholar] [CrossRef] [Green Version]
- Manieri, E.; Folgueira, C.; Rodriguez, M.E.; Leiva-Vega, L.; Esteban-Lafuente, L.; Chen, C.; Cubero, F.J.; Barrett, T.; Cavanagh-Kyros, J.; Seruggia, D.; et al. Jnk-mediated disruption of bile acid homeostasis promotes intrahepatic cholangiocarcinoma. Proc. Natl. Acad. Sci. USA 2020, 117, 16492–16499. [Google Scholar] [CrossRef]
- Muller, K.; Honcharova-Biletska, H.; Koppe, C.; Egger, M.; Chan, L.K.; Schneider, A.T.; Kusgens, L.; Bohm, F.; Boege, Y.; Healy, M.E.; et al. Jnk signaling prevents biliary cyst formation through a caspase-8-dependent function of ripk1 during aging. Proc. Natl. Acad. Sci. USA 2021, 118, e2007194118. [Google Scholar] [CrossRef]
- Cnossen, W.R.; Drenth, J.P. Polycystic liver disease: An overview of pathogenesis, clinical manifestations and management. Orphanet J. Rare Dis. 2014, 9, 69. [Google Scholar] [CrossRef] [Green Version]
- Drenth, J.P.; Chrispijn, M.; Bergmann, C. Congenital fibrocystic liver diseases. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 573–584. [Google Scholar] [CrossRef]
- Strazzabosco, M.; Fabris, L. Development of the bile ducts: Essentials for the clinical hepatologist. J. Hepatol. 2012, 56, 1159–1170. [Google Scholar] [CrossRef]
- Perugorria, M.J.; Banales, J.M. Genetics: Novel causative genes for polycystic liver disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 391–392. [Google Scholar] [CrossRef]
- Santos-Laso, A.; Izquierdo-Sanchez, L.; Rodrigues, P.M.; Huang, B.Q.; Azkargorta, M.; Lapitz, A.; Munoz-Garrido, P.; Arbelaiz, A.; Caballero-Camino, F.J.; Fernandez-Barrena, M.G.; et al. Proteostasis disturbances and endoplasmic reticulum stress contribute to polycystic liver disease: New therapeutic targets. Liver Int. 2020, 40, 1670–1685. [Google Scholar] [CrossRef]
- Strazzabosco, M.; Somlo, S. Polycystic liver diseases: Congenital disorders of cholangiocyte signaling. Gastroenterology 2011, 140, 1855–1859.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee-Law, P.Y.; Olaizola, P.; Caballero-Camino, F.J.; Izquierdo-Sanchez, L.; Rodrigues, P.M.; Santos-Laso, A.; Azkargorta, M.; Elortza, F.; Martinez-Chantar, M.L.; Perugorria, M.J.; et al. Targeting ubc9-mediated protein hyper-sumoylation in cystic cholangiocytes halts polycystic liver disease in experimental models. J. Hepatol. 2021, 74, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Jiang, F.; Sluss, H.K.; Zhang, C.; Shokat, K.M.; Flavell, R.A.; Davis, R.J. Suppression of p53-dependent senescence by the jnk signal transduction pathway. Proc. Natl. Acad. Sci. USA 2007, 104, 15759–15764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, M.; Sabio, G.; Jiang, F.; Rincon, M.; Flavell, R.A.; Davis, R.J. Induction of hepatitis by jnk-mediated expression of tnf-alpha. Cell 2009, 136, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Colyn, L.; Barcena-Varela, M.; Alvarez-Sola, G.; Latasa, M.U.; Uriarte, I.; Santamaria, E.; Herranz, J.M.; Santos-Laso, A.; Arechederra, M.; Ruiz de Gauna, M.; et al. Dual targeting of g9a and DNA methyltransferase-1 for the treatment of experimental cholangiocarcinoma. Hepatology 2021, 73, 2380–2396. [Google Scholar] [CrossRef]
- San Jose-Eneriz, E.; Agirre, X.; Rabal, O.; Vilas-Zornoza, A.; Sanchez-Arias, J.A.; Miranda, E.; Ugarte, A.; Roa, S.; Paiva, B.; Estella-Hermoso de Mendoza, A.; et al. Discovery of first-in-class reversible dual small molecule inhibitors against g9a and dnmts in hematological malignancies. Nat. Commun. 2017, 8, 15424. [Google Scholar] [CrossRef] [Green Version]
- Ambrogio, C.; Carmona, F.J.; Vidal, A.; Falcone, M.; Nieto, P.; Romero, O.A.; Puertas, S.; Vizoso, M.; Nadal, E.; Poggio, T.; et al. Modeling lung cancer evolution and preclinical response by orthotopic mouse allografts. Cancer Res. 2014, 74, 5978–5988. [Google Scholar] [CrossRef] [Green Version]
- An, L.; Wang, X.; Cederbaum, A.I. Cytokines in alcoholic liver disease. Arch. Toxicol. 2012, 86, 1337–1348. [Google Scholar] [CrossRef]
- Mirzayans, R.; Murray, D. Do tunel and other apoptosis assays detect cell death in preclinical studies? Int. J. Mol. Sci. 2020, 21, 9090. [Google Scholar] [CrossRef]
- Lee, D.H.; Park, J.O.; Kim, T.S.; Kim, S.K.; Kim, T.H.; Kim, M.C.; Park, G.S.; Kim, J.H.; Kuninaka, S.; Olson, E.N.; et al. Lats-yap/taz controls lineage specification by regulating tgfbeta signaling and hnf4alpha expression during liver development. Nat. Commun. 2016, 7, 11961. [Google Scholar] [CrossRef]
- Fischer, A.; Schumacher, N.; Maier, M.; Sendtner, M.; Gessler, M. The notch target genes hey1 and hey2 are required for embryonic vascular development. Genes Dev. 2004, 18, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Kropski, J.A.; Blackwell, T.S. Endoplasmic reticulum stress in the pathogenesis of fibrotic disease. J. Clin. Investig. 2018, 128, 64–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. Chop induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Shen, X.; Wu, J.; Sakaki, K.; Saunders, T.; Rutkowski, D.T.; Back, S.H.; Kaufman, R.J. Endoplasmic reticulum stress activates cleavage of crebh to induce a systemic inflammatory response. Cell 2006, 124, 587–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwyer, B.J.; Jarman, E.J.; Gogoi-Tiwari, J.; Ferreira-Gonzalez, S.; Boulter, L.; Guest, R.V.; Kendall, T.J.; Kurian, D.; Kilpatrick, A.M.; Robson, A.J.; et al. Tweak/fn14 signalling promotes cholangiocarcinoma niche formation and progression. J. Hepatol. 2021, 74, 860–872. [Google Scholar] [CrossRef] [PubMed]
- Delire, B.; Starkel, P.; Leclercq, I. Animal models for fibrotic liver diseases: What we have, what we need, and what is under development. J. Clin. Transl. Hepatol. 2015, 3, 53–66. [Google Scholar] [PubMed]
- Khan, S.A.; Tavolari, S.; Brandi, G. Cholangiocarcinoma: Epidemiology and risk factors. Liver Int. 2019, 39 (Suppl. S1), 19–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadd, V.L.; Aleksieva, N.; Forbes, S.J. Epithelial plasticity during liver injury and regeneration. Cell Stem Cell 2020, 27, 557–573. [Google Scholar] [CrossRef] [PubMed]
- Lasagni, A.; Cadamuro, M.; Morana, G.; Fabris, L.; Strazzabosco, M. Fibrocystic liver disease: Novel concepts and translational perspectives. Transl. Gastroenterol. Hepatol. 2021, 6, 26. [Google Scholar] [CrossRef]
- Fard-Aghaie, M.H.; Makridis, G.; Reese, T.; Feyerabend, B.; Wagner, K.C.; Schnitzbauer, A.; Bechstein, W.O.; Oldhafer, F.; Kleine, M.; Klempnauer, J.; et al. The rate of cholangiocarcinoma in caroli disease a german multicenter study. HPB 2021, in press. [Google Scholar] [CrossRef]
- Chen, I.Y.; Whitney-Miller, C.L.; Liao, X. Congenital hepatic fibrosis and its mimics: A clinicopathologic study of 19 cases at a single institution. Diagn. Pathol. 2021, 16, 81. [Google Scholar] [CrossRef]
- Hur, K.Y.; So, J.S.; Ruda, V.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Iwawaki, T.; Glimcher, L.H.; Lee, A.H. Ire1alpha activation protects mice against acetaminophen-induced hepatotoxicity. J. Exp. Med. 2012, 209, 307–318. [Google Scholar] [CrossRef] [Green Version]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Nevzorova, Y.A.; Boyer-Diaz, Z.; Cubero, F.J.; Gracia-Sancho, J. Animal models for liver disease—A practical approach for translational research. J. Hepatol. 2020, 73, 423–440. [Google Scholar] [CrossRef] [PubMed]
- Choe, J.Y.; Kim, H. Intrahepatic cholangiocarcinoma with predominant ductal plate malformation pattern. Clin. Mol. Hepatol. 2014, 20, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Peng, B.; Chen, G.; Pes, M.G.; Ribback, S.; Ament, C.; Xu, H.; Pal, R.; Rodrigues, P.M.; Banales, J.M.; et al. Yap accelerates notch-driven cholangiocarcinogenesis via mtorc1 in mice. Am. J. Pathol. 2021, 191, 1651–1667. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Song, X.; Liao, H.; Wang, P.; Zhang, Y.; Che, L.; Zhang, J.; Zhou, Y.; Cigliano, A.; Ament, C.; et al. Overexpression of mothers against decapentaplegic homolog 7 activates the yes-associated protein/notch cascade and promotes liver carcinogenesis in mice and humans. Hepatology 2021, 74, 248–263. [Google Scholar] [CrossRef]
- Esfandiari, F.; Medici, V.; Wong, D.H.; Jose, S.; Dolatshahi, M.; Quinlivan, E.; Dayal, S.; Lentz, S.R.; Tsukamoto, H.; Zhang, Y.H.; et al. Epigenetic regulation of hepatic endoplasmic reticulum stress pathways in the ethanol-fed cystathionine beta synthase-deficient mouse. Hepatology 2010, 51, 932–941. [Google Scholar] [CrossRef] [Green Version]
- Duvigneau, J.C.; Luis, A.; Gorman, A.M.; Samali, A.; Kaltenecker, D.; Moriggl, R.; Kozlov, A.V. Crosstalk between inflammatory mediators and endoplasmic reticulum stress in liver diseases. Cytokine 2019, 124, 154577. [Google Scholar] [CrossRef] [PubMed]
- Barcena-Varela, M.; Paish, H.; Alvarez, L.; Uriarte, I.; Latasa, M.U.; Santamaria, E.; Recalde, M.; Garate, M.; Claveria, A.; Colyn, L.; et al. Epigenetic mechanisms and metabolic reprogramming in fibrogenesis: Dual targeting of g9a and dnmt1 for the inhibition of liver fibrosis. Gut 2021, 70, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Chi, L.; Ahmed, A.; Roy, A.R.; Vuong, S.; Cahill, L.S.; Caporiccio, L.; Sled, J.G.; Caniggia, I.; Wilson, M.D.; Delgado-Olguin, P. G9a controls placental vascular maturation by activating the notch pathway. Development 2017, 144, 1976–1987. [Google Scholar] [CrossRef] [Green Version]
- Fabris, L.; Fiorotto, R.; Spirli, C.; Cadamuro, M.; Mariotti, V.; Perugorria, M.J.; Banales, J.M.; Strazzabosco, M. Pathobiology of inherited biliary diseases: A roadmap to understand acquired liver diseases. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 497–511. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.; Wu, H.; Ye, H.; Tortajada, A.; Rodríguez-Perales, S.; Torres-Ruiz, R.; Vidal, A.; Peligros, M.I.; Reissing, J.; Bruns, T.; et al. Activation of the Unfolded Protein Response (UPR) Is Associated with Cholangiocellular Injury, Fibrosis and Carcinogenesis in an Experimental Model of Fibropolycystic Liver Disease. Cancers 2022, 14, 78. https://doi.org/10.3390/cancers14010078
Chen C, Wu H, Ye H, Tortajada A, Rodríguez-Perales S, Torres-Ruiz R, Vidal A, Peligros MI, Reissing J, Bruns T, et al. Activation of the Unfolded Protein Response (UPR) Is Associated with Cholangiocellular Injury, Fibrosis and Carcinogenesis in an Experimental Model of Fibropolycystic Liver Disease. Cancers. 2022; 14(1):78. https://doi.org/10.3390/cancers14010078
Chicago/Turabian StyleChen, Chaobo, Hanghang Wu, Hui Ye, Agustín Tortajada, Sandra Rodríguez-Perales, Raúl Torres-Ruiz, August Vidal, Maria Isabel Peligros, Johanna Reissing, Tony Bruns, and et al. 2022. "Activation of the Unfolded Protein Response (UPR) Is Associated with Cholangiocellular Injury, Fibrosis and Carcinogenesis in an Experimental Model of Fibropolycystic Liver Disease" Cancers 14, no. 1: 78. https://doi.org/10.3390/cancers14010078
APA StyleChen, C., Wu, H., Ye, H., Tortajada, A., Rodríguez-Perales, S., Torres-Ruiz, R., Vidal, A., Peligros, M. I., Reissing, J., Bruns, T., Mohamed, M. R., Zheng, K., Lujambio, A., Iraburu, M. J., Colyn, L., Latasa, M. U., Arechederra, M., Fernández-Barrena, M. G., Berasain, C., ... Cubero, F. J. (2022). Activation of the Unfolded Protein Response (UPR) Is Associated with Cholangiocellular Injury, Fibrosis and Carcinogenesis in an Experimental Model of Fibropolycystic Liver Disease. Cancers, 14(1), 78. https://doi.org/10.3390/cancers14010078