
Natural and Synthetic Estrogens in Chronic Inflammation and Breast Cancer

 , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
2. Estrogen Signaling
2.1. Canonical Estrogen Signaling within the Nucleus
2.2. Noncanonical Estrogen Signaling on the Surface
2.3. Mitochondrial ER Signaling
2.4. Differential ERα and ERβ Signaling
2.5. Estrogen Signaling in Breast Cancer
3. Estrogen, Inflammation, and Breast Cancer
3.1. Estrogen and Inflammation—The Positive Feedback
3.1.1. IL-6 and Estrogen
3.1.2. PGE2 and Estrogen
3.1.3. Adipokines and Estrogen
3.2. Estrogen in Breast Cancer and the Involvement of Inflammation
3.3. Anti-Inflammatory Role of Estrogens
3.4. Endocrine Therapy for Breast Cancer and Inflammation
4. Xenoestrogens in Inflammation and Breast Cancer
4.1. Phytoestrogens and Cancer
4.2. Role of Phytoestrogens in Inflammation
4.2.1. Reduced Expression and Activity of NF-κB and AP-1 Signaling
4.2.2. Antioxidative Activity

{kind=link}
{kind=link}
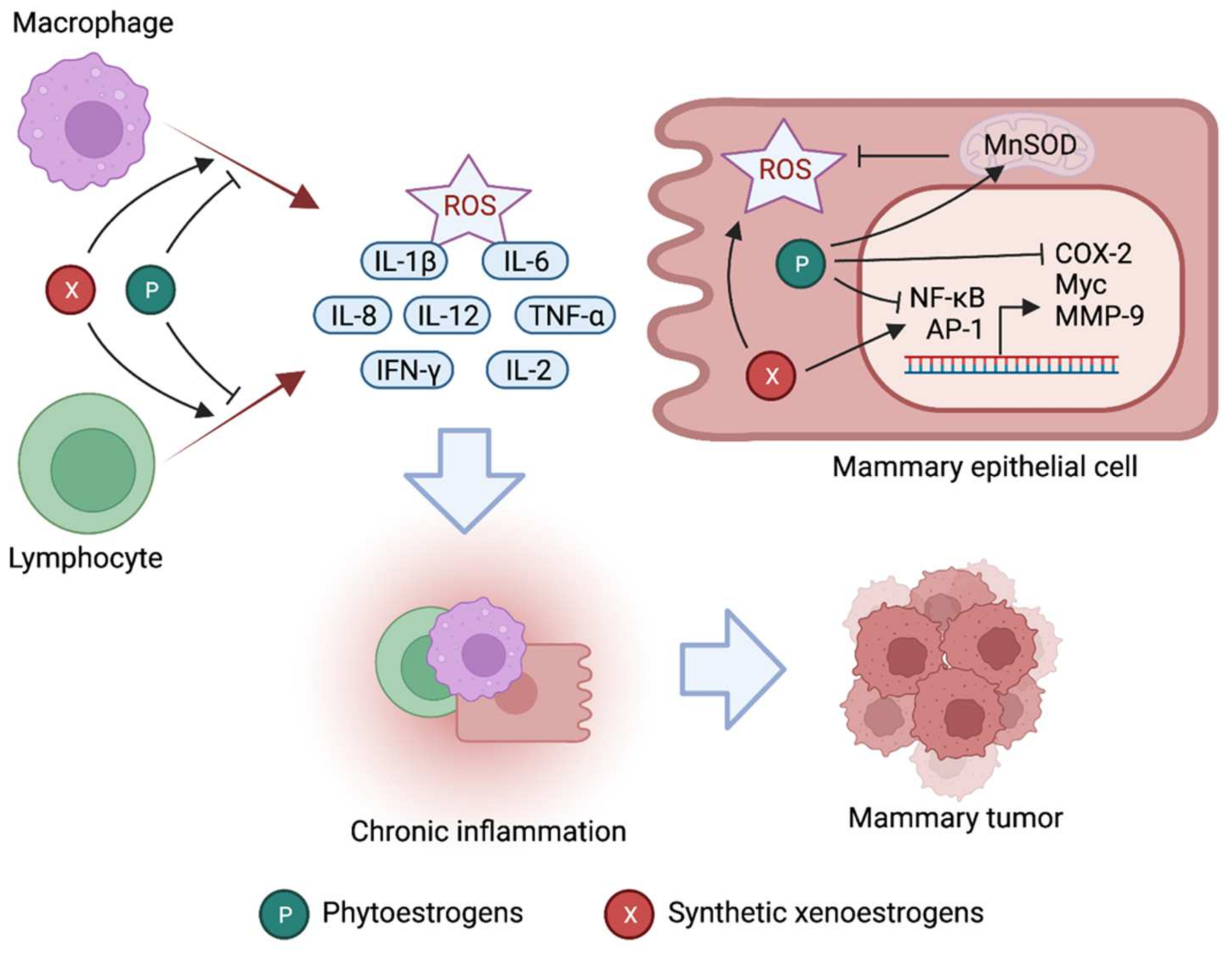{kind=link}
| Mechanisms | Key Findings | References |
|---|---|---|
| Inhibition of NF-κB and AP-1 signaling | Suppression of DMBA-induced mammary carcinogenesis by resveratrol correlates with the inhibition of NF-κB | [177] |
| Isoflavone genistein suppresses TNF-α-induced NF-κB activation in peripheral blood lymphocytes and oxidative DNA damage | [182] | |
| Genistein inhibits LPS-induced NF-κB activation in RAW 264.7 macrophages | [191] | |
| Antioxidative activity | Genistein reduces oxidative stress in breast cancer cell with low ERα-to-ERβ ratio | [188] |
| Isoflavones reduce reactive nitrogen species in LPS-challenged rats | [189] | |
| Resveratrol suppresses oxidative stress and inflammatory response related to hypoxic-ischemic brain injury in rats via Nrf2/HO-1 pathway | [190] | |
| Reduced proinflammatory cytokine and chemokine generation | Isoflavones, including genistein, suppress the LPS-stimulated overproduction of IL-6, TNF-α, and IL-1β | [191,192,193] |
| Isoflavone-rich diet reduces proinflammatory cytokines and immunosuppressive cells in pancreatic cancer patients | [194] | |
| Resveratrol attenuates lymphocytic IL-2 and IFN-γ production, as well as macrophageal IL-1β, IL-6, and TNF-α production | [195] | |
| Suppressed COX-2 activity | Resveratrol attenuates DMBA-induced mammary carcinogenesis, which correlates with suppression of COX-2 | [177] |
| Resveratrol reduces COX-2 expression and activity in PMA-treated mammary epithelial cells | [196] | |
| Genistein and daidzein attenuates PMA-induced COX-2 expression in MCF-7 breast cancer cells | [197] | |
| Resveratrol suppresses lung and colorectal cancer cell proliferation via COX-2 downregulation | [198,199] |
4.2.3. Suppressed Proinflammatory Cytokines and Chemokine Production
4.2.4. Reduced Cyclooxygenase-2 Activity
4.3. Synthetic Xenoestrogens and Cancer
4.4. Specific Synthetic Xenoestrogens in Inflammation and Breast Cancer
4.4.1. Bisphenol A (BPA)
| Xenoestrogen | Key Findings | References |
|---|---|---|
| Bisphenol A (BPA) | Induces CD4+ T lymphocyte differentiation into the proinflammatory Th1 or Th17 subsets | [233,234,235] |
| Induces CD4+ T lymphocyte differentiation into the anti-inflammatory Th2 subsets | [236,237,238] | |
| Reduces regulatory T cell number in prenatal and adult mice | [229] | |
| Increases the number and immunoglobulin production of B cells in vivo | [230] | |
| Either suppresses or enhances LPS-induced NO production | [231,232] | |
| Dichloro-diphenyl-trichloroethane (DDT) | Upregulates COX-2 and prostaglandins in breast cancer cells | [239] |
| Elevates inflammatory and oxidative stress marker genes, CXCL8, HMO-1, and TNF, in MCF-7 breast cancer cells | [240] | |
| Induces oxidative DNA damage culminating in hepatic neoplasia | [241] | |
| Suppresses antigen-induced serum γ-globulin levels | [242] | |
| Upregulates inducible iNOS and proinflammatory cytokines in NF-κB-dependent manner | [243] | |
| Polychlorinated biphenyls (PCBs) | PCB 126 induces macrophage polarization to the proinflammatory M1 and enhances the secretion of TNF-α and IL-1β. It also induces oxidative stress marker genes. | [244] |
| PCB 126 disrupts gut microbiota, as well as promotes intestinal and systemic inflammation | [245] | |
| PCB 153 promotes NF-κB-mediated inflammation and oxidative stress | [246] | |
| Implicated in immunosuppression and delayed immune response | [247,248] | |
| Polycyclic aromatic hydrocarbons (PAHs) | Induces oxidative stress | [249,250] |
| Elicits serious immunotoxic and immunosuppressive effects in mice | [251,252,253] | |
| Promotes Th17 differentiation of T lymphocytes and increases IFN-γ-positive dendritic cell population | [254] | |
| 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) | Induces inflammatory cytokine production by dendritic cells and macrophages Elicits immunosuppressive effects in mice by disrupting peripheral T lymphocyte development | [255] |
| Activates AhR to increase ROS generation and proteolytic maturation of IL-1β | [256] |
4.4.2. Dichloro-Diphenyl-Trichloroethane (DDT)
4.4.3. Polychlorinated Biphenyls (PCBs)
4.4.4. Polycyclic Aromatic Hydrocarbons (PAHs)
4.4.5. 2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD)
5. Concluding Thoughts and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, H.-R.; Kim, T.-H.; Choi, K.-C. Functions and physiological roles of two types of estrogen receptors, ERα and ERβ, identified by estrogen receptor knockout mouse. Lab. Anim. Res. 2012, 28, 71–76. [Google Scholar] [CrossRef]
- Liang, J.; Shang, Y. Estrogen and cancer. Ann. Rev. Physiol. 2013, 75, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Levitz, M.; Young, B.K. Estrogens in pregnancy. Vitam. Horm. 1977, 35, 109–147. [Google Scholar] [CrossRef] [PubMed]
- Cooke, P.S.; Nanjappa, M.K.; Ko, C.; Prins, G.S.; Hess, R.A. Estrogens in Male Physiology. Physiol. Rev. 2017, 97, 995–1043. [Google Scholar] [CrossRef]
- Miller, W.L.; Auchus, R.J. The Molecular Biology, Biochemistry, and Physiology of Human Steroidogenesis and Its Disorders. Endocr. Rev. 2011, 32, 81–151. [Google Scholar] [CrossRef] [PubMed]
- Beatson, G.T. On the Treatment of Inoperable Cases of Carcinoma of the Mamma: Suggestions for a New Method of Treatment, with Illustrative Cases. Trans. Med. Chir. Soc. 1896, 15, 153–179. [Google Scholar]
- Shang, Y. Hormones and cancer. Cell Res. 2007, 17, 277–279. [Google Scholar] [CrossRef][Green Version]
- Folkerd, E.J.; Dowsett, M. Influence of sex hormones on cancer progression. J. Clin. Oncol. 2010, 28, 4038–4044. [Google Scholar] [CrossRef]
- Straub, R.H. The Complex Role of Estrogens in Inflammation. Endocr. Rev. 2007, 28, 521–574. [Google Scholar] [CrossRef]
- Naugler, W.E.; Sakurai, T.; Kim, S.; Maeda, S.; Kim, K.; Elsharkawy, A.M.; Karin, M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 2007, 317, 121–124. [Google Scholar] [CrossRef]
- Singh, N.; Baby, D.; Rajguru, J.P.; Patil, P.B.; Thakkannavar, S.S.; Pujari, V.B. Inflammation and cancer. Ann. Afr. Med. 2019, 18, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A. Cancer: Inflammation by remote control. Nature 2005, 435, 752–753. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Yaşar, P.; Ayaz, G.; User, S.D.; Güpür, G.; Muyan, M. Molecular mechanism of estrogen–estrogen receptor signaling. Reprod. Med. Biol. 2017, 16, 4–20. [Google Scholar] [CrossRef]
- Razandi, M.; Pedram, A.; Merchenthaler, I.; Greene, G.L.; Levin, E.R. Plasma Membrane Estrogen Receptors Exist and Functions as Dimers. Mol. Endocrinol. 2004, 18, 2854–2865. [Google Scholar] [CrossRef]
- Yang, S.H.; Liu, R.; Perez, E.J.; Wen, Y.; Stevens, S.M., Jr.; Valencia, T.; Brun-Zinkernagel, A.M.; Prokai, L.; Will, Y.; Dykens, J.; et al. Mitochondrial localization of estrogen receptor beta. Proc. Natl. Acad. Sci. USA 2004, 101, 4130–4135. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.M.; McDonnell, D.P. The estrogen receptor beta-isoform (ERbeta) of the human estrogen receptor modulates ERalpha transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology 1999, 140, 5566–5578. [Google Scholar] [CrossRef] [PubMed]
- Mak, H.Y.; Hoare, S.; Henttu, P.M.; Parker, M.G. Molecular determinants of the estrogen receptor-coactivator interface. Mol. Cell. Biol. 1999, 19, 3895–3903. [Google Scholar] [CrossRef]
- Bai, Y.; Giguére, V. Isoform-selective interactions between estrogen receptors and steroid receptor coactivators promoted by estradiol and ErbB-2 signaling in living cells. Mol. Endocrinol. 2003, 17, 589–599. [Google Scholar] [CrossRef][Green Version]
- Driscoll, M.D.; Sathya, G.; Muyan, M.; Klinge, C.M.; Hilf, R.; Bambara, R.A. Sequence requirements for estrogen receptor binding to estrogen response elements. J. Biol. Chem. 1998, 273, 29321–29330. [Google Scholar] [CrossRef]
- Wang, L.H.; Chen, L.R.; Chen, K.H. In Vitro and Vivo Identification, Metabolism and Action of Xenoestrogens: An Overview. Int. J. Mol. Sci. 2021, 22, 84013. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J. Epidermal growth factor receptor (EGFR) transactivation by estrogen via the G-protein-coupled receptor, GPR30: A novel signaling pathway with potential significance for breast cancer. J. Steroid Biochem. Mol. Biol. 2002, 80, 231–238. [Google Scholar] [CrossRef]
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Aronica, S.M.; Kraus, W.L.; Katzenellenbogen, B.S. Estrogen action via the cAMP signaling pathway: Stimulation of adenylate cyclase and cAMP-regulated gene transcription. Proc. Natl. Acad. Sci. USA 1994, 91, 8517–8521. [Google Scholar] [CrossRef]
- Le Mellay, V.; Grosse, B.; Lieberherr, M. Phospholipase Cβ; and Membrane Action of Calcitriol and Estradiol. J. Biol. Chem. 1997, 272, 11902–11907. [Google Scholar] [CrossRef]
- Morley, P.; Whitfield, J.F.; Vanderhyden, B.C.; Tsang, B.K.; Schwartz, J.L. A new, nongenomic estrogen action: The rapid release of intracellular calcium. Endocrinology 1992, 131, 1305–1312. [Google Scholar] [CrossRef]
- Klinge, C.M. Estrogenic control of mitochondrial function and biogenesis. J. Cell. Biochem. 2008, 105, 1342–1351. [Google Scholar] [CrossRef]
- Simpkins, J.W.; Yang, S.-H.; Sarkar, S.N.; Pearce, V. Estrogen actions on mitochondria--physiological and pathological implications. Mol. Cell. Endocrinol. 2008, 290, 51–59. [Google Scholar] [CrossRef]
- Monje, P.; Boland, R. Subcellular distribution of native estrogen receptor alpha and beta isoforms in rabbit uterus and ovary. J. Cell. Biochem. 2001, 82, 467–479. [Google Scholar] [CrossRef]
- Chen, J.Q.; Eshete, M.; Alworth, W.L.; Yager, J.D. Binding of MCF-7 cell mitochondrial proteins and recombinant human estrogen receptors alpha and beta to human mitochondrial DNA estrogen response elements. J. Cell. Biochem. 2004, 93, 358–373. [Google Scholar] [CrossRef]
- Mattingly, K.A.; Ivanova, M.M.; Riggs, K.A.; Wickramasinghe, N.S.; Barch, M.J.; Klinge, C.M. Estradiol stimulates transcription of nuclear respiratory factor-1 and increases mitochondrial biogenesis. Mol. Endocrinol. 2008, 22, 609–622. [Google Scholar] [CrossRef]
- Thomas, C.; Gustafsson, J. The different roles of ER subtypes in cancer biology and therapy. Nat. Rev. Cancer 2011, 11, 597–608. [Google Scholar] [CrossRef]
- Routledge, E.J.; White, R.; Parker, M.G.; Sumpter, J.P. Differential effects of xenoestrogens on coactivator recruitment by estrogen receptor (ER) alpha and ERbeta. J. Biol. Chem. 2000, 275, 35986–35993. [Google Scholar] [CrossRef]
- Ström, A.; Hartman, J.; Foster, J.S.; Kietz, S.; Wimalasena, J.; Gustafsson, J.A. Estrogen receptor beta inhibits 17beta-estradiol-stimulated proliferation of the breast cancer cell line T47D. Proc. Natl. Acad. Sci. USA 2004, 101, 1566–1571. [Google Scholar] [CrossRef]
- Marotti, J.D.; Collins, L.C.; Hu, R.; Tamimi, R.M. Estrogen receptor-beta expression in invasive breast cancer in relation to molecular phenotype: Results from the Nurses’ Health Study. Mod. Pathol. 2010, 23, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Skliris, G.P.; Leygue, E.; Watson, P.H.; Murphy, L.C. Estrogen receptor alpha negative breast cancer patients: Estrogen receptor beta as a therapeutic target. J. Steroid Biochem. Mol. Biol. 2008, 109, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Novelli, F.; Milella, M.; Melucci, E.; Di Benedetto, A.; Sperduti, I.; Perrone-Donnorso, R.; Perracchio, L.; Venturo, I.; Nisticò, C.; Fabi, A.; et al. A divergent role for estrogen receptor-beta in node-positive and node-negative breast cancer classified according to molecular subtypes: An observational prospective study. Breast Cancer Res. 2008, 10, R74. [Google Scholar] [CrossRef]
- Shaaban, A.M.; Green, A.R.; Karthik, S.; Alizadeh, Y.; Hughes, T.A.; Harkins, L.; Ellis, I.O.; Robertson, J.F.; Paish, E.C.; Saunders, P.T.K.; et al. Nuclear and Cytoplasmic Expression of ERβ1, ERβ2, and ERβ5 Identifies Distinct Prognostic Outcome for Breast Cancer Patients. Clin. Cancer Res. 2008, 14, 5228–5235. [Google Scholar] [CrossRef]
- Honma, N.; Horii, R.; Iwase, T.; Saji, S.; Younes, M.; Takubo, K.; Matsuura, M.; Ito, Y.; Akiyama, F.; Sakamoto, G. Clinical importance of estrogen receptor-beta evaluation in breast cancer patients treated with adjuvant tamoxifen therapy. J. Clin. Oncol. 2008, 26, 3727–3734. [Google Scholar] [CrossRef]
- Bozovic, A.; Mandusic, V.; Todorovic, L.; Krajnovic, M. Estrogen Receptor Beta: The Promising Biomarker and Potential Target in Metastases. Int. J. Mol. Sci. 2021, 22, 1565. [Google Scholar] [CrossRef] [PubMed]
- Hilton, H.N.; Clarke, C.L.; Graham, J.D. Estrogen and progesterone signalling in the normal breast and its implications for cancer development. Mol. Cell. Endocrinol. 2018, 466, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Kohler, B.A.; Sherman, R.L.; Howlader, N.; Jemal, A.; Ryerson, A.B.; Henry, K.A.; Boscoe, F.P.; Cronin, K.A.; Lake, A.; Noone, A.M.; et al. Annual Report to the Nation on the Status of Cancer, 1975-2011, Featuring Incidence of Breast Cancer Subtypes by Race/Ethnicity, Poverty, and State. J. Natl. Cancer Inst. 2015, 107, djv048. [Google Scholar] [CrossRef] [PubMed]
- Onitilo, A.A.; Engel, J.M.; Greenlee, R.T.; Mukesh, B.N. Breast cancer subtypes based on ER/PR and Her2 expression: Comparison of clinicopathologic features and survival. Clin. Med. Res. 2009, 7, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Shoker, B.S.; Jarvis, C.; Clarke, R.B.; Anderson, E.; Hewlett, J.; Davies, M.P.; Sibson, D.R.; Sloane, J.P. Estrogen receptor-positive proliferating cells in the normal and precancerous breast. Am. J. Pathol. 1999, 155, 1811–1815. [Google Scholar] [CrossRef]
- Khan, S.A.; Rogers, M.A.; Obando, J.A.; Tamsen, A. Estrogen receptor expression of benign breast epithelium and its association with breast cancer. Cancer Res. 1994, 54, 993–997. [Google Scholar] [PubMed]
- Dubik, D.; Shiu, R.P. Mechanism of estrogen activation of c-myc oncogene expression. Oncogene 1992, 7, 1587–1594. [Google Scholar]
- Planas-Silva, M.D.; Shang, Y.; Donaher, J.L.; Brown, M.; Weinberg, R.A. AIB1 Enhances Estrogen-dependent Induction of Cyclin D1 Expression. Cancer Res. 2001, 61, 3858–3862. [Google Scholar]
- Lindberg, M.K.; Movérare, S.; Skrtic, S.; Gao, H.; Dahlman-Wright, K.; Gustafsson, J.-A.k.; Ohlsson, C. Estrogen Receptor (ER)-β Reduces ERα-Regulated Gene Transcription, Supporting a “Ying Yang” Relationship between ERα and ERβ in Mice. Mol. Endocrinol. 2003, 17, 203–208. [Google Scholar] [CrossRef]
- Williams, C.; Edvardsson, K.; Lewandowski, S.A.; Ström, A.; Gustafsson, J.Å. A genome-wide study of the repressive effects of estrogen receptor beta on estrogen receptor alpha signaling in breast cancer cells. Oncogene 2008, 27, 1019–1032. [Google Scholar] [CrossRef]
- Hartman, J.; Lindberg, K.; Morani, A.; Inzunza, J.; Ström, A.; Gustafsson, J.A. Estrogen receptor beta inhibits angiogenesis and growth of T47D breast cancer xenografts. Cancer Res. 2006, 66, 11207–11213. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Ström, A.; Li Kong, S.; Kietz, S.; Thomsen, J.S.; Tee, J.B.S.; Vega, V.B.; Miller, L.D.; Smeds, J.; Bergh, J.; et al. Inhibitory effects of estrogen receptor beta on specific hormone-responsive gene expression and association with disease outcome in primary breast cancer. Breast Cancer Res. 2007, 9, R25. [Google Scholar] [CrossRef]
- Métivier, R.; Penot, G.; Hübner, M.R.; Reid, G.; Brand, H.; Koš, M.; Gannon, F. Estrogen Receptor-α Directs Ordered, Cyclical, and Combinatorial Recruitment of Cofactors on a Natural Target Promoter. Cell 2003, 115, 751–763. [Google Scholar] [CrossRef]
- Tyson, J.J.; Baumann, W.T.; Chen, C.; Verdugo, A.; Tavassoly, I.; Wang, Y.; Weiner, L.M.; Clarke, R. Dynamic modelling of oestrogen signalling and cell fate in breast cancer cells. Nat. Rev. Cancer 2011, 11, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Gompel, A.; Somaï, S.; Chaouat, M.; Kazem, A.; Kloosterboer, H.J.; Beusman, I.; Forgez, P.; Mimoun, M.; Rostène, W. Hormonal regulation of apoptosis in breast cells and tissues. Steroids 2000, 65, 593–598. [Google Scholar] [CrossRef]
- Crawford, A.C.; Riggins, R.B.; Shajahan, A.N.; Zwart, A.; Clarke, R. Co-Inhibition of BCL-W and BCL2 Restores Antiestrogen Sensitivity through BECN1 and Promotes an Autophagy-Associated Necrosis. PLoS ONE 2010, 5, e8604. [Google Scholar] [CrossRef] [PubMed]
- Kanda, N.; Watanabe, S. 17beta-estradiol inhibits oxidative stress-induced apoptosis in keratinocytes by promoting Bcl-2 expression. J. Investig. Dermatol. 2003, 121, 1500–1509. [Google Scholar] [CrossRef] [PubMed]
- Pratt, M.A.; Bishop, T.E.; White, D.; Yasvinski, G.; Ménard, M.; Niu, M.Y.; Clarke, R. Estrogen withdrawal-induced NF-kappaB activity and bcl-3 expression in breast cancer cells: Roles in growth and hormone independence. Mol. Cell. Biol. 2003, 23, 6887–6900. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Barton, M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat. Rev. Endocrinol. 2011, 7, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Prenzel, N.; Zwick, E.; Daub, H.; Leserer, M.; Abraham, R.; Wallasch, C.; Ullrich, A. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 1999, 402, 884–888. [Google Scholar] [CrossRef]
- Saha Roy, S.; Vadlamudi, R.K. Role of Estrogen Receptor Signaling in Breast Cancer Metastasis. Int. J. Breast Cancer 2012, 2012, 654698. [Google Scholar] [CrossRef]
- Wang, X.; Belguise, K.; Kersual, N.; Kirsch, K.H.; Mineva, N.D.; Galtier, F.; Chalbos, D.; Sonenshein, G.E. Oestrogen signalling inhibits invasive phenotype by repressing RelB and its target BCL2. Nat. Cell. Biol. 2007, 9, 470–478. [Google Scholar] [CrossRef]
- Guttilla, I.K.; Adams, B.D.; White, B.A. ERα, microRNAs, and the epithelial–mesenchymal transition in breast cancer. Trends Endocrinol. Metab. 2012, 23, 73–82. [Google Scholar] [CrossRef]
- Anderson, E. The role of oestrogen and progesterone receptors in human mammary development and tumorigenesis. Breast Cancer Res. 2002, 4, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Rosen, J.M.; Roarty, K. Paracrine signaling in mammary gland development: What can we learn about intratumoral heterogeneity? Breast Cancer Res. 2014, 16, 202. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Zhang, W.; Strasner, A.; Grivennikov, S.; Cheng, J.Q.; Hoffman, R.M.; Karin, M. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature 2011, 470, 548–553. [Google Scholar] [CrossRef]
- Jones, D.H.; Nakashima, T.; Sanchez, O.H.; Kozieradzki, I.; Komarova, S.V.; Sarosi, I.; Morony, S.; Rubin, E.; Sarao, R.; Hojilla, C.V.; et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature 2006, 440, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Borcherding, N.; Kusner, D.; Kolb, R.; Xie, Q.; Li, W.; Yuan, F.; Velez, G.; Askeland, R.; Weigel, R.J.; Zhang, W. Paracrine WNT5A Signaling Inhibits Expansion of Tumor-Initiating Cells. Cancer Res. 2015, 75, 1972–1982. [Google Scholar] [CrossRef]
- Lyons, T.R.; Borges, V.F.; Betts, C.B.; Guo, Q.; Kapoor, P.; Martinson, H.A.; Jindal, S.; Schedin, P. Cyclooxygenase-2-dependent lymphangiogenesis promotes nodal metastasis of postpartum breast cancer. J. Clin. Investig. 2014, 124, 3901–3912. [Google Scholar] [CrossRef]
- Reed, M.J.; Purohit, A. Breast Cancer and the Role of Cytokines in Regulating Estrogen Synthesis: An Emerging Hypothesis. Endocr. Rev. 1997, 18, 701–715. [Google Scholar] [CrossRef]
- Kolb, R.; Zhang, W. Obesity and Breast Cancer: A Case of Inflamed Adipose Tissue. Cancers 2020, 12, 1686. [Google Scholar] [CrossRef]
- Kolb, R.; Phan, L.; Borcherding, N.; Liu, Y.; Yuan, F.; Janowski, A.M.; Xie, Q.; Markan, K.R.; Li, W.; Potthoff, M.J.; et al. Obesity-associated NLRC4 inflammasome activation drives breast cancer progression. Nat. Commun. 2016, 7, 13007. [Google Scholar] [CrossRef]
- Subbaramaiah, K.; Howe, L.R.; Bhardwaj, P.; Du, B.; Gravaghi, C.; Yantiss, R.K.; Zhou, X.K.; Blaho, V.A.; Hla, T.; Yang, P.; et al. Obesity Is Associated with Inflammation and Elevated Aromatase Expression in the Mouse Mammary Gland. Cancer Prev. Res. 2011, 4, 329–346. [Google Scholar] [CrossRef]
- Purohit, A.; Newman, S.P.; Reed, M.J. The role of cytokines in regulating estrogen synthesis: Implications for the etiology of breast cancer. Breast Cancer Res. 2002, 4, 65–69. [Google Scholar] [CrossRef]
- Zhu, P.; Baek, S.H.; Bourk, E.M.; Ohgi, K.A.; Garcia-Bassets, I.; Sanjo, H.; Akira, S.; Kotol, P.F.; Glass, C.K.; Rosenfeld, M.G.; et al. Macrophage/cancer cell interactions mediate hormone resistance by a nuclear receptor derepression pathway. Cell 2006, 124, 615–629. [Google Scholar] [CrossRef]
- Honma, S.; Shimodaira, K.; Shimizu, Y.; Tsuchiya, N.; Saito, H.; Yanaihara, T.; Okai, T. The influence of inflammatory cytokines on estrogen production and cell proliferation in human breast cancer cells. Endocr. J. 2002, 49, 371–377. [Google Scholar] [CrossRef]
- Sofi, M.; Young, M.J.; Papamakarios, T.; Simpson, E.R.; Clyne, C.D. Role of CRE-binding protein (CREB) in aromatase expression in breast adipose. Breast Cancer Res. Treat. 2003, 79, 399–407. [Google Scholar] [CrossRef]
- Chen, D.; Reierstad, S.; Lu, M.; Lin, Z.; Ishikawa, H.; Bulun, S.E. Regulation of breast cancer-associated aromatase promoters. Cancer Lett. 2009, 273, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zhou, L.; Shangguan, A.J.; Bulun, S.E. Aromatase expression and regulation in breast and endometrial cancer. J. Mol. Endocrinol. 2016, 57, R19–R33. [Google Scholar] [CrossRef] [PubMed]
- Subbaramaiah, K.; Iyengar, N.M.; Morrow, M.; Elemento, O.; Zhou, X.K.; Dannenberg, A.J. Prostaglandin E(2) down-regulates sirtuin 1 (SIRT1), leading to elevated levels of aromatase, providing insights into the obesity-breast cancer connection. J. Biol. Chem. 2019, 294, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Brodie, A.M.; Lu, Q.; Long, B.J.; Fulton, A.; Chen, T.; Macpherson, N.; DeJong, P.C.; Blankenstein, M.A.; Nortier, J.W.; Slee, P.H.; et al. Aromatase and COX-2 expression in human breast cancers. J. Steroid Biochem. Mol. Biol. 2001, 79, 41–47. [Google Scholar] [CrossRef]
- Gonçalves, R.M.; Delgobo, M.; Agnes, J.P.; das Neves, R.N.; Falchetti, M.; Casagrande, T.; Garcia, A.P.V.; Vieira, T.C.; Somensi, N.; Bruxel, M.A.; et al. COX-2 promotes mammary adipose tissue inflammation, local estrogen biosynthesis, and carcinogenesis in high-sugar/fat diet treated mice. Cancer Lett. 2021, 502, 44–57. [Google Scholar] [CrossRef]
- Lyons, T.R.; O’Brien, J.; Borges, V.F.; Conklin, M.W.; Keely, P.J.; Eliceiri, K.W.; Marusyk, A.; Tan, A.C.; Schedin, P. Postpartum mammary gland involution drives progression of ductal carcinoma in situ through collagen and COX-2. Nat. Med. 2011, 17, 1109–1115. [Google Scholar] [CrossRef]
- Chung, H.H.; Or, Y.Z.; Shrestha, S.; Loh, J.T.; Lim, C.L.; Ong, Z.; Woo, A.R.E.; Su, I.H.; Lin, V.C.L. Estrogen reprograms the activity of neutrophils to foster protumoral microenvironment during mammary involution. Sci. Rep. 2017, 7, 46485. [Google Scholar] [CrossRef]
- Lim, C.L.; Or, Y.Z.; Ong, Z.; Chung, H.H.; Hayashi, H.; Shrestha, S.; Chiba, S.; Lin, F.; Lin, V.C.L. Estrogen exacerbates mammary involution through neutrophil-dependent and -independent mechanism. eLife 2020, 9, e57274. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Andò, S.; Barone, I.; Giordano, C.; Bonofiglio, D.; Catalano, S. The Multifaceted Mechanism of Leptin Signaling within Tumor Microenvironment in Driving Breast Cancer Growth and Progression. Front. Oncol. 2014, 4, 340. [Google Scholar] [CrossRef] [PubMed]
- Jardé, T.; Caldefie-Chézet, F.; Damez, M.; Mishellany, F.; Penault-Llorca, F.; Guillot, J.; Vasson, M.P. Leptin and leptin receptor involvement in cancer development: A study on human primary breast carcinoma. Oncol. Rep. 2008, 19, 905–911. [Google Scholar] [CrossRef]
- Tessitore, L.; Vizio, B.; Pesola, D.; Cecchini, F.; Mussa, A.; Argiles, J.M.; Benedetto, C. Adipocyte expression and circulating levels of leptin increase in both gynaecological and breast cancer patients. Int. J. Oncol. 2004, 24, 1529–1535. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.A.; McInnes, K.J.; Hunger, N.I.; Oakhill, J.S.; Steinberg, G.R.; Simpson, E.R. Subcellular Localization of Cyclic AMP-Responsive Element Binding Protein-Regulated Transcription Coactivator 2 Provides a Link between Obesity and Breast Cancer in Postmenopausal Women. Cancer Res. 2009, 69, 5392–5399. [Google Scholar] [CrossRef]
- Catalano, S.; Marsico, S.; Giordano, C.; Mauro, L.; Rizza, P.; Panno, M.L.; Andò, S. Leptin enhances, via AP-1, expression of aromatase in the MCF-7 cell line. J. Biol. Chem. 2003, 278, 28668–28676. [Google Scholar] [CrossRef]
- Gashaw, D.; Ayelign, B.; Akalu, Y.; Shibabaw, T.; Molla, M.D. Effect of Leptin on Chronic Inflammatory Disorders: Insights to Therapeutic Target to Prevent Further Cardiovascular Complication. Diabetes Metab. Syndr. Obes. 2021, 14, 3307–3322. [Google Scholar] [CrossRef]
- Garonna, E.; Botham, K.M.; Birdsey, G.M.; Randi, A.M.; Gonzalez-Perez, R.R.; Wheeler-Jones, C.P.D. Vascular Endothelial Growth Factor Receptor-2 Couples Cyclo-Oxygenase-2 with Pro-Angiogenic Actions of Leptin on Human Endothelial Cells. PLoS ONE 2011, 6, e18823. [Google Scholar] [CrossRef]
- Iikuni, N.; Lam, Q.L.K.; Lu, L.; Matarese, G.; La Cava, A. Leptin and Inflammation. Curr. Immunol. Rev. 2008, 4, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, C.; Koda, M.; Cascio, S.; Sulkowska, M.; Kanczuga-Koda, L.; Golaszewska, J.; Russo, A.; Sulkowski, S.; Surmacz, E. Increased expression of leptin and the leptin receptor as a marker of breast cancer progression: Possible role of obesity-related stimuli. Clin. Cancer Res. 2006, 12, 1447–1453. [Google Scholar] [CrossRef]
- Stofkova, A. Leptin and adiponectin: From energy and metabolic dysbalance to inflammation and autoimmunity. Endocr. Regul. 2009, 43, 157–168. [Google Scholar]
- Makki, K.; Froguel, P.; Wolowczuk, I. Adipose tissue in obesity-related inflammation and insulin resistance: Cells, cytokines, and chemokines. ISRN Inflamm. 2013, 2013, 139239. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Role of adiponectin and PBEF/visfatin as regulators of inflammation: Involvement in obesity-associated diseases. Clin. Sci. 2008, 114, 275–288. [Google Scholar] [CrossRef]
- Ouchi, N.; Walsh, K. Adiponectin as an anti-inflammatory factor. Clin. Chim. Acta 2007, 380, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Behl, S.; Adem, A.; Hussain, A.; Singh, J. Effects of rilpivirine, 17β-estradiol and β-naphthoflavone on the inflammatory status of release of adipocytokines in 3T3-L1 adipocytes in vitro. Mol. Biol. Rep. 2019, 46, 2643–2655. [Google Scholar] [CrossRef]
- Miyatani, Y.; Yasui, T.; Uemura, H.; Yamada, M.; Matsuzaki, T.; Kuwahara, A.; Tsuchiya, N.; Yuzurihara, M.; Kase, Y.; Irahara, M. Associations of circulating adiponectin with estradiol and monocyte chemotactic protein-1 in postmenopausal women. Menopause 2008, 15, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Shukla, S.; Sinha, S.; Meeran, S.M. Role of adipokines and cytokines in obesity-associated breast cancer: Therapeutic targets. Cytokine Growth Factor Rev. 2013, 24, 503–513. [Google Scholar] [CrossRef]
- Bhardwaj, P.; Du, B.; Zhou, X.K.; Sue, E.; Giri, D.; Harbus, M.D.; Falcone, D.J.; Hudis, C.A.; Subbaramaiah, K.; Dannenberg, A.J. Estrogen Protects against Obesity-Induced Mammary Gland Inflammation in Mice. Cancer Prev. Res. 2015, 8, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Kolb, R.; Sutterwala, F.S.; Zhang, W. Obesity and cancer: Inflammation bridges the two. Curr. Opin. Pharmacol. 2016, 29, 77–89. [Google Scholar] [CrossRef]
- Chan, D.S.M.; Vieira, A.R.; Aune, D.; Bandera, E.V.; Greenwood, D.C.; McTiernan, A.; Navarro Rosenblatt, D.; Thune, I.; Vieira, R.; Norat, T. Body mass index and survival in women with breast cancer-systematic literature review and meta-analysis of 82 follow-up studies. Ann. Oncol. 2014, 25, 1901–1914. [Google Scholar] [CrossRef] [PubMed]
- Protani, M.; Coory, M.; Martin, J.H. Effect of obesity on survival of women with breast cancer: Systematic review and meta-analysis. Breast Cancer Res. Treat. 2010, 123, 627–635. [Google Scholar] [CrossRef]
- Chang, S.-L.; Tchernof, A.; Durocher, F.; Diorio, C. Associations of Biomarkers of Inflammation and Breast Cancer in the Breast Adipose Tissue of Women with Combined Measures of Adiposity. J. Obes. 2021, 2021, 3620147. [Google Scholar] [CrossRef]
- Tawara, K.; Scott, H.; Emathinger, J.; Wolf, C.; LaJoie, D.; Hedeen, D.; Bond, L.; Montgomery, P.; Jorcyk, C. HIGH expression of OSM and IL-6 are associated with decreased breast cancer survival: Synergistic induction of IL-6 secretion by OSM and IL-1β. Oncotarget 2019, 10, 2068. [Google Scholar] [CrossRef]
- McAndrew, N.P.; Bottalico, L.; Mesaros, C.; Blair, I.A.; Tsao, P.Y.; Rosado, J.M.; Ganguly, T.; Song, S.J.; Gimotty, P.A.; Mao, J.J.; et al. Effects of systemic inflammation on relapse in early breast cancer. NPJ Breast Cancer 2021, 7, 7. [Google Scholar] [CrossRef]
- Madeddu, C.; Gramignano, G.; Floris, C.; Murenu, G.; Sollai, G.; Macciò, A. Role of inflammation and oxidative stress in post-menopausal oestrogen-dependent breast cancer. J. Cell Mol. Med. 2014, 18, 2519–2529. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Garduño, A.M.; Mendoza-Rodríguez, M.G.; Urrutia-Cabrera, D.; Domínguez-Robles, M.C.; Pérez-Yépez, E.A.; Ayala-Sumuano, J.T.; Meza, I. IL-1β induced methylation of the estrogen receptor ERα gene correlates with EMT and chemoresistance in breast cancer cells. Biochem. Biophys. Res. Commun. 2017, 490, 780–785. [Google Scholar] [CrossRef]
- Kim, S.; Campbell, J.; Yoo, W.; Taylor, J.A.; Sandler, D.P. Systemic Levels of Estrogens and PGE(2) Synthesis in Relation to Postmenopausal Breast Cancer Risk. Cancer Epidemiol. Biomark. Prev. 2017, 26, 383–388. [Google Scholar] [CrossRef]
- Gupta, P.B.; Proia, D.; Cingoz, O.; Weremowicz, J.; Naber, S.P.; Weinberg, R.A.; Kuperwasser, C. Systemic Stromal Effects of Estrogen Promote the Growth of Estrogen Receptor–Negative Cancers. Cancer Res. 2007, 67, 2062–2071. [Google Scholar] [CrossRef] [PubMed]
- Svensson, S.; Abrahamsson, A.; Rodriguez, G.V.; Olsson, A.-K.; Jensen, L.; Cao, Y.; Dabrosin, C. CCL2 and CCL5 Are Novel Therapeutic Targets for Estrogen-Dependent Breast Cancer. Clin. Cancer Res. 2015, 21, 3794–3805. [Google Scholar] [CrossRef]
- Marquez-Garban, D.C.; Deng, G.; Comin-Anduix, B.; Garcia, A.J.; Xing, Y.; Chen, H.W.; Cheung-Lau, G.; Hamilton, N.; Jung, M.E.; Pietras, R.J. Antiestrogens in combination with immune checkpoint inhibitors in breast cancer immunotherapy. J. Steroid Biochem. Mol. Biol. 2019, 193, 105415. [Google Scholar] [CrossRef] [PubMed]
- Svoronos, N.; Perales-Puchalt, A.; Allegrezza, M.J.; Rutkowski, M.R.; Payne, K.K.; Tesone, A.J.; Nguyen, J.M.; Curiel, T.J.; Cadungog, M.G.; Singhal, S.; et al. Tumor Cell-Independent Estrogen Signaling Drives Disease Progression through Mobilization of Myeloid-Derived Suppressor Cells. Cancer Discov. 2017, 7, 72–85. [Google Scholar] [CrossRef]
- Matthews, S.B.; Thompson, H.J. The Obesity-Breast Cancer Conundrum: An Analysis of the Issues. Int. J. Mol. Sci. 2016, 17, 989. [Google Scholar] [CrossRef]
- Lin, Y.; Huang, R.; Chen, L.; Li, S.; Shi, Q.; Jordan, C.; Huang, R.P. Identification of interleukin-8 as estrogen receptor-regulated factor involved in breast cancer invasion and angiogenesis by protein arrays. Int. J. Cancer 2004, 109, 507–515. [Google Scholar] [CrossRef]
- Guo, S.; Gärtner, F.; Schmitt, F.; Russo, J. 17 -Estradiol-Mediated Vessel Assembly and Stabilization in Tumor Angiogenesis Requires TGF and EGFR Crosstalk. Angiogenesis 2003, 6, 271–281. [Google Scholar] [CrossRef]
- Inadera, H.; Sekiya, T.; Yoshimura, T.; Matsushima, K. Molecular Analysis of the Inhibition of Monocyte Chemoattractant Protein-1 Gene Expression by Estrogens and Xenoestrogens in MCF-7 Cells1. Endocrinology 2000, 141, 50–59. [Google Scholar] [CrossRef]
- Quigley, D.A.; Tahiri, A.; Lüders, T.; Riis, M.H.; Balmain, A.; Børresen-Dale, A.-L.; Bukholm, I.; Kristensen, V. Age, estrogen, and immune response in breast adenocarcinoma and adjacent normal tissue. Oncoimmunology 2017, 6, e1356142. [Google Scholar] [CrossRef]
- Goldberg, J.; Pastorello, R.G.; Vallius, T.; Davis, J.; Cui, Y.X.; Agudo, J.; Waks, A.G.; Keenan, T.; McAllister, S.S.; Tolaney, S.M.; et al. The Immunology of Hormone Receptor Positive Breast Cancer. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Tse, L.A.; Wang, D.; Koka, H.; Zhang, T.; Abubakar, M.; Lee, P.; Wang, F.; Wu, C.; Tsang, K.H.; et al. Immune gene expression profiling reveals heterogeneity in luminal breast tumors. Breast Cancer Res. 2019, 21, 147. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.M.; Moulder-Thompson, S.L. Neoadjuvant treatment of breast cancer. Ann. Oncol. 2012, 23 Suppl. S10, 231–236. [Google Scholar] [CrossRef]
- Burstein, H.J.; Lacchetti, C.; Anderson, H.; Buchholz, T.A.; Davidson, N.E.; Gelmon, K.A.; Giordano, S.H.; Hudis, C.A.; Solky, A.J.; Stearns, V.; et al. Adjuvant Endocrine Therapy for Women With Hormone Receptor-Positive Breast Cancer: ASCO Clinical Practice Guideline Focused Update. J. Clin. Oncol. 2019, 37, 423–438. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.R. Aromatase inhibitors: Mechanism of action and role in the treatment of breast cancer. Semin. Oncol. 2003, 30, 3–11. [Google Scholar] [CrossRef]
- Kharb, R.; Haider, K.; Neha, K.; Yar, M.S. Aromatase inhibitors: Role in postmenopausal breast cancer. Arch. Pharm. 2020, 353, e2000081. [Google Scholar] [CrossRef]
- Osborne, C.K.; Zhao, H.; Fuqua, S.A. Selective estrogen receptor modulators: Structure, function, and clinical use. J. Clin. Oncol. 2000, 18, 3172–3186. [Google Scholar] [CrossRef]
- Osborne, C.K. Tamoxifen in the treatment of breast cancer. N. Engl. J. Med. 1998, 339, 1609–1618. [Google Scholar] [CrossRef]
- Love, R.R.; Cameron, L.; Connell, B.L.; Leventhal, H. Symptoms associated with tamoxifen treatment in postmenopausal women. Arch. Intern. Med. 1991, 151, 1842–1847. [Google Scholar] [CrossRef] [PubMed]
- Love, R.R.; Mazess, R.B.; Barden, H.S.; Epstein, S.; Newcomb, P.A.; Jordan, V.C.; Carbone, P.P.; DeMets, D.L. Effects of tamoxifen on bone mineral density in postmenopausal women with breast cancer. N. Engl. J. Med. 1992, 326, 852–856. [Google Scholar] [CrossRef]
- Williams, J.K.; Wagner, J.D.; Li, Z.; Golden, D.L.; Adams, M.R. Tamoxifen inhibits arterial accumulation of LDL degradation products and progression of coronary artery atherosclerosis in monkeys. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 403–408. [Google Scholar] [CrossRef]
- Scott, J.S.; Moss, T.A.; Balazs, A.; Barlaam, B.; Breed, J.; Carbajo, R.J.; Chiarparin, E.; Davey, P.R.J.; Delpuech, O.; Fawell, S.; et al. Discovery of AZD9833, a Potent and Orally Bioavailable Selective Estrogen Receptor Degrader and Antagonist. J. Med. Chem. 2020, 63, 14530–14559. [Google Scholar] [CrossRef]
- Perey, L.; Paridaens, R.; Hawle, H.; Zaman, K.; Nole, F.; Wildiers, H.; Fiche, M.; Dietrich, D.; Clement, P.; Koberle, D.; et al. Clinical benefit of fulvestrant in postmenopausal women with advanced breast cancer and primary or acquired resistance to aromatase inhibitors: Final results of phase II Swiss Group for Clinical Cancer Research Trial (SAKK 21/00). Ann. Oncol. 2007, 18, 64–69. [Google Scholar] [CrossRef]
- Pepermans, R.A.; Prossnitz, E.R. ERalpha-targeted endocrine therapy, resistance and the role of GPER. Steroids 2019, 152, 108493. [Google Scholar] [CrossRef]
- Pierdominici, M.; Maselli, A.; Colasanti, T.; Giammarioli, A.M.; Delunardo, F.; Vacirca, D.; Sanchez, M.; Giovannetti, A.; Malorni, W.; Ortona, E. Estrogen receptor profiles in human peripheral blood lymphocytes. Immunol. Lett. 2010, 132, 79–85. [Google Scholar] [CrossRef]
- Cunningham, M.; Gilkeson, G. Estrogen receptors in immunity and autoimmunity. Clin. Rev. Allergy Immunol. 2011, 40, 66–73. [Google Scholar] [CrossRef]
- Huang, H.; Zhou, J.; Chen, H.; Li, J.; Zhang, C.; Jiang, X.; Ni, C. The immunomodulatory effects of endocrine therapy in breast cancer. J. Exp. Clin. Cancer Res. 2021, 40, 19. [Google Scholar] [CrossRef]
- Ferrarelli, L.K. Tamoxifen as an immunotherapy. Sci. Signal. 2017, 10, eaam9611. [Google Scholar] [CrossRef]
- Segovia-Mendoza, M.; Morales-Montor, J. Immune Tumor Microenvironment in Breast Cancer and the Participation of Estrogen and Its Receptors in Cancer Physiopathology. Front. Immunol. 2019, 10, 348. [Google Scholar] [CrossRef]
- Wu, D.; Tang, S.; Ye, R.; Li, D.; Gu, D.; Chen, R.; Zhang, H.; Sun, J.; Chen, Z. Case Report: Long-Term Response to Pembrolizumab Combined With Endocrine Therapy in Metastatic Breast Cancer Patients With Hormone Receptor Expression. Front. Immunol. 2021, 12, 610149. [Google Scholar] [CrossRef]
- Tanwar, A.K.; Dhiman, N.; Kumar, A.; Jaitak, V. Engagement of phytoestrogens in breast cancer suppression: Structural classification and mechanistic approach. Eur. J. Med. Chem. 2021, 213, 113037. [Google Scholar] [CrossRef]
- Mense, S.M.; Hei, T.K.; Ganju, R.K.; Bhat, H.K. Phytoestrogens and breast cancer prevention: Possible mechanisms of action. Environ. Health Perspect. 2008, 116, 426–433. [Google Scholar] [CrossRef]
- Wang, Y.H.; Tsay, Y.G.; Tan, B.C.M.; Lo, W.Y.; Lee, S.C. Identification and characterization of a novel p300-mediated p53 acetylation site, lysine 305. J. Biol. Chem. 2003, 278, 25568–25576. [Google Scholar] [CrossRef]
- Brzezinski, A.; Debi, A. Phytoestrogens: The “natural” selective estrogen receptor modulators? Eur. J. Obstet. Gynecol. Reprod. Biol. 1999, 85, 47–51. [Google Scholar] [CrossRef]
- Torrens-Mas, M.; Roca, P. Phytoestrogens for Cancer Prevention and Treatment. Biology 2020, 9, 9120427. [Google Scholar] [CrossRef]
- Martin, P.M.; Horwitz, K.B.; Ryan, D.S.; McGuire, W.L. Phytoestrogen Interaction with Estrogen Receptors in Human Breast Cancer Cells. Endocrinology 1978, 103, 1860–1867. [Google Scholar] [CrossRef]
- Zava, D.T.; Blen, M.; Duwe, G. Estrogenic activity of natural and synthetic estrogens in human breast cancer cells in culture. Environ. Health Perspect. 1997, 105 Suppl 3, 637–645. [Google Scholar] [CrossRef]
- Adeel, M.; Song, X.; Wang, Y.; Francis, D.; Yang, Y. Environmental impact of estrogens on human, animal and plant life: A critical review. Environ. Int. 2017, 99, 107–119. [Google Scholar] [CrossRef]
- Adlercreutz, H.; Hämäläinen, E.; Gorbach, S.; Goldin, B. Dietary phyto-oestrogens and the menopause in Japan. Lancet 1992, 339, 1233. [Google Scholar] [CrossRef]
- Parkin, D.M. Cancers of the breast, endometrium and ovary: Geographic correlations. Eur. J. Cancer Clin. Oncol. 1989, 25, 1917–1925. [Google Scholar] [CrossRef]
- Ingram, D.; Sanders, K.; Kolybaba, M.; Lopez, D. Case-control study of phyto-oestrogens and breast cancer. Lancet 1997, 350, 990–994. [Google Scholar] [CrossRef]
- Cotterchio, M.; Boucher, B.A.; Kreiger, N.; Mills, C.A.; Thompson, L.U. Dietary phytoestrogen intake--lignans and isoflavones--and breast cancer risk (Canada). Cancer Causes Control 2008, 19, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-García, C.; Sánchez-Quesada, C.; Toledo, E.; Delgado-Rodríguez, M.; Gaforio, J.J. Naturally Lignan-Rich Foods: A Dietary Tool for Health Promotion? Molecules 2019, 24, 917. [Google Scholar] [CrossRef]
- Jensen, S.N.; Cady, N.M.; Shahi, S.K.; Peterson, S.R.; Gupta, A.; Gibson-Corley, K.N.; Mangalam, A.K. Isoflavone diet ameliorates experimental autoimmune encephalomyelitis through modulation of gut bacteria depleted in patients with multiple sclerosis. Sci. Adv. 2021, 7, eabd4595. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Lemmen, J.G.; Carlsson, B.; Corton, J.C.; Safe, S.H.; van der Saag, P.T.; van der Burg, B.; Gustafsson, J.A. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology 1998, 139, 4252–4263. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Carlsson, B.; Grandien, K.; Enmark, E.; Häggblad, J.; Nilsson, S.; Gustafsson, J.A. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology 1997, 138, 863–870. [Google Scholar] [CrossRef]
- Bowers, J.L.; Tyulmenkov, V.V.; Jernigan, S.C.; Klinge, C.M. Resveratrol acts as a mixed agonist/antagonist for estrogen receptors alpha and beta. Endocrinology 2000, 141, 3657–3667. [Google Scholar] [CrossRef]
- Maggiolini, M.; Bonofiglio, D.; Marsico, S.; Panno, M.L.; Cenni, B.; Picard, D.; Andò, S. Estrogen receptor alpha mediates the proliferative but not the cytotoxic dose-dependent effects of two major phytoestrogens on human breast cancer cells. Mol. Pharmacol. 2001, 60, 595–602. [Google Scholar]
- Duncan, A.M.; Underhill, K.E.; Xu, X.; Lavalleur, J.; Phipps, W.R.; Kurzer, M.S. Modest hormonal effects of soy isoflavones in postmenopausal women. J. Clin. Endocrinol. Metab. 1999, 84, 3479–3484. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.B.; Cantor, A.; Allen, K.; Riccardi, D.; Cox, C.E. The specific role of isoflavones on estrogen metabolism in premenopausal women. Cancer 2002, 94, 1166–1174. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.J.; Anderson, K.E.; Grady, J.J.; Kohen, F.; Nagamani, M. Decreased ovarian hormones during a soya diet: Implications for breast cancer prevention. Cancer Res. 2000, 60, 4112–4121. [Google Scholar]
- Rice, S.; Whitehead, S.A. Phytoestrogens and breast cancer –promoters or protectors? Endocr. Relat. Cancer 2006, 13, 995–1015. [Google Scholar] [CrossRef]
- Rice, S.; Mason, H.D.; Whitehead, S.A. Phytoestrogens and their low dose combinations inhibit mRNA expression and activity of aromatase in human granulosa-luteal cells. J. Steroid Biochem. Mol. Biol. 2006, 101, 216–225. [Google Scholar] [CrossRef]
- Pozo-Guisado, E.; Alvarez-Barrientos, A.; Mulero-Navarro, S.; Santiago-Josefat, B.; Fernandez-Salguero, P.M. The antiproliferative activity of resveratrol results in apoptosis in MCF-7 but not in MDA-MB-231 human breast cancer cells: Cell-specific alteration of the cell cycle. Biochem. Pharmacol. 2002, 64, 1375–1386. [Google Scholar] [CrossRef]
- Pozo-Guisado, E.; Merino, J.M.; Mulero-Navarro, S.; Lorenzo-Benayas, M.J.; Centeno, F.; Alvarez-Barrientos, A.; Fernandez-Salguero, P.M. Resveratrol-induced apoptosis in MCF-7 human breast cancer cells involves a caspase-independent mechanism with downregulation of Bcl-2 and NF-kappaB. Int. J. Cancer 2005, 115, 74–84. [Google Scholar] [CrossRef]
- Balabhadrapathruni, S.; Thomas, T.J.; Yurkow, E.; Amenta, P. Effects of genistein and structurally related phytoestrogens on cell cycle kinetics and apoptosis in MDA-MB-468 human breast cancer cells. Oncol. Rep. 2000, 7, 3–12. [Google Scholar] [CrossRef]
- Le Corre, L.; Chalabi, N.; Delort, L.; Bignon, Y.J.; Bernard-Gallon, D.J. Differential expression of genes induced by resveratrol in human breast cancer cell lines. Nutr. Cancer 2006, 56, 193–203. [Google Scholar] [CrossRef]
- Shao, Z.M.; Wu, J.; Shen, Z.Z.; Barsky, S.H. Genistein exerts multiple suppressive effects on human breast carcinoma cells. Cancer Res. 1998, 58, 4851–4857. [Google Scholar]
- Hsieh, C.Y.; Santell, R.C.; Haslam, S.Z.; Helferich, W.G. Estrogenic effects of genistein on the growth of estrogen receptor-positive human breast cancer (MCF-7) cells in vitro and in vivo. Cancer Res. 1998, 58, 3833–3838. [Google Scholar]
- Barnes, S.; Grubbs, C.; Setchell, K.D.; Carlson, J. Soybeans inhibit mammary tumors in models of breast cancer. Prog. Clin. Biol. Res. 1990, 347, 239–253. [Google Scholar]
- Thompson, L.U.; Rickard, S.E.; Orcheson, L.J.; Seidl, M.M. Flaxseed and its lignan and oil components reduce mammary tumor growth at a late stage of carcinogenesis. Carcinogenesis 1996, 17, 1373–1376. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Virk-Baker, M.K.; Nagy, T.R.; Barnes, S. Role of phytoestrogens in cancer therapy. Planta Med. 2010, 76, 1132–1142. [Google Scholar] [CrossRef]
- Moris, D.; Kontos, M.; Spartalis, E.; Fentiman, I.S. The Role of NSAIDs in Breast Cancer Prevention and Relapse: Current Evidence and Future Perspectives. Breast Care 2016, 11, 339–344. [Google Scholar] [CrossRef]
- Yu, J.; Bi, X.; Yu, B.; Chen, D. Isoflavones: Anti-Inflammatory Benefit and Possible Caveats. Nutrients 2016, 8, 361. [Google Scholar] [CrossRef]
- Meng, T.; Xiao, D.; Muhammed, A.; Deng, J.; Chen, L.; He, J. Anti-Inflammatory Action and Mechanisms of Resveratrol. Molecules 2021, 26, 229. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Bueso-Ramos, C.; Aggarwal, B.B. Suppression of 7,12-dimethylbenz(a)anthracene-induced mammary carcinogenesis in rats by resveratrol: Role of nuclear factor-kappaB, cyclooxygenase 2, and matrix metalloprotease 9. Cancer Res. 2002, 62, 4945–4954. [Google Scholar]
- Kundu, J.K.; Surh, Y.J. Molecular basis of chemoprevention by resveratrol: NF-kappaB and AP-1 as potential targets. Mutat. Res. 2004, 555, 65–80. [Google Scholar] [CrossRef]
- Vanden Berghe, W.; Dijsselbloem, N.; Vermeulen, L.; Ndlovu, M.N.; Boone, E.; Haegeman, G. Attenuation of Mitogen- and Stress-Activated Protein Kinase-1–Driven Nuclear Factor-κB Gene Expression by Soy Isoflavones Does Not Require Estrogenic Activity. Cancer Res. 2006, 66, 4852. [Google Scholar] [CrossRef]
- Yu, R.; Hebbar, V.; Kim, D.W.; Mandlekar, S.; Pezzuto, J.M.; Kong, A.N. Resveratrol inhibits phorbol ester and UV-induced activator protein 1 activation by interfering with mitogen-activated protein kinase pathways. Mol. Pharmacol. 2001, 60, 217–224. [Google Scholar] [CrossRef]
- Ditsworth, D.; Zong, W.-X. NF-kappaB: Key mediator of inflammation-associated cancer. Cancer Biol. Ther. 2004, 3, 1214–1216. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.N.; Kucuk, O.; Djuric, Z.; Sarkar, F.H. Soy isoflavone supplementation in healthy men prevents NF-kappa B activation by TNF-alpha in blood lymphocytes. Free Radic. Biol. Med. 2001, 30, 1293–1302. [Google Scholar] [CrossRef]
- Karin, M. The regulation of AP-1 activity by mitogen-activated protein kinases. Philos. Trans. R Soc. Lond. B Biol. Sci. 1996, 351, 127–134. [Google Scholar] [CrossRef]
- Robb, E.L.; Stuart, J.A. Resveratrol interacts with estrogen receptor-β to inhibit cell replicative growth and enhance stress resistance by upregulating mitochondrial superoxide dismutase. Free Radic. Biol. Med. 2011, 50, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Kampkötter, A.; Chovolou, Y.; Kulawik, A.; Röhrdanz, E.; Weber, N.; Proksch, P.; Wätjen, W. Isoflavone daidzein possesses no antioxidant activities in cell-free assays but induces the antioxidant enzyme catalase. Nutr. Res. 2008, 28, 620–628. [Google Scholar] [CrossRef]
- Surico, D.; Ercoli, A.; Farruggio, S.; Raina, G.; Filippini, D.; Mary, D.; Minisini, R.; Surico, N.; Pirisi, M.; Grossini, E. Modulation of Oxidative Stress by 17 β-Estradiol and Genistein in Human Hepatic Cell Lines In Vitro. Cell. Physiol. Biochem. 2017, 42, 1051–1062. [Google Scholar] [CrossRef]
- Behrend, L.; Henderson, G.; Zwacka, R.M. Reactive oxygen species in oncogenic transformation. Biochem. Soc. Trans. 2003, 31, 1441–1444. [Google Scholar] [CrossRef]
- Nadal-Serrano, M.; Pons, D.G.; Sastre-Serra, J.; Blanquer-Rosselló Mdel, M.; Roca, P.; Oliver, J. Genistein modulates oxidative stress in breast cancer cell lines according to ERα/ERβ ratio: Effects on mitochondrial functionality, sirtuins, uncoupling protein 2 and antioxidant enzymes. Int. J. Biochem. Cell. Biol. 2013, 45, 2045–2051. [Google Scholar] [CrossRef] [PubMed]
- Yen, G.C.; Lai, H.H. Inhibition of reactive nitrogen species effects in vitro and in vivo by isoflavones and soy-based food extracts. J. Agric. Food Chem. 2003, 51, 7892–7900. [Google Scholar] [CrossRef]
- Gao, Y.; Fu, R.; Wang, J.; Yang, X.; Wen, L.; Feng, J. Resveratrol mitigates the oxidative stress mediated by hypoxic-ischemic brain injury in neonatal rats via Nrf2/HO-1 pathway. Pharm. Biol. 2018, 56, 440–449. [Google Scholar] [CrossRef]
- Ji, G.; Zhang, Y.; Yang, Q.; Cheng, S.; Hao, J.; Zhao, X.; Jiang, Z. Genistein suppresses LPS-induced inflammatory response through inhibiting NF-κB following AMP kinase activation in RAW 264.7 macrophages. PLoS ONE 2012, 7, e53101. [Google Scholar] [CrossRef]
- Blay, M.; Espinel, A.E.; Delgado, M.A.; Baiges, I.; Bladé, C.; Arola, L.; Salvadó, J. Isoflavone effect on gene expression profile and biomarkers of inflammation. J. Pharm. Biomed. Anal. 2010, 51, 382–390. [Google Scholar] [CrossRef]
- Gao, Y.; Fang, L.; Liu, F.; Zong, C.; Cai, R.; Chen, X.; Qi, Y. Suppressive effects of irisflorentin on LPS-induced inflammatory responses in RAW 264.7 macrophages. Exp. Biol. Med. 2014, 239, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Lesinski, G.B.; Reville, P.K.; Mace, T.A.; Young, G.S.; Ahn-Jarvis, J.; Thomas-Ahner, J.; Vodovotz, Y.; Ameen, Z.; Grainger, E.; Riedl, K.; et al. Consumption of soy isoflavone enriched bread in men with prostate cancer is associated with reduced proinflammatory cytokines and immunosuppressive cells. Cancer Prev. Res. 2015, 8, 1036–1044. [Google Scholar] [CrossRef]
- Gao, X.; Xu, Y.X.; Janakiraman, N.; Chapman, R.A.; Gautam, S.C. Immunomodulatory activity of resveratrol: Suppression of lymphocyte proliferation, development of cell-mediated cytotoxicity, and cytokine production. Biochem. Pharmacol. 2001, 62, 1299–1308. [Google Scholar] [CrossRef]
- Subbaramaiah, K.; Chung, W.J.; Michaluart, P.; Telang, N.; Tanabe, T.; Inoue, H.; Jang, M.; Pezzuto, J.M.; Dannenberg, A.J. Resveratrol inhibits cyclooxygenase-2 transcription and activity in phorbol ester-treated human mammary epithelial cells. J. Biol. Chem. 1998, 273, 21875–21882. [Google Scholar] [CrossRef] [PubMed]
- Lau, T.Y.; Leung, L.K. Soya isoflavones suppress phorbol 12-myristate 13-acetate-induced COX-2 expression in MCF-7 cells. Br. J. Nutr. 2006, 96, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, F.; Wang, F.; Li, J.; Lin, C.; Du, J. Resveratrol inhibits the proliferation of A549 cells by inhibiting the expression of COX-2. Onco. Targets Ther. 2018, 11, 2981–2989. [Google Scholar] [CrossRef]
- Gong, W.H.; Zhao, N.; Zhang, Z.M.; Zhang, Y.X.; Yan, L.; Li, J.B. The inhibitory effect of resveratrol on COX-2 expression in human colorectal cancer: A promising therapeutic strategy. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 1136–1143. [Google Scholar] [PubMed]
- Goldberg, J.E.; Schwertfeger, K.L. Proinflammatory cytokines in breast cancer: Mechanisms of action and potential targets for therapeutics. Curr. Drug Targets 2010, 11, 1133–1146. [Google Scholar] [CrossRef]
- Rundhaug, J.E.; Fischer, S.M. Cyclo-oxygenase-2 plays a critical role in UV-induced skin carcinogenesis. Photochem. Photobiol. 2008, 84, 322–329. [Google Scholar] [CrossRef]
- Kundu, J.K.; Shin, Y.K.; Kim, S.H.; Surh, Y.J. Resveratrol inhibits phorbol ester-induced expression of COX-2 and activation of NF-kappaB in mouse skin by blocking IkappaB kinase activity. Carcinogenesis 2006, 27, 1465–1474. [Google Scholar] [CrossRef]
- Yellayi, S.; Zakroczymski, M.; Selvaraj, V.; Valli, V.; V, G.; Helferich, W.; Cooke, P. The phytoestrogen genistein suppresses cell-mediated immunity in mice. J. Endocrinol. 2003, 176, 267–274. [Google Scholar] [CrossRef]
- Yellayi, S.; Naaz, A.; Szewczykowski, M.A.; Sato, T.; Woods, J.A.; Chang, J.; Segre, M.; Allred, C.D.; Helferich, W.G.; Cooke, P.S. The phytoestrogen genistein induces thymic and immune changes: A human health concern? Proc. Natl. Acad. Sci. USA 2002, 99, 7616–7621. [Google Scholar] [CrossRef]
- Portman, M.A. Kawasaki disease and soy: Potential role for isoflavone interaction with Fcγ receptors. Pediatric Res. 2013, 73, 130–134. [Google Scholar] [CrossRef]
- Andrade, J.E.; Ju, Y.H.; Baker, C.; Doerge, D.R.; Helferich, W.G. Long-term exposure to dietary sources of genistein induces estrogen-independence in the human breast cancer (MCF-7) xenograft model. Mol. Nutr. Food Res. 2015, 59, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Calaf, G.M.; Ponce-Cusi, R.; Aguayo, F.; Muñoz, J.P.; Bleak, T.C. Endocrine disruptors from the environment affecting breast cancer. Oncol. Lett. 2020, 20, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Davis, D.L.; Bradlow, H.L.; Wolff, M.; Woodruff, T.; Hoel, D.G.; Anton-Culver, H. Medical hypothesis: Xenoestrogens as preventable causes of breast cancer. Environ. Health Perspect. 1993, 101, 372–377. [Google Scholar] [CrossRef]
- Eve, L.; Fervers, B.; Le Romancer, M.; Etienne-Selloum, N. Exposure to Endocrine Disrupting Chemicals and Risk of Breast Cancer. Int. J. Mol. Sci. 2020, 21, 9139. [Google Scholar] [CrossRef] [PubMed]
- Davis, D.L.; Bradlow, H.L. Can environmental estrogens cause breast cancer? Sci. Am. 1995, 273, 167–172. [Google Scholar] [CrossRef]
- Ardies, C.M.; Dees, C. Xenoestrogens significantly enhance risk for breast cancer during growth and adolescence. Med. Hypotheses 1998, 50, 457–464. [Google Scholar] [CrossRef]
- Fernandez, S.V.; Russo, J. Estrogen and xenoestrogens in breast cancer. Toxicol. Pathol. 2010, 38, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Troisi, R.; Hatch, E.E.; Titus-Ernstoff, L.; Hyer, M.; Palmer, J.R.; Robboy, S.J.; Strohsnitter, W.C.; Kaufman, R.; Herbst, A.L.; Hoover, R.N. Cancer risk in women prenatally exposed to diethylstilbestrol. Int J. Cancer 2007, 121, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.; Lee, S.-O.; Jin, U.-H. Role of the Aryl Hydrocarbon Receptor in Carcinogenesis and Potential as a Drug Target. Toxicol. Sci. 2013, 135, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.O.; Kling, M.; Arifin Firzani, P.; Mecky, A.; Duranti, E.; Shields-Botella, J.; Delansorne, R.; Broschard, T.; Kramer, P.J. Activation of estrogen receptor alpha and ERbeta by 4-methylbenzylidene-camphor in human and rat cells: Comparison with phyto- and xenoestrogens. Toxicol. Lett. 2003, 142, 89–101. [Google Scholar] [CrossRef]
- MacMahon, B. Pesticide Residues and Breast Cancer? JNCI J. Natl. Cancer Inst. 1994, 86, 572–573. [Google Scholar] [CrossRef]
- Dewailly, E.; Dodin, S.; Verreault, R.; Ayotte, P.; Sauvé, L.; Morin, J.; Brisson, J. High organochlorine body burden in women with estrogen receptor-positive breast cancer. J. Natl. Cancer Inst. 1994, 86, 232–234. [Google Scholar] [CrossRef]
- Krieger, N.; Wolff, M.S.; Hiatt, R.A.; Rivera, M.; Vogelman, J.; Orentreich, N. Breast cancer and serum organochlorines: A prospective study among white, black, and Asian women. J. Natl. Cancer Inst. 1994, 86, 589–599. [Google Scholar] [CrossRef]
- Wolff, M.S.; Toniolo, P.G.; Lee, E.W.; Rivera, M.; Dubin, N. Blood levels of organochlorine residues and risk of breast cancer. J. Natl. Cancer Inst. 1993, 85, 648–652. [Google Scholar] [CrossRef]
- La Merrill, M.A.; Vandenberg, L.N.; Smith, M.T.; Goodson, W.; Browne, P.; Patisaul, H.B.; Guyton, K.Z.; Kortenkamp, A.; Cogliano, V.J.; Woodruff, T.J.; et al. Consensus on the key characteristics of endocrine-disrupting chemicals as a basis for hazard identification. Nat. Rev.Endocrinol. 2020, 16, 45–57. [Google Scholar] [CrossRef]
- Rogers, J.A.; Metz, L.; Yong, V.W. Review: Endocrine disrupting chemicals and immune responses: A focus on bisphenol-A and its potential mechanisms. Mol. Immunol. 2013, 53, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Sprague, B.L.; Trentham-Dietz, A.; Hedman, C.J.; Wang, J.; Hemming, J.D.C.; Hampton, J.M.; Buist, D.S.M.; Aiello Bowles, E.J.; Sisney, G.S.; Burnside, E.S. Circulating serum xenoestrogens and mammographic breast density. Breast Cancer Res. 2013, 15, R45. [Google Scholar] [CrossRef] [PubMed]
- Binder, A.M.; Corvalan, C.; Pereira, A.; Calafat, A.M.; Ye, X.; Shepherd, J.; Michels, K.B. Prepubertal and Pubertal Endocrine-Disrupting Chemical Exposure and Breast Density among Chilean Adolescents. Cancer Epidemiol. Biomarkers Prev. 2018, 27, 1491–1499. [Google Scholar] [CrossRef]
- Yang, M.; Ryu, J.-H.; Jeon, R.; Kang, D.; Yoo, K.-Y. Effects of bisphenol A on breast cancer and its risk factors. Arch. Toxicol. 2009, 83, 281–285. [Google Scholar] [CrossRef]
- Trabert, B.; Falk, R.T.; Figueroa, J.D.; Graubard, B.I.; Garcia-Closas, M.; Lissowska, J.; Peplonska, B.; Fox, S.D.; Brinton, L.A. Urinary bisphenol A-glucuronide and postmenopausal breast cancer in Poland. Cancer Causes Control 2014, 25, 1587–1593. [Google Scholar] [CrossRef]
- Lang, I.A.; Galloway, T.S.; Scarlett, A.; Henley, W.E.; Depledge, M.; Wallace, R.B.; Melzer, D. Association of Urinary Bisphenol A Concentration With Medical Disorders and Laboratory Abnormalities in Adults. JAMA 2008, 300, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Durando, M.; Kass, L.; Piva, J.; Sonnenschein, C.; Soto, A.M.; Luque, E.H.; Muñoz-de-Toro, M. Prenatal Bisphenol A Exposure Induces Preneoplastic Lesions in the Mammary Gland in Wistar Rats. Environ. Health Perspect. 2007, 115, 80–86. [Google Scholar] [CrossRef]
- Murray, T.J.; Maffini, M.V.; Ucci, A.A.; Sonnenschein, C.; Soto, A.M. Induction of mammary gland ductal hyperplasias and carcinoma in situ following fetal bisphenol A exposure. Reprod. Toxicol. 2007, 23, 383–390. [Google Scholar] [CrossRef]
- Yan, H.; Takamoto, M.; Sugane, K. Exposure to Bisphenol A prenatally or in adulthood promotes T(H)2 cytokine production associated with reduction of CD4CD25 regulatory T cells. Environ. Health Perspect. 2008, 116, 514–519. [Google Scholar] [CrossRef]
- GOTO, M.; TAKANO-ISHIKAWA, Y.; ONO, H.; YOSHIDA, M.; YAMAKI, K.; SHINMOTO, H. Orally Administered Bisphenol A Disturbed Antigen Specific Immunoresponses in the Naïve Condition. Biosci. Biotechnol. Biochem. 2007, 71, 2136–2143. [Google Scholar] [CrossRef]
- Yoshitake, J.; Kato, K.; Yoshioka, D.; Sueishi, Y.; Sawa, T.; Akaike, T.; Yoshimura, T. Suppression of NO production and 8-nitroguanosine formation by phenol-containing endocrine-disrupting chemicals in LPS-stimulated macrophages: Involvement of estrogen receptor-dependent or -independent pathways. Nitric Oxide 2008, 18, 223–228. [Google Scholar] [CrossRef]
- Hong, C.C.; Shimomura-Shimizu, M.; Muroi, M.; Tanamoto, K. Effect of endocrine disrupting chemicals on lipopolysaccharide-induced tumor necrosis factor-alpha and nitric oxide production by mouse macrophages. Biol. Pharm. Bull. 2004, 27, 1136–1139. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.Y.; Park, H.-Y.; Lee, J.-W.; Jung, I.-O.; Choi, K.-H.; Kim, K.; Cho, K.-H. Evaluation of the immune response following exposure of mice to bisphenol A: Induction of th1 cytokine and prolactin by BPA exposure in the mouse spleen cells. Arch. Pharmacol. Res. 2002, 25, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, S.; Yamaki, K.; Yanagisawa, R.; Takano, H.; Hayashi, H.; Mori, Y. Effects of bisphenol A on antigen-specific antibody production, proliferative responses of lymphoid cells, and TH1 and TH2 immune responses in mice. Br. J. Pharmacol. 2003, 138, 1271–1276. [Google Scholar] [CrossRef]
- Yoshino, S.; Yamaki, K.; Li, X.; Sai, T.; Yanagisawa, R.; Takano, H.; Taneda, S.; Hayashi, H.; Mori, Y. Prenatal exposure to bisphenol A up-regulates immune responses, including T helper 1 and T helper 2 responses, in mice. Immunology 2004, 112, 489–495. [Google Scholar] [CrossRef]
- Guo, H.; Liu, T.; Uemura, Y.; Jiao, S.; Wang, D.; Lin, Z.; Narita, Y.; Suzuki, M.; Hirosawa, N.; Ichihara, Y.; et al. Bisphenol A in combination with TNF-alpha selectively induces Th2 cell-promoting dendritic cells in vitro with an estrogen-like activity. Cell. Mol. Immunol. 2010, 7, 227–234. [Google Scholar] [CrossRef]
- Lee, J.; Lim, K.T. Plant-originated glycoprotein (36 kDa) suppresses interleukin-4 and -10 in bisphenol A-stimulated primary cultured mouse lymphocytes. Drug Chem. Toxicol. 2010, 33, 421–429. [Google Scholar] [CrossRef]
- Lee, M.H.; Chung, S.W.; Kang, B.Y.; Park, J.; Lee, C.H.; Hwang, S.Y.; Kim, T.S. Enhanced interleukin-4 production in CD4+ T cells and elevated immunoglobulin E levels in antigen-primed mice by bisphenol A and nonylphenol, endocrine disruptors: Involvement of nuclear factor-AT and Ca2+. Immunology 2003, 109, 76–86. [Google Scholar] [CrossRef]
- Han, E.H.; Kim, H.G.; Hwang, Y.P.; Choi, J.H.; Im, J.H.; Park, B.; Yang, J.H.; Jeong, T.C.; Jeong, H.G. The role of cyclooxygenase-2-dependent signaling via cyclic AMP response element activation on aromatase up-regulation by o,p′-DDT in human breast cancer cells. Toxicol. Lett. 2010, 198, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.A.; Ikenaka, Y.; Sobhy Darwish, W.; Nakayama, S.M.M.; Mizukawa, H.; Ishizuka, M. Effects of the organochlorine p,p’-DDT on MCF-7 cells: Investigating metabolic and immune modulatory transcriptomic changes. Environ. Toxicol. Pharmacol. 2019, 72, 103249. [Google Scholar] [CrossRef]
- Harada, T.; Takeda, M.; Kojima, S.; Tomiyama, N. Toxicity and Carcinogenicity of Dichlorodiphenyltrichloroethane (DDT). Toxicol. Res. 2016, 32, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Street, J.C.; Sharma, R.P. Alteration of induced cellular and humoral immune responses by pesticides and chemicals of environmental concern: Quantitative studies of immunosuppression by DDT, aroclor 1254, carbaryl, carbofuran, and methylparathion. Toxicol. Appl. Pharmacol. 1975, 32, 587–602. [Google Scholar] [CrossRef]
- Kim, J.Y.; Choi, C.Y.; Lee, K.J.; Shin, D.W.; Jung, K.S.; Chung, Y.C.; Lee, S.S.; Shin, J.G.; Jeong, H.G. Induction of inducible nitric oxide synthase and proinflammatory cytokines expression by o,p’-DDT in macrophages. Toxicol. Lett. 2004, 147, 261–269. [Google Scholar] [CrossRef]
- Wang, C.; Petriello, M.C.; Zhu, B.; Hennig, B. PCB 126 induces monocyte/macrophage polarization and inflammation through AhR and NF-κB pathways. Toxicol. Appl. Pharmacol. 2019, 367, 71–81. [Google Scholar] [CrossRef]
- Petriello, M.C.; Hoffman, J.B.; Vsevolozhskaya, O.; Morris, A.J.; Hennig, B. Dioxin-like PCB 126 increases intestinal inflammation and disrupts gut microbiota and metabolic homeostasis. Environ. Pollut. 2018, 242, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Yu, W.; Meng, F.; Mi, J.; Peng, J.; Liu, J.; Zhang, X.; Hai, C.; Wang, X. Polychlorinated biphenyls-153 induces metabolic dysfunction through activation of ROS/NF-κB signaling via downregulation of HNF1b. Redox. Biol. 2017, 12, 300–310. [Google Scholar] [CrossRef]
- Kimbrough, R.D. Human Health Effects of Polychlorinated Biphenyls (PCBs) and Polybrominated Biphenyls (PBBs). Annu. Rev. Pharmacol. Toxicol. 1987, 27, 87–111. [Google Scholar] [CrossRef]
- Selgrade, M.K. Immunotoxicity—The Risk is Real. Toxicol. Sci. 2007, 100, 328–332. [Google Scholar] [CrossRef]
- Zhu, H.; Li, Y.; Trush, M.A. Characterization of benzo[a]pyrene quinone-induced toxicity to primary cultured bone marrow stromal cells from DBA/2 mice: Potential role of mitochondrial dysfunction. Toxicol. Appl. Pharmacol. 1995, 130, 108–120. [Google Scholar] [CrossRef]
- Smith, J.; Neupane, R.; McAmis, W.; Singh, U.; Chatterjee, S.; Raychoudhury, S. Toxicity of polycyclic aromatic hydrocarbons involves NOX2 activation. Toxicol. Rep. 2019, 6, 1176–1181. [Google Scholar] [CrossRef]
- Thurmond, L.M.; Lauer, L.D.; House, R.V.; Cook, J.C.; Dean, J.H. Immunosuppression following exposure to 7,12-dimethylbenz[a]anthracene (DMBA) in Ah-responsive and Ah-nonresponsive mice. Toxicol. Appl. Pharmacol. 1987, 91, 450–460. [Google Scholar] [CrossRef]
- Heidel, S.M.; MacWilliams, P.S.; Baird, W.M.; Dashwood, W.M.; Buters, J.T.; Gonzalez, F.J.; Larsen, M.C.; Czuprynski, C.J.; Jefcoate, C.R. Cytochrome P4501B1 mediates induction of bone marrow cytotoxicity and preleukemia cells in mice treated with 7,12-dimethylbenz[a]anthracene. Cancer Res. 2000, 60, 3454–3460. [Google Scholar] [PubMed]
- De Jong, W.H.; Kroese, E.D.; Vos, J.G.; Van Loveren, H. Detection of immunotoxicity of benzo[a]pyrene in a subacute toxicity study after oral exposure in rats. Toxicol. Sci 1999, 50, 214–220. [Google Scholar] [CrossRef]
- O’Driscoll, C.A.; Gallo, M.E.; Hoffmann, E.J.; Fechner, J.H.; Schauer, J.J.; Bradfield, C.A.; Mezrich, J.D. Polycyclic aromatic hydrocarbons (PAHs) present in ambient urban dust drive proinflammatory T cell and dendritic cell responses via the aryl hydrocarbon receptor (AHR) in vitro. PLoS ONE 2018, 13, e0209690. [Google Scholar] [CrossRef]
- Veldhoen, M.; Duarte, J.H. The aryl hydrocarbon receptor: Fine-tuning the immune-response. Curr. Opin. Immunol. 2010, 22, 747–752. [Google Scholar] [CrossRef]
- Martinon, F. Signaling by ROS drives inflammasome activation. Eur. J. Immunol. 2010, 40, 616–619. [Google Scholar] [CrossRef]
- Jaga, K. What are the implications of the interaction between DDT and estrogen receptors in the body? Med. Hypotheses 2000, 54, 18–25. [Google Scholar] [CrossRef]
- Scribner, J.D.; Mottet, N.K. DDT acceleration of mammary gland tumors induced in the male Sprague-Dawley rat by 2-acetamidophenanthrene. Carcinogenesis 1981, 2, 1235–1239. [Google Scholar] [CrossRef]
- Ikeda, M.; Kuratsune, M.; Nakamura, Y.; Hirohata, T. A cohort study on mortality of yusho patients—A preliminary report. Fukuoka Igaku Zasshi 1987, 78, 297–300. [Google Scholar] [PubMed]
- Falck, F., Jr.; Ricci, A., Jr.; Wolff, M.S.; Godbold, J.; Deckers, P. Pesticides and polychlorinated biphenyl residues in human breast lipids and their relation to breast cancer. Arch. Environ. Health 1992, 47, 143–146. [Google Scholar]
- Bertazzi, P.A.; Zocchetti, C.; Pesatori, A.C.; Guercilena, S.; Sanarico, M.; Radice, L. Ten-year mortality study of the population involved in the Seveso incident in 1976. Am. J. Epidemiol. 1989, 129, 1187–1200. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-H.; Jahan, S.A.; Kabir, E.; Brown, R.J.C. A review of airborne polycyclic aromatic hydrocarbons (PAHs) and their human health effects. Environ. Int. 2013, 60, 71–80. [Google Scholar] [CrossRef]
- Ranadive, K.J.; Karande, K.A. Studies on 1,2:5,6-Dibenzanthracene Induced Mammary Carcinogenesis in Mice. Br. J. Cancer 1963, 17, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Bohácová, S.; Borská, L.; Fiala, Z.; Andrýs, C. Effect of polycyclic aromatic hydrocarbons on the immune system. Acta Med. Hradec Kral. Suppl. 1999, 42, 17–23. [Google Scholar] [PubMed]
- Burchiel, S.W. Polycyclic Aromatic Hydrocarbons (PAHs) and the Immune System. In Encyclopedic Reference of Immunotoxicology; Vohr, H.-W., Ed.; Springer: Berlin/Heidelberg, Germany, 2005; pp. 515–518. [Google Scholar]
- Salian, K.; Strezov, V.; Evans, T.J.; Taylor, M.; Nelson, P.F. Application of national pollutant inventories for monitoring trends on dioxin emissions from stationary industrial sources in Australia, Canada and European Union. PLoS ONE 2019, 14, e0224328. [Google Scholar] [CrossRef]
- Manz, A.; Berger, J.; Dwyer, J.H.; Flesch-Janys, D.; Nagel, S.; Waltsgott, H. Cancer mortality among workers in chemical plant contaminated with dioxin. Lancet 1991, 338, 959–964. [Google Scholar] [CrossRef]
- Warner, M.; Eskenazi, B.; Mocarelli, P.; Gerthoux, P.M.; Samuels, S.; Needham, L.; Patterson, D.; Brambilla, P. Serum dioxin concentrations and breast cancer risk in the Seveso Women’s Health Study. Environ. Health Perspect. 2002, 110, 625–628. [Google Scholar] [CrossRef] [PubMed]
- Danjou, A.M.; Fervers, B.; Boutron-Ruault, M.C.; Philip, T.; Clavel-Chapelon, F.; Dossus, L. Estimated dietary dioxin exposure and breast cancer risk among women from the French E3N prospective cohort. Breast Cancer Res. 2015, 17, 39. [Google Scholar] [CrossRef]
- Danjou, A.M.N.; Coudon, T.; Praud, D.; Lévêque, E.; Faure, E.; Salizzoni, P.; Le Romancer, M.; Severi, G.; Mancini, F.R.; Leffondré, K.; et al. Long-term airborne dioxin exposure and breast cancer risk in a case-control study nested within the French E3N prospective cohort. Environ. Int. 2019, 124, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Bekki, K.; Vogel, H.; Li, W.; Ito, T.; Sweeney, C.; Haarmann-Stemmann, T.; Matsumura, F.; Vogel, C.F. The aryl hydrocarbon receptor (AhR) mediates resistance to apoptosis induced in breast cancer cells. Pestic. Biochem. Physiol. 2015, 120, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Ahn, N.S.; Hu, H.; Park, J.S.; Park, J.S.; Kim, J.S.; An, S.; Kong, G.; Aruoma, O.I.; Lee, Y.S.; Kang, K.S. Molecular mechanisms of the 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced inverted U-shaped dose responsiveness in anchorage independent growth and cell proliferation of human breast epithelial cells with stem cell characteristics. Mutat. Res. 2005, 579, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Brunnberg, S.; Andersson, P.; Poellinger, L.; Hanberg, A. The constitutively active Ah receptor (CA-AhR) mouse as a model for dioxin exposure—Effects in reproductive organs. Chemosphere 2011, 85, 1701–1706. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Hung, C.-M.; Kay, N.; Chen, C.-C.; Kao, Y.-H.; Yuan, S.-S. Progesterone receptor is involved in 2,3,7,8-tetrachlorodibenzo-p-dioxin-stimulated breast cancer cells proliferation. Cancer Lett. 2012, 319, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Frericks, M.; Meissner, M.; Esser, C. Microarray analysis of the AHR system: Tissue-specific flexibility in signal and target genes. Toxicol. Appl. Pharmacol. 2007, 220, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Huhn, D.; Marti-Rodrigo, P.; Mouron, S.; Hansel, C.; Tschapalda, K.; Porebski, B.; Haggblad, M.; Lidemalm, L.; Quintela-Fandino, M.; Carreras-Puigvert, J.; et al. Prolonged estrogen deprivation triggers a broad immunosuppressive phenotype in breast cancer cells. Mol. Oncol. 2021. [Google Scholar] [CrossRef]
- Joffroy, C.M.; Buck, M.B.; Stope, M.B.; Popp, S.L.; Pfizenmaier, K.; Knabbe, C. Antiestrogens induce transforming growth factor beta-mediated immunosuppression in breast cancer. Cancer Res. 2010, 70, 1314–1322. [Google Scholar] [CrossRef]
- Shu, X.O.; Zheng, Y.; Cai, H.; Gu, K.; Chen, Z.; Zheng, W.; Lu, W. Soy food intake and breast cancer survival. JAMA 2009, 302, 2437–2443. [Google Scholar] [CrossRef]



| Inflammatory Marker | Pearson r | p-Values |
|---|---|---|
| TNF-α | −0.077 | 0.069 |
| IL-6 | −0.635 | <0.0001 * |
| IL-1β | −0.084 | 0.060 |
| IL-1α | −0.011 | 0.710 |
| IL-17 | −0.020 | 0.024 * |
| TGF-β | −0.171 | <0.0001 * |
| CXCL-1 | −0.484 | <0.0001 * |
| Leptin | −0.704 | <0.0001 * |
| CD Molecules | Pearson r | p-Values |
| CD4 | −0.319 | <0.0001 * |
| CD5 | −0.255 | <0.0001 * |
| CD83 | −0.270 | <0.0001 * |
| CD163 | −0.268 | <0.0001 * |
| CD40 | −0.421 | <0.0001 * |
| CD34 | −0.380 | <0.0001 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maharjan, C.K.; Mo, J.; Wang, L.; Kim, M.-C.; Wang, S.; Borcherding, N.; Vikas, P.; Zhang, W. Natural and Synthetic Estrogens in Chronic Inflammation and Breast Cancer. Cancers 2022, 14, 206. https://doi.org/10.3390/cancers14010206
Maharjan CK, Mo J, Wang L, Kim M-C, Wang S, Borcherding N, Vikas P, Zhang W. Natural and Synthetic Estrogens in Chronic Inflammation and Breast Cancer. Cancers. 2022; 14(1):206. https://doi.org/10.3390/cancers14010206
Chicago/Turabian StyleMaharjan, Chandra K., Jiao Mo, Lei Wang, Myung-Chul Kim, Sameul Wang, Nicholas Borcherding, Praveen Vikas, and Weizhou Zhang. 2022. "Natural and Synthetic Estrogens in Chronic Inflammation and Breast Cancer" Cancers 14, no. 1: 206. https://doi.org/10.3390/cancers14010206
APA StyleMaharjan, C. K., Mo, J., Wang, L., Kim, M.-C., Wang, S., Borcherding, N., Vikas, P., & Zhang, W. (2022). Natural and Synthetic Estrogens in Chronic Inflammation and Breast Cancer. Cancers, 14(1), 206. https://doi.org/10.3390/cancers14010206