
Genomic Correlates of DNA Damage in Breast Cancer Subtypes

 and
and
Abstract
Simple Summary
Abstract
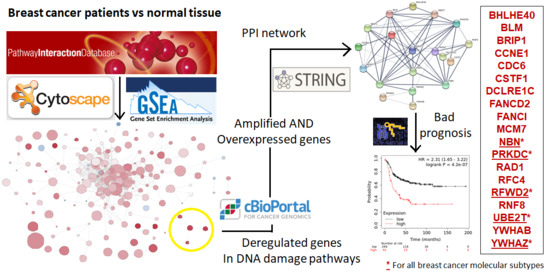
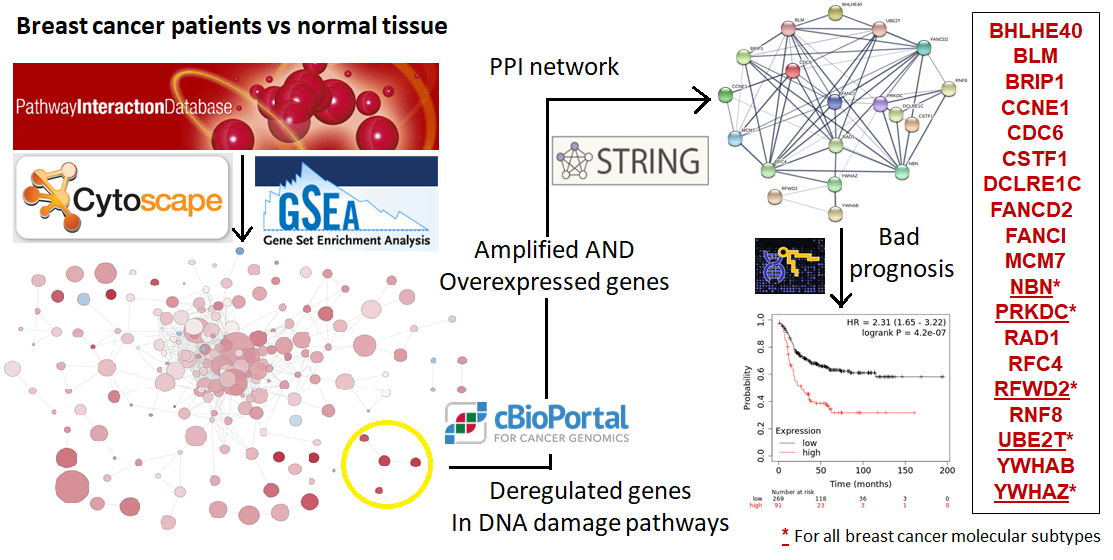{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
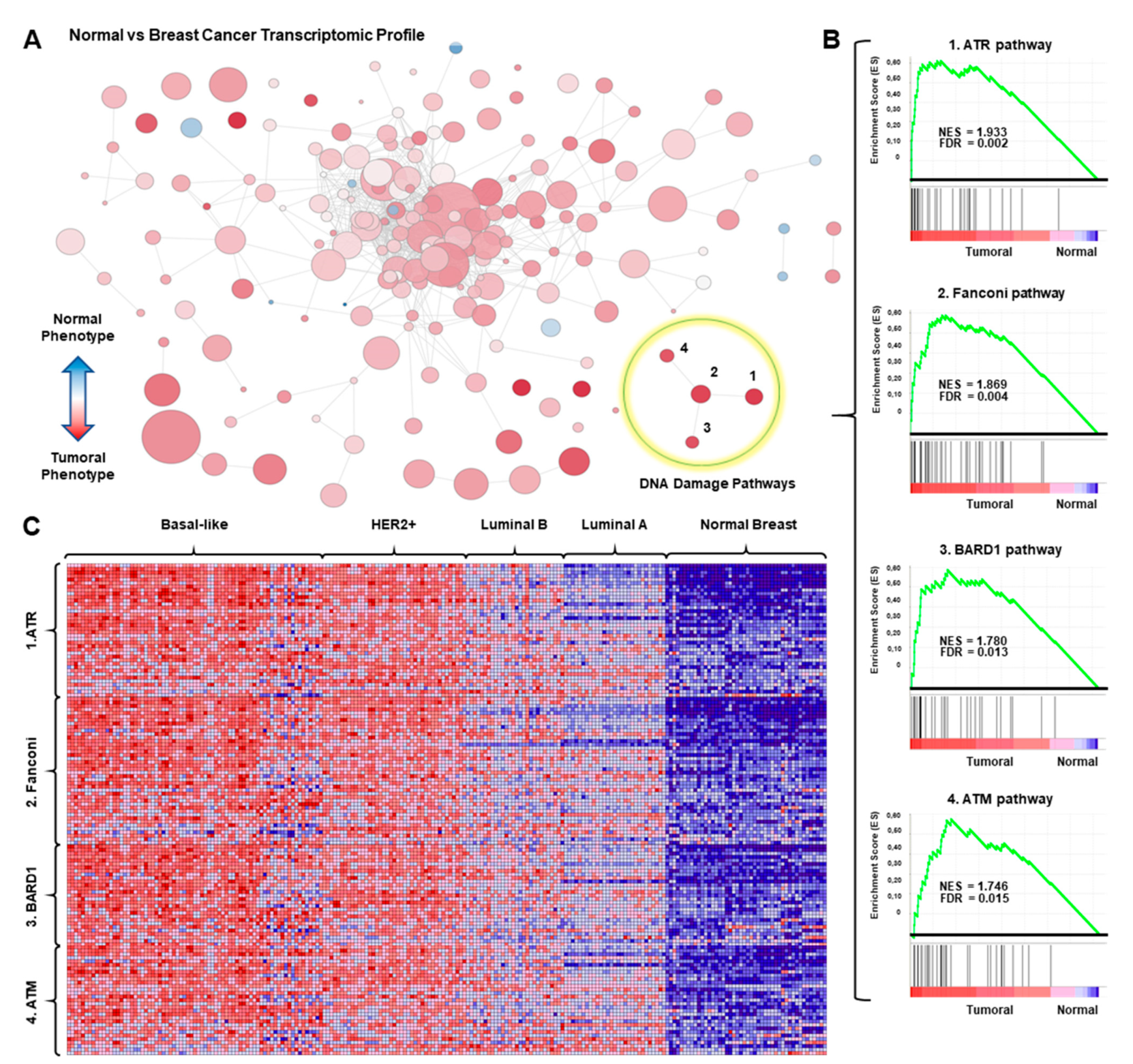
2.1. Whole Genome Transcription Profiling and Gene Set Enrichment Analyses
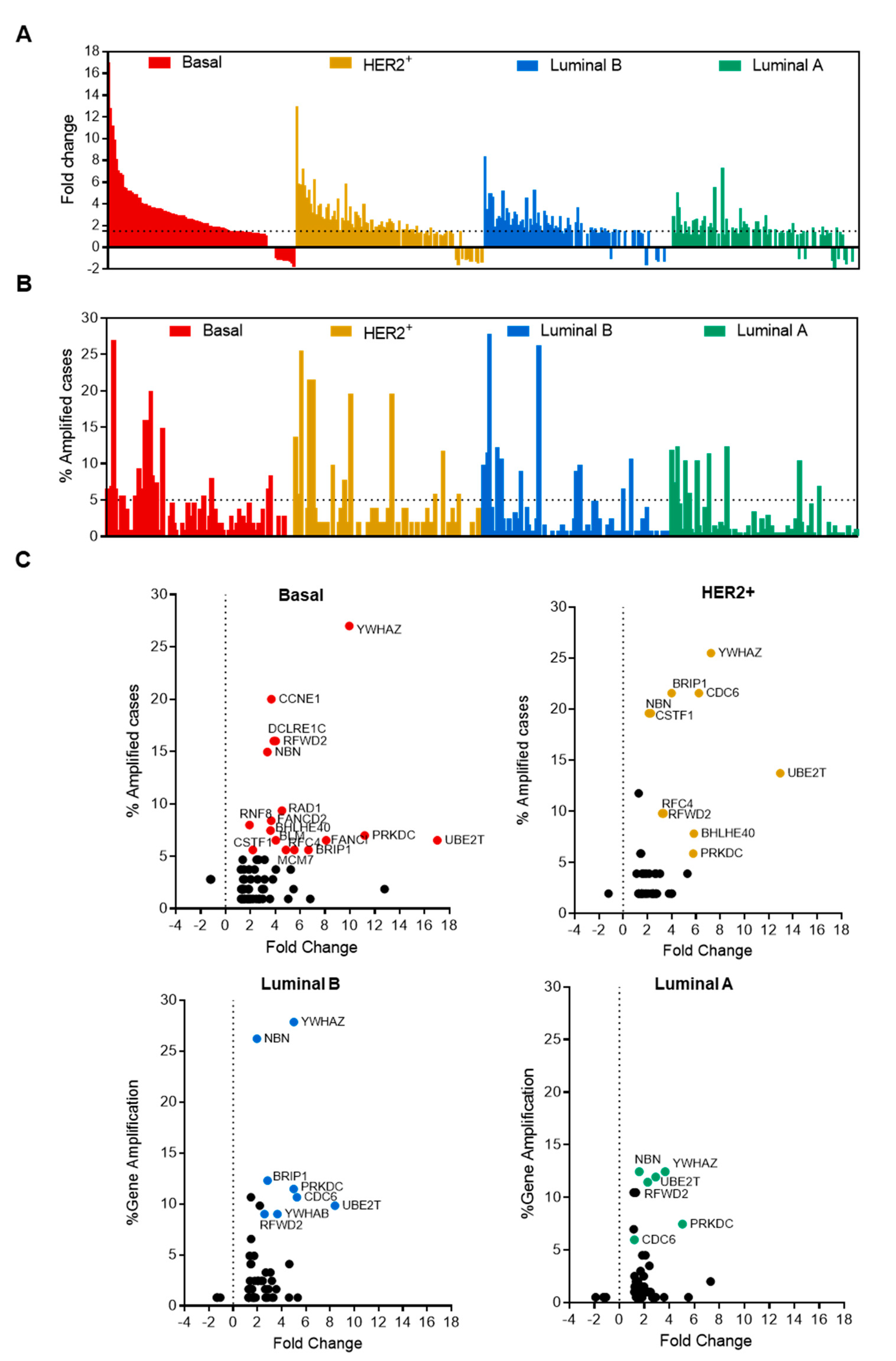
2.2. Evaluation of Molecular Alterations
2.3. Construction and Analysis of PPI Networks and Functional Annotation
2.4. Expression Analyses
2.5. Outcome Analyses
2.6. Data Availability
3. Results
3.1. Transcriptomic Mapping and Gene Set Enrichment Analyses Identify DNA Repair Pathways as Upregulated in Breast Cancer
3.2. Analysis of Breast Cancer Subtypes Display Different Expression of Transcripts and Amplification of Genes
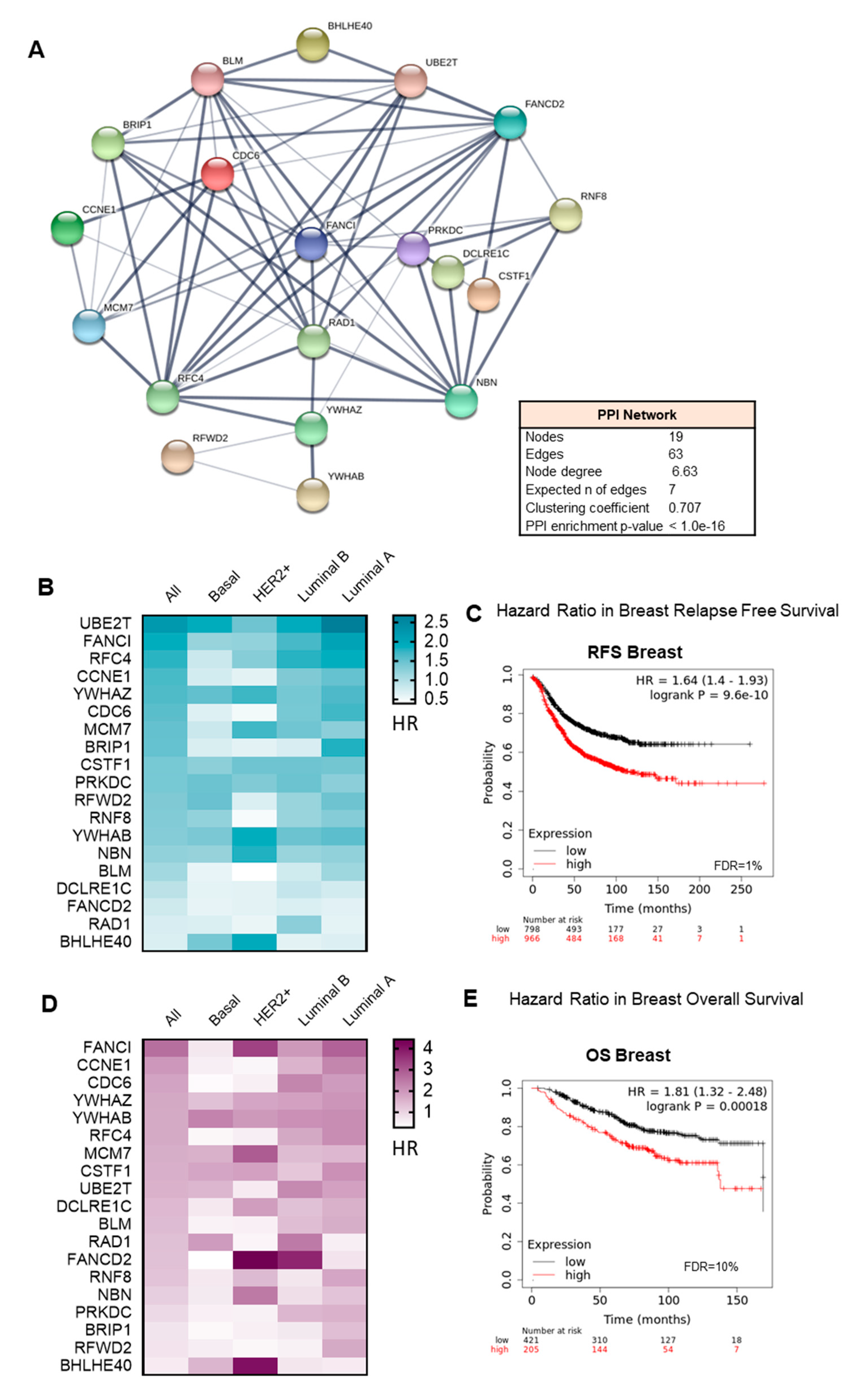
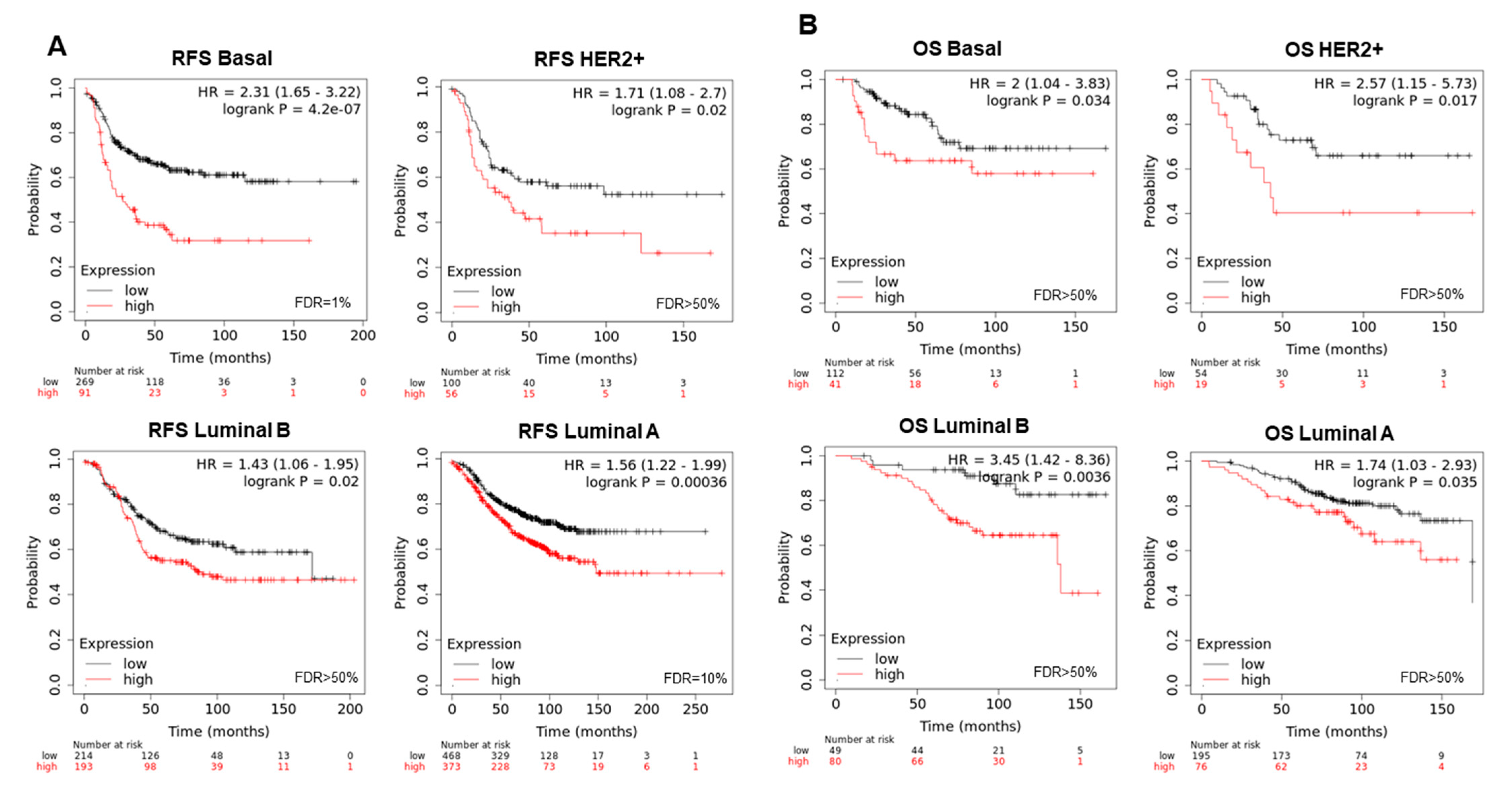
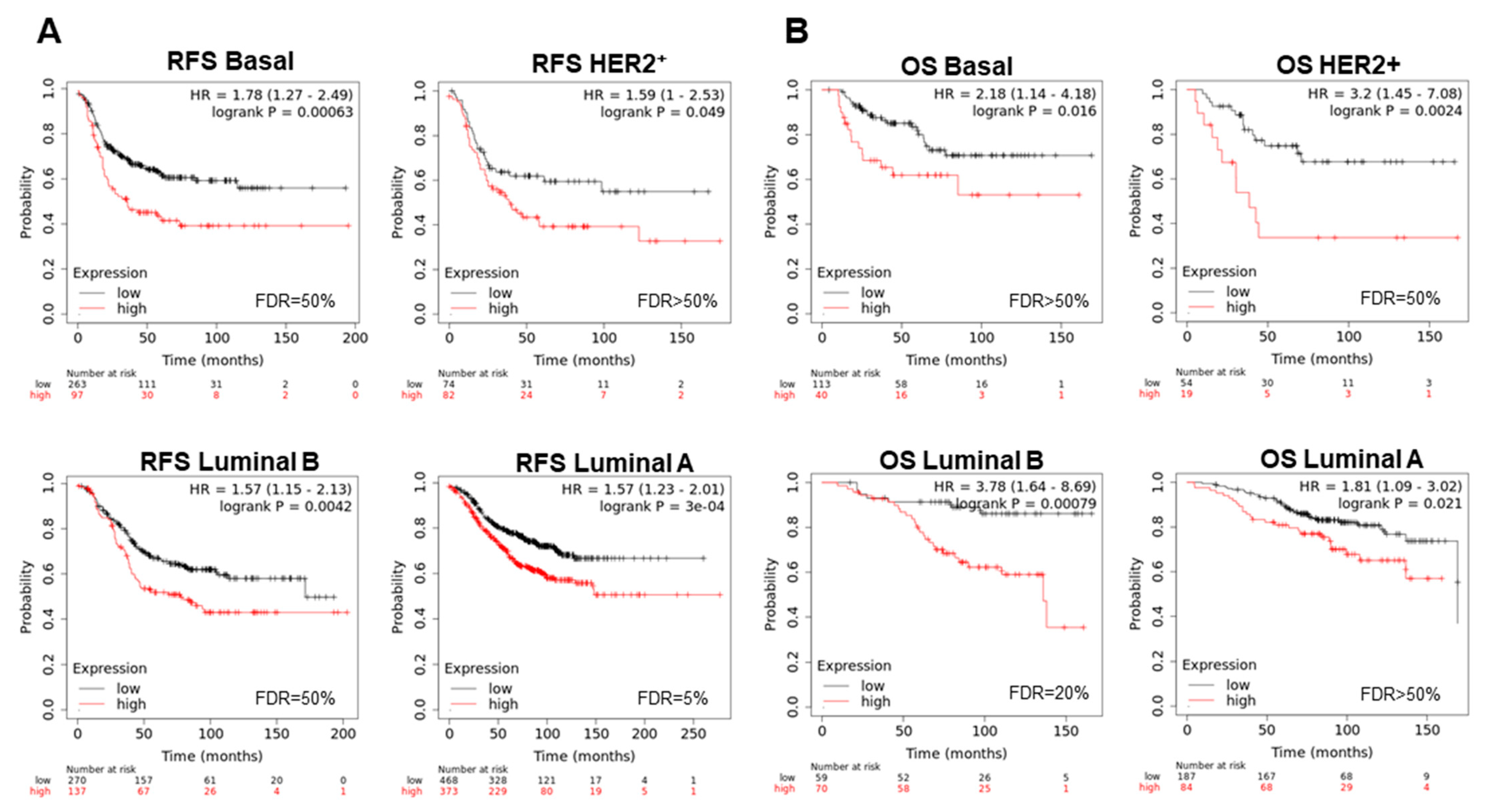
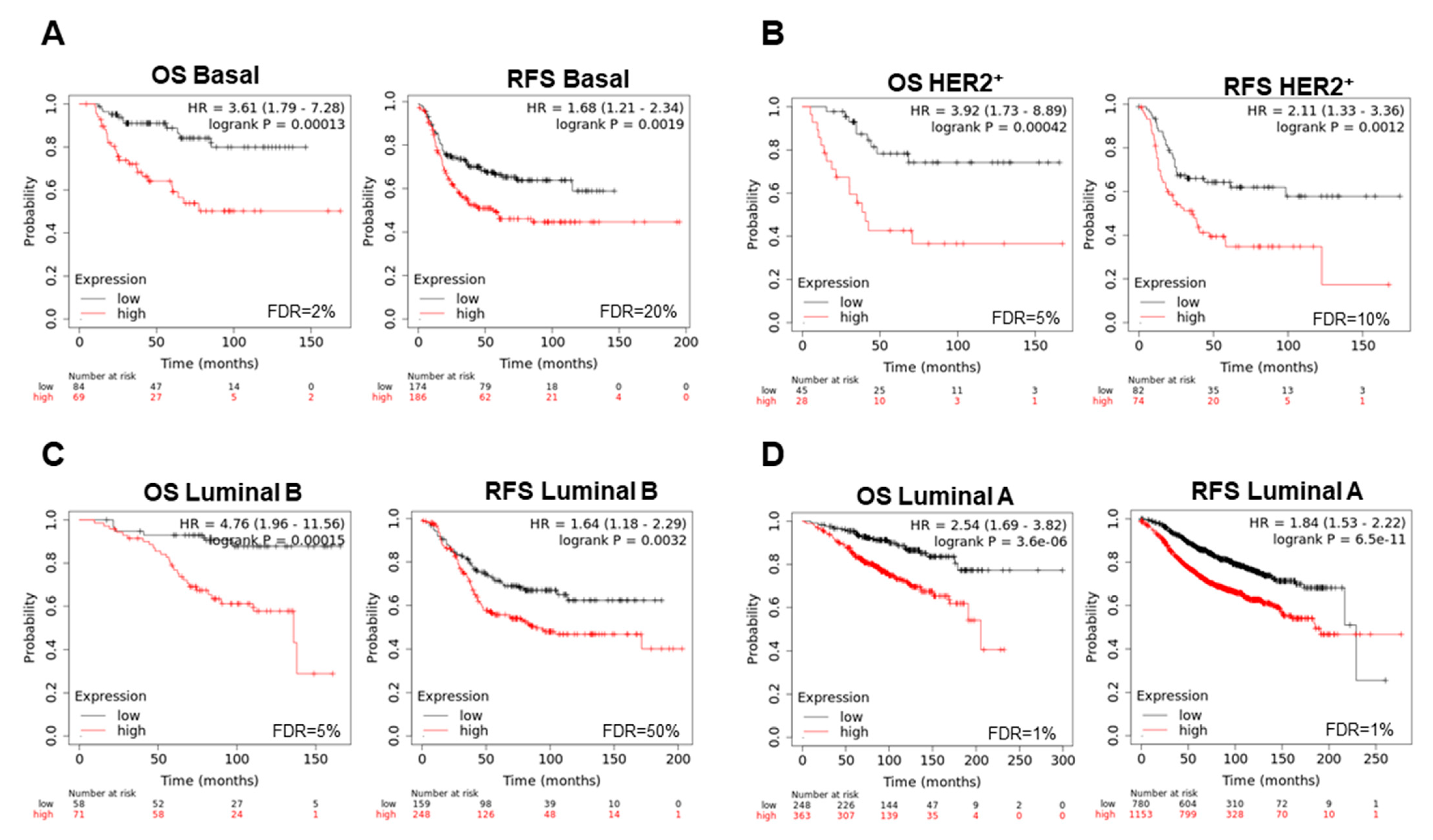
3.3. Interacting DNA Damage Network Associates with Detrimental Prognosis
3.4. Amplified Genes Correlated with Poor Prognosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baliu-Piqué, M.; Pandiella, A.; Ocana, A. Breast Cancer Heterogeneity and Response to Novel Therapeutics. Cancers 2020, 12, 3271. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Fujimoto, J.; Wedge, D.C.; Song, X.; Seth, S.; Chow, C.-W.; Cao, Y.; Gumbs, C.; Gold, K.A.; Kalhor, N.; et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 2014, 346, 256–259. [Google Scholar] [CrossRef]
- Reiter, J.G.; Baretti, M.; Gerold, J.M.; Makohon-Moore, A.P.; Daud, A.; Iacobuzio-Donahue, C.A.; Azad, N.S.; Kinzler, K.W.; Nowak, M.A.; Vogelstein, B. An analysis of genetic heterogeneity in untreated cancers. Nat. Rev. Cancer 2019, 19, 639–650. [Google Scholar] [CrossRef]
- Amir, E.; Šeruga, B.; Serrano, R.; Ocaña, A. Targeting DNA repair in breast cancer: A clinical and translational update. Cancer Treat. Rev. 2010, 36, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J.; Szabo, C. Poly(ADP-ribose) polymerase inhibition: Past, present and future. Nat. Rev. Drug Discov. 2020, 19, 711–736. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Patnaik, A.; Campbell, D.; Shapiro, J.; Bryce, A.H.; McDermott, R.; Sautois, B.; Vogelzang, N.J.; Bambury, R.M.; Voog, E.; et al. Rucaparib in Men With Metastatic Castration-Resistant Prostate Cancer Harboring a BRCA1 or BRCA2 Gene Alteration. J. Clin. Oncol. 2020, 38, 3763–3772. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Jackson, H.W.; Fischer, J.R.; Zanotelli, V.R.T.; Ali, H.R.; Mechera, R.; Soysal, S.D.; Moch, H.; Muenst, S.; Varga, Z.; Weber, W.P.; et al. The single-cell pathology landscape of breast cancer. Nature 2020, 578, 615–620. [Google Scholar] [CrossRef]
- Mateo, J.; Chakravarty, D.; Dienstmann, R.; Jezdic, S.; Gonzalez-Perez, A.; Lopez-Bigas, N.; Ng, C.; Bedard, P.; Tortora, G.; Douillard, J.-Y.; et al. A framework to rank genomic alterations as targets for cancer precision medicine: The ESMO Scale for Clinical Actionability of molecular Targets (ESCAT). Ann. Oncol. 2018, 29, 1895–1902. [Google Scholar] [CrossRef]
- Ocaña, A.; Pandiella, A. Targeting oncogenic vulnerabilities in triple negative breast cancer: Biological bases and ongoing clinical studies. Oncotarget 2017, 8, 22218–22234. [Google Scholar] [CrossRef] [PubMed]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.M.; Robson, M.E.; Ventz, S.; Santa-Maria, C.A.; Nanda, R.; Marcom, P.K.; Shah, P.D.; Ballinger, T.J.; Yang, E.S.; Vinayak, S.; et al. TBCRC 048: Phase II Study of Olaparib for Metastatic Breast Cancer and Mutations in Homologous Recombination-Related Genes. J. Clin. Oncol. 2020, 38, 4274–4282. [Google Scholar] [CrossRef]
- Telli, M.L.; Gradishar, W.J.; Ward, J.H. NCCN Guidelines Updates: Breast Cancer. J. Natl. Compr. Cancer Netw. 2019, 17, 552–555. [Google Scholar]
- Sparano, J.A.; Gray, R.J.; Makower, D.F.; Pritchard, K.I.; Albain, K.S.; Hayes, D.F.; Geyer, C.E., Jr.; Dees, E.C.; Goetz, M.P.; Olson, J.A., Jr.; et al. Adjuvant Chemotherapy Guided by a 21-Gene Expression Assay in Breast Cancer. N. Engl. J. Med. 2018, 379, 111–121. [Google Scholar] [CrossRef]
- Schaefer, C.F.; Anthony, K.; Krupa, S.; Buchoff, J.; Day, M.; Hannay, T.; Buetow, K.H. PID: The Pathway Interaction Database. Nucleic Acids Res. 2008, 37, D674–D679. [Google Scholar] [CrossRef]
- Pratt, D.; Chen, J.; Pillich, R.; Rynkov, V.; Gary, A.; Demchak, B.; Ideker, T. NDEx 2.0: A Clearinghouse for Research on Cancer Pathways. Cancer Res. 2017, 77, e58–e61. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteome Res. 2019, 18, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed]
- Györffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 2009, 123, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, K.M.; Yuan, S.-S.F.; Lee, E.Y.-H.P.; Sung, P. Nuclease Activities in a Complex of Human Recombination and DNA Repair Factors Rad50, Mre11, and p95. J. Biol. Chem. 1998, 273, 21447–21450. [Google Scholar] [CrossRef] [PubMed]
- Stiff, T.; Reis, C.; Alderton, G.K.; Woodbine, L.; O’Driscoll, M.; A Jeggo, P. Nbs1 is required for ATR-dependent phosphorylation events. EMBO J. 2004, 24, 199–208. [Google Scholar] [CrossRef]
- Bian, L.; Meng, Y.; Zhang, M.; Li, D. MRE11-RAD50-NBS1 complex alterations and DNA damage response: Implications for cancer treatment. Mol. Cancer 2019, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hartley, K.; Gell, D.; Smith, G.C.; Zhang, H.; Divecha, N.; A Connelly, M.; Admon, A.; Lees-Miller, S.P.; Anderson, C.W.; Jackson, S.P. DNA-dependent protein kinase catalytic subunit: A relative of phosphatidylinositol 3-kinase and the ataxia telangiectasia gene product. Cell 1995, 82, 849–856. [Google Scholar] [CrossRef]
- Ma, Y.; Pannicke, U.; Schwarz, K.; Lieber, M.R. Hairpin Opening and Overhang Processing by an Artemis/DNA-Dependent Protein Kinase Complex in Nonhomologous End Joining and V(D)J Recombination. Cell 2002, 108, 781–794. [Google Scholar] [CrossRef]
- Ma, Y.; Lu, H.; Tippin, B.; Goodman, M.F.; Shimazaki, N.; Koiwai, O.; Hsieh, C.-L.; Schwarz, K.; Lieber, M.R. A Biochemically Defined System for Mammalian Nonhomologous DNA End Joining. Mol. Cell 2004, 16, 701–713. [Google Scholar] [CrossRef]
- Shintani, S.; Mihara, M.; Li, C.; Nakahara, Y.; Hino, S.; Nakashiro, K.-I.; Hamakawa, H. Up-regulation of DNA-dependent protein kinase correlates with radiation resistance in oral squamous cell carcinoma. Cancer Sci. 2003, 94, 894–900. [Google Scholar] [CrossRef]
- Beskow, C.; Skikuniene, J.; Holgersson, Å.; Nilsson, B.; Lewensohn, R.; Kanter, L.; Viktorsson, K. Radioresistant cervical cancer shows upregulation of the NHEJ proteins DNA-PKcs, Ku70 and Ku86. Br. J. Cancer 2009, 101, 816–821. [Google Scholar] [CrossRef]
- Bouchaert, P.; Guerif, S.; Debiais, C.; Irani, J.; Fromont, G. DNA-PKcs Expression Predicts Response to Radiotherapy in Prostate Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2012, 84, 1179–1185. [Google Scholar] [CrossRef]
- Sun, G.; Yang, L.; Dong, C.; Ma, B.; Shan, M.; Ma, B. PRKDC regulates chemosensitivity and is a potential prognostic and predictive marker of response to adjuvant chemotherapy in breast cancer patients. Oncol. Rep. 2017, 37, 3536–3542. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, W.; Wen, G.; Tang, H.; Wu, C.; Wu, Y.; Jing, Z.; Tang, M.; Liu, G.; Li, D.; et al. High expression of PRKDC promotes breast cancer cell growth via p38 MAPK signaling and is associated with poor survival. Mol. Genet. Genom. Med. 2019, 7, e908. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, E.; Denti, S.; Catena, R.; Rossetti, G.; Polo, S.; Gasparian, S.; Putignano, S.; Rogge, L.; Pardi, R. Characterization of Human Constitutive Photomorphogenesis Protein 1, a RING Finger Ubiquitin Ligase That Interacts with Jun Transcription Factors and Modulates Their Transcriptional Activity. J. Biol. Chem. 2003, 278, 19682–19690. [Google Scholar] [CrossRef] [PubMed]
- Dornan, D.; Wertz, I.; Shimizu, H.; Arnott, D.; Frantz, G.D.; Dowd, P.; Rourke, K.O.; Koeppen, H.; Dixit, V.M. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature 2004, 429, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Liu, Y.; Pan, S.; Xie, S.; Wang, Z.-W.; Zhu, X. Role of the COP1 protein in cancer development and therapy. Semin. Cancer Biol. 2020, 67, 43–52. [Google Scholar] [CrossRef]
- Machida, Y.J.; Machida, Y.; Chen, Y.; Gurtan, A.M.; Kupfer, G.M.; D’Andrea, A.D.; Dutta, A. UBE2T Is the E2 in the Fanconi Anemia Pathway and Undergoes Negative Autoregulation. Mol. Cell 2006, 23, 589–596. [Google Scholar] [CrossRef]
- Alpi, A.F.; Pace, P.E.; Babu, M.M.; Patel, K.J. Mechanistic Insight into Site-Restricted Monoubiquitination of FANCD2 by Ube2t, FANCL, and FANCI. Mol. Cell 2008, 32, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Ueki, T.; Park, J.-H.; Nishidate, T.; Kijima, K.; Hirata, K.; Nakamura, Y.; Katagiri, T. Ubiquitination and Downregulation of BRCA1 by Ubiquitin-Conjugating Enzyme E2T Overexpression in Human Breast Cancer Cells. Cancer Res. 2009, 69, 8752–8760. [Google Scholar] [CrossRef]
- Jones, D.H.; Ley, S.; Aitken, A. Isoforms of 14-3-3 protein can form homo- and heterodimers in vivo and in vitro: Implications for function as adapter proteins. FEBS Lett. 1995, 368, 55–58. [Google Scholar] [CrossRef]
- Qi, W.; Liu, X.; Qiao, D.; Martinez, J.D. Isoform-specific expression of 14-3-3 proteins in human lung cancer tissues. Int. J. Cancer 2004, 113, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.-F.; Zhao, Q.; Hu, H.; Liao, J.-Z.; Lin, J.-S.; Xia, C.; Chang, Y.; Liu, J.; Guo, A.-Y.; He, X.-X. The ASH1-miR-375-YWHAZ Signaling Axis Regulates Tumor Properties in Hepatocellular Carcinoma. Mol. Ther.-Nucleic Acids 2018, 11, 538–553. [Google Scholar] [CrossRef]
- Gan, Y.; Ye, F.; He, X.-X. The role of YWHAZ in cancer: A maze of opportunities and challenges. J. Cancer 2020, 11, 2252–2264. [Google Scholar] [CrossRef]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef]
- Alcaraz-Sanabria, A.; Baliu-Piqué, M.; Saiz-Ladera, C.; Rojas, K.; Manzano, A.; Marquina, G.; Casado, A.; Cimas, F.J.; Pérez-Segura, P.; Pandiella, A.; et al. Genomic Signatures of Immune Activation Predict Outcome in Advanced Stages of Ovarian Cancer and Basal-Like Breast Tumors. Front. Oncol. 2020, 9, 1486. [Google Scholar] [CrossRef]
- Pérez-Pena, J.; Fekete, J.T.; Páez, R.; Baliu-Piqué, M.; García-Saenz, J. Ángel; García-Barberán, V.; Manzano, A.; Pérez-Segura, P.; Esparis-Ogando, A.; Pandiella, A.; et al. A Transcriptomic Immunologic Signature Predicts Favorable Outcome in Neoadjuvant Chemotherapy Treated Triple Negative Breast Tumors. Front. Immunol. 2019, 10, 2802. [Google Scholar] [CrossRef]
- Miller, R.; Leary, A.; Scott, C.; Serra, V.; Lord, C.; Bowtell, D.; Chang, D.; Garsed, D.; Jonkers, J.; Ledermann, J.; et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann. Oncol. 2020, 31, 1606–1622. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabañas Morafraile, E.; Pérez-Peña, J.; Fuentes-Antrás, J.; Manzano, A.; Pérez-Segura, P.; Pandiella, A.; Galán-Moya, E.M.; Ocaña, A. Genomic Correlates of DNA Damage in Breast Cancer Subtypes. Cancers 2021, 13, 2117. https://doi.org/10.3390/cancers13092117
Cabañas Morafraile E, Pérez-Peña J, Fuentes-Antrás J, Manzano A, Pérez-Segura P, Pandiella A, Galán-Moya EM, Ocaña A. Genomic Correlates of DNA Damage in Breast Cancer Subtypes. Cancers. 2021; 13(9):2117. https://doi.org/10.3390/cancers13092117
Chicago/Turabian StyleCabañas Morafraile, Esther, Javier Pérez-Peña, Jesús Fuentes-Antrás, Aránzazu Manzano, Pedro Pérez-Segura, Atanasio Pandiella, Eva M. Galán-Moya, and Alberto Ocaña. 2021. "Genomic Correlates of DNA Damage in Breast Cancer Subtypes" Cancers 13, no. 9: 2117. https://doi.org/10.3390/cancers13092117
APA StyleCabañas Morafraile, E., Pérez-Peña, J., Fuentes-Antrás, J., Manzano, A., Pérez-Segura, P., Pandiella, A., Galán-Moya, E. M., & Ocaña, A. (2021). Genomic Correlates of DNA Damage in Breast Cancer Subtypes. Cancers, 13(9), 2117. https://doi.org/10.3390/cancers13092117