
Gene Regulation Network Analysis on Human Prostate Orthografts Highlights a Potential Role for the JMJD6 Regulon in Clinical Prostate Cancer

 ,
,  , ,
, ,  , , , and
, , , and
Abstract
Simple Summary
Abstract
1. Background
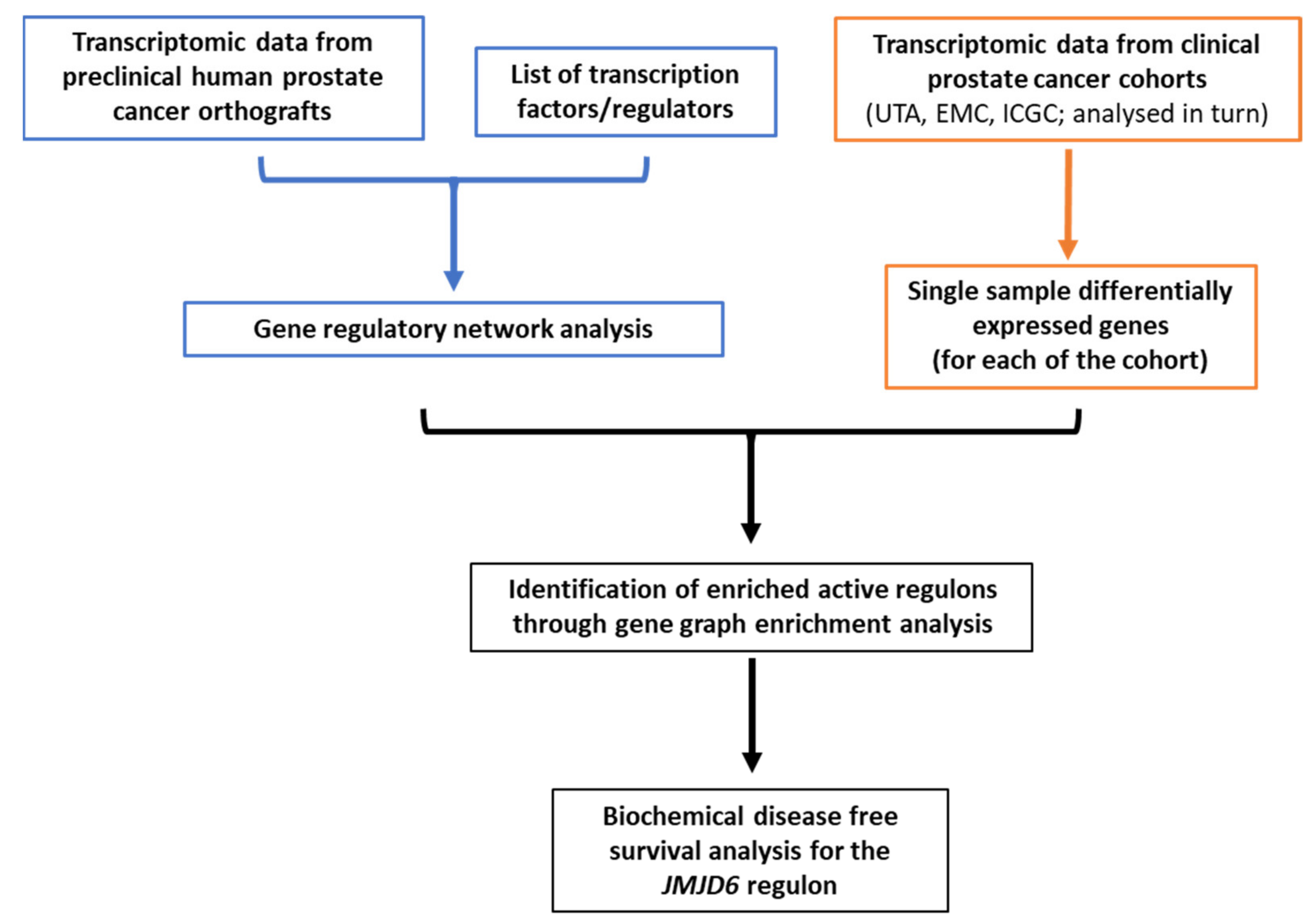
2. Materials and Methods
2.1. Datasets
2.2. Regulons Identification and Filtering
2.3. Gene Regulatory Network Metrics
2.4. Regulons Enrichment
2.5. Statistical Analyses
3. Results
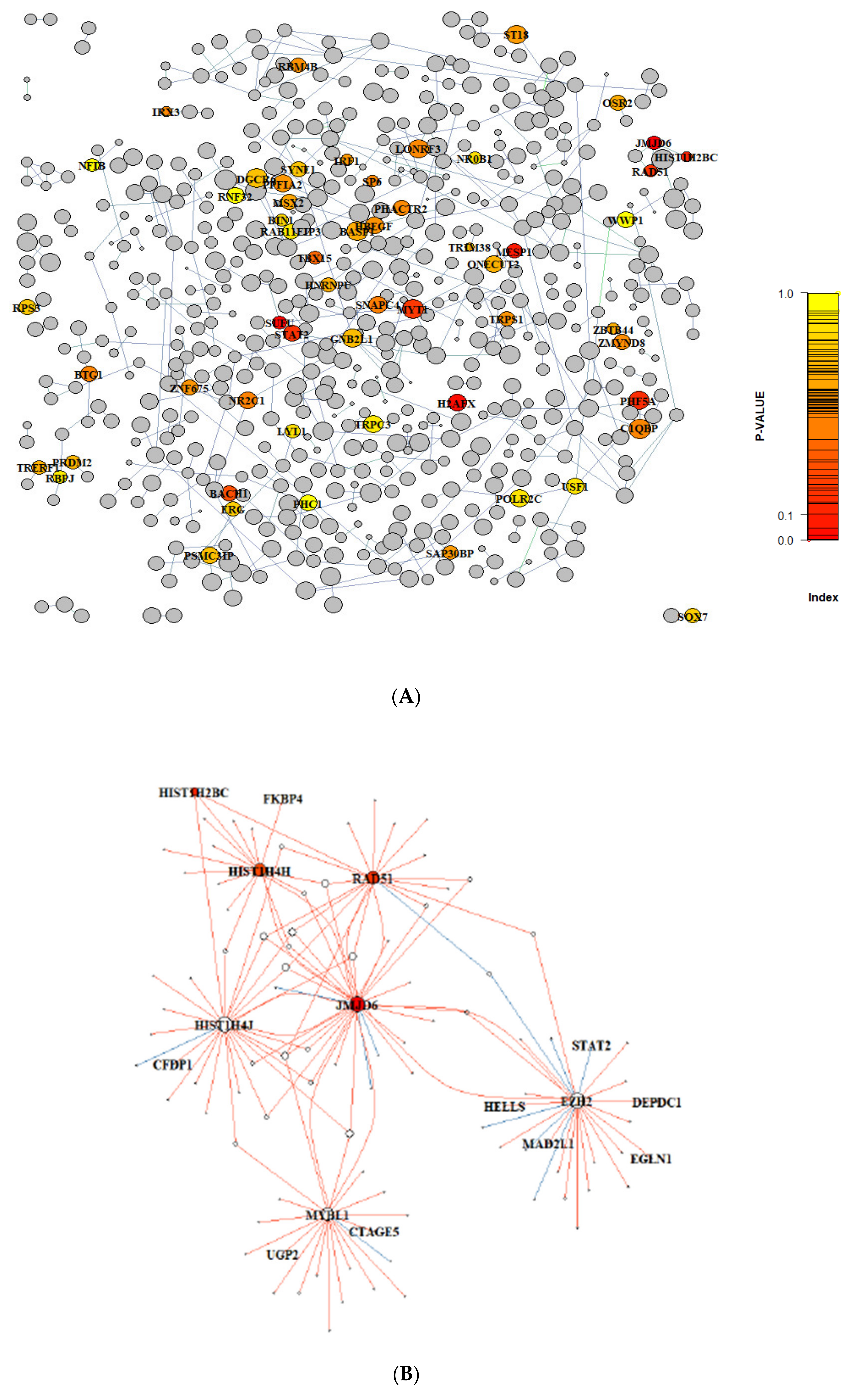
3.1. Gene Regulatory Network Inferred from Preclinical Prostate Orthograft Models
3.2. Analysis of Differentially Expressed Genes (DEG) in Clinical PCa Patient Cohorts
3.3. Gene Graph Enrichment Analysis
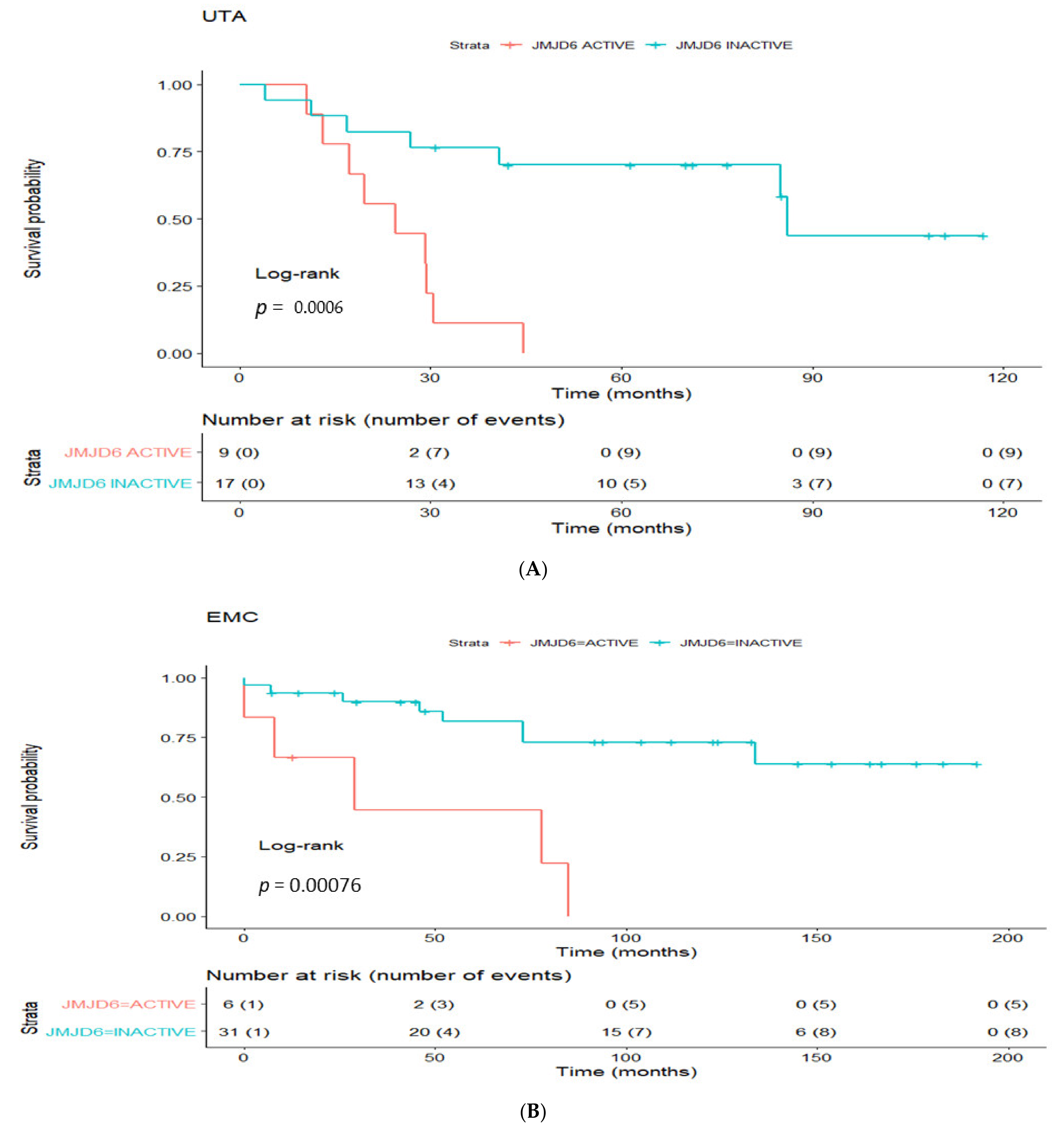
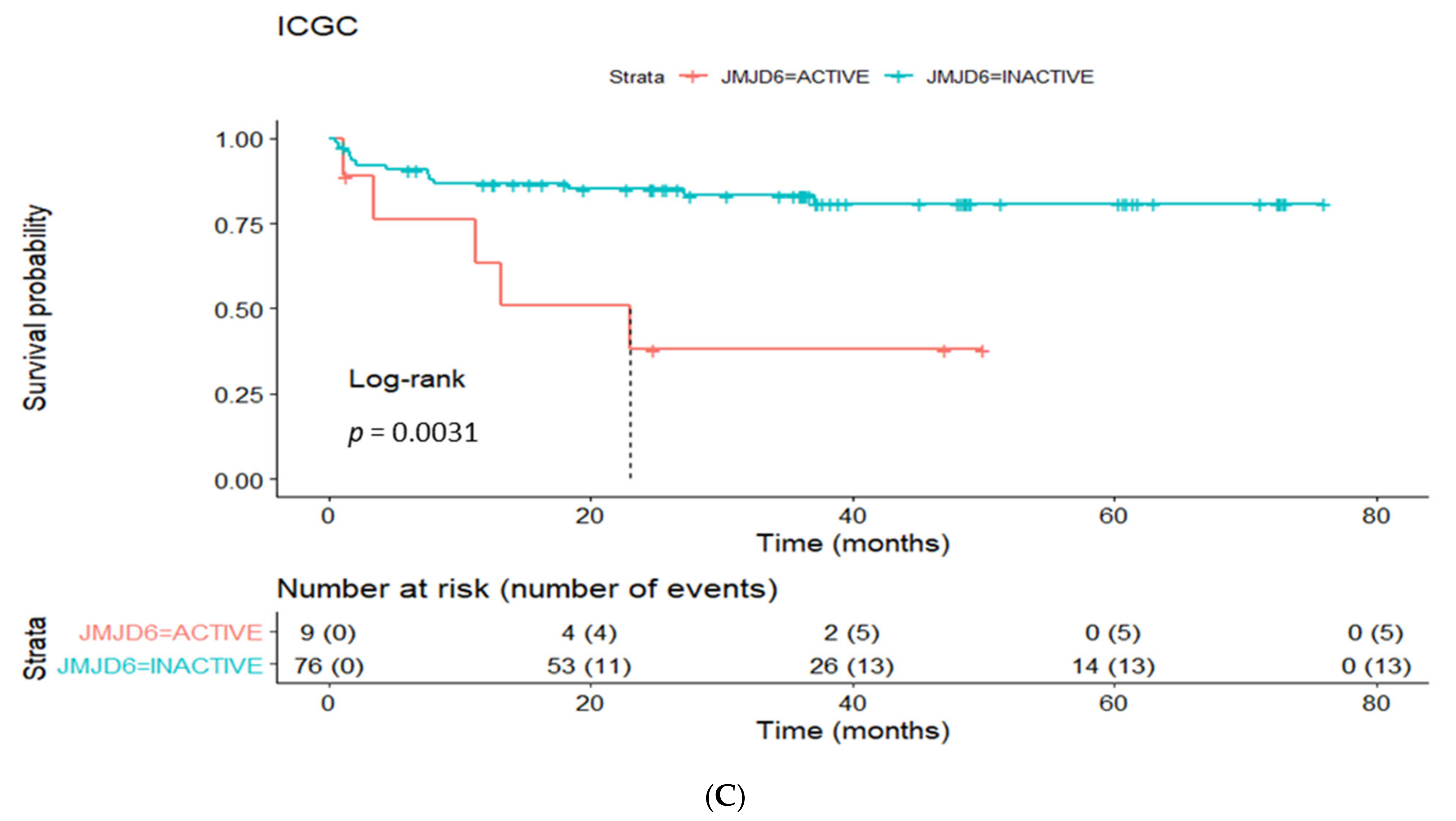
3.4. Prognostic Utility of Regulon Activity Status in Radical Prostatectomy Clinical Cohorts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PCa | Prostate Cancer |
| GRN | Gene regulatory network |
| AR | Androgen Receptor |
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Ciccarese, C.; Massari, F.; Iacovelli, R.; Fiorentino, M.; Montironi, R.; Di Nunno, V.; Giunchi, F.; Brunelli, M.; Tortora, G. Prostate cancer heterogeneity: Discovering novel molecular targets for therapy. Cancer Treat. Rev. 2017, 54, 68–73. [Google Scholar] [CrossRef]
- Cornford, P.; Bellmunt, J.; Bolla, M.; Briers, E.; De Santis, M.; Gross, T.; Henry, A.M.; Joniau, S.; Lam, T.B.; Mason, M.D.; et al. EAU-ESTRO-SIOG Guidelines on Prostate Cancer. Part II: Treatment of Relapsing, Metastatic, and Castration-Resistant Prostate Cancer. Eur. Urol. 2017, 71, 630–642. [Google Scholar] [CrossRef]
- Amaral, T.M.S.; Macedo, D.; Fernandes, I.; Costa, L. Castration-Resistant Prostate Cancer: Mechanisms, Targets, and Treatment. Prostate Cancer 2012, 2012, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Parimi, V.; Goyal, R.; Poropatich, K.; Yang, X.J. Neuroendocrine differentiation of prostate cancer: A review. Am. J. Clin. Exp. Urol. 2014, 2, 273–285. [Google Scholar]
- Myers, J.S.; Von Lersner, A.K.; Robbins, C.J.; Sang, Q.-X.A. Differentially Expressed Genes and Signature Pathways of Human Prostate Cancer. PLoS ONE 2015, 10, e0145322. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Sun, S.-Y.; Wang, Y.; Zhu, H.; Chen, L. Network biomarkers reveal dysfunctional gene regulations during disease progression. FEBS J. 2013, 280, 5682–5695. [Google Scholar] [CrossRef]
- Prolaris Cell Cycle Progression Test for Localized Prostate Cancer: A Health Technology Assessment. Ont. Health Technol. Assess. Ser. 2017, 17, 1–75.
- Dalela, D.; Löppenberg, B.; Sood, A.; Sammon, J.; Abdollah, F. Contemporary Role of the Decipher® Test in Prostate Cancer Management: Current Practice and Future Perspectives. Rev. Urol. 2016, 18, 1–9. [Google Scholar]
- Eeden, S.K.V.D.; Lu, R.; Zhang, N.; Quesenberry, C.P.; Shan, J.; Han, J.S.; Tsiatis, A.C.; Leimpeter, A.D.; Lawrence, H.J.; Febbo, P.G.; et al. A Biopsy-based 17-gene Genomic Prostate Score as a Predictor of Metastases and Prostate Cancer Death in Surgically Treated Men with Clinically Localized Disease. Eur. Urol. 2018, 73, 129–138. [Google Scholar] [CrossRef]
- Dempster, J.; Krill-Burger, J.; Warren, A.; McFarland, J.; Golub, T.; Tsherniak, A. Gene expression has more power for predicting in vitro cancer cell vulnerabilities than genomics. bioRxiv 2020. [Google Scholar] [CrossRef]
- Litwin, M.S.; Tan, H.J. The diagnosis and treatment of prostate cancer: A review. JAMA 2017, 317, 2532–2542. [Google Scholar] [CrossRef] [PubMed]
- Creed, J.H.; Berglund, A.E.; Rounbehler, R.J.; Awasthi, S.; Cleveland, J.L.; Park, J.Y.; Yamoah, K.; Gerke, T.A. Commercial Gene Expression Tests for Prostate Cancer Prognosis Provide Paradoxical Estimates of Race-Specific Risk. Cancer Epidemiol. Biomark. Prev. 2020, 29, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-H.; Wu, S.-Q.; Su, H.; Zhao, X.-K. Role of tumor-associated immune cells in prostate cancer: Angel or devil? Asian J. Androl. 2019, 21, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Krušlin, B.; Ulamec, M.; Tomas, D. Prostate cancer stroma: An important factor in cancer growth and progression. Bosn. J. Basic Med. Sci. 2015, 15, 1–8. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Evans, J.C.; McCarthy, J.; Guo, J.; Prencipe, M.; Kearney, M.; Watson, W.R.; O’Driscoll, C.M. The role of transcription factors in prostate cancer and potential for future RNA interference therapy. Expert Opin. Ther. Targets 2014, 18, 633–649. [Google Scholar] [CrossRef] [PubMed]
- Grimes, T.; Potter, S.S.; Datta, S. Integrating gene regulatory pathways into differential network analysis of gene expression data. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Mochida, K.; Koda, S.; Inoue, K.; Nishii, R. Statistical and Machine Learning Approaches to Predict Gene Regulatory Networks from Transcriptome Datasets. Front. Plant Sci. 2018, 9, 1770. [Google Scholar] [CrossRef]
- Martinez, R.S.; Salji, L.M.; Ntala, R.C.; Blanco, G.B.R.; Hedley, A.; Clark, W.; Peixito, P.; Hervouet, E.; Renaude, E. Schlafen family member 5 (SLFN5) regulates LAT1-mediated mTOR activation in castration-resistant prostate cancer. bioRxiv 2020. [Google Scholar] [CrossRef]
- Ylipää, A.; Kivinummi, K.; Kohvakka, A.; Annala, M.; Latonen, L.; Scaravilli, M.; Kartasalo, K.; Leppänen, S.-P.; Karakurt, S.; Seppälä, J.; et al. Transcriptome Sequencing Reveals PCAT5 as a Novel ERG-Regulated Long Noncoding RNA in Prostate Cancer. Cancer Res. 2015, 75, 4026–4031. [Google Scholar] [CrossRef]
- Chen, S.; Huang, V.; Xu, X.; Livingstone, J.; Soares, F.; Jeon, J.; Zeng, Y.; Hua, J.T.; Petricca, J.; Guo, H.; et al. Widespread and Functional RNA Circularization in Localized Prostate Cancer. Cell 2019, 176, 831–843.e22. [Google Scholar] [CrossRef]
- Hendriksen, P.J.; Dits, N.F.; Kokame, K.; Veldhoven, A.; Van Weerden, W.M.; Bangma, C.H.; Trapman, J.; Jenster, G. Evolution of the Androgen Receptor Pathway during Progression of Prostate Cancer. Cancer Res. 2006, 66, 5012–5020. [Google Scholar] [CrossRef] [PubMed]
- Gerhauser, C.; Favero, F.; Risch, T.; Simon, R.; Feuerbach, L.; Assenov, Y.; Heckmann, D.; Sidiropoulos, N.; Waszak, S.M.; Hübschmann, D.; et al. Molecular Evolution of Early-Onset Prostate Cancer Identifies Molecular Risk Markers and Clinical Trajectories. Cancer Cell 2018, 34, 996–1011.e8. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, M.N.C.; Castro, M.A.A.; Wang, X.; De Santiago, I.; O’Reilly, M.; Chin, S.-F.; Rueda, O.M.; Caldas, C.; Ponder, B.A.J.; Markowetz, F.; et al. Master regulators of FGFR2 signalling and breast cancer risk. Nat. Commun. 2013, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Alvarez, M.J.; Shen, M.J.A.Y.; Giorgi, F.M.; Lachmann, A.; Ding, B.B.; Ye, B.H.; Califano, A. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat. Genet. 2016, 48, 838–847. [Google Scholar] [CrossRef]
- Geistlinger, L.; Csaba, G.; Zimmer, R. Bioconductor’s EnrichmentBrowser: Seamless navigation through combined results of set-& network-based enrichment analysis. BMC Bioinform. 2016, 17, 1–11. [Google Scholar]
- Georgescu, C.; Corbin, J.M.; Thibivilliers, S.; Webb, Z.D.; Zhao, Y.D.; Koster, J.; Fung, K.-M.; Asch, A.S.; Wren, J.D.; Ruiz-Echevarría, M.J. A TMEFF2-regulated cell cycle derived gene signature is prognostic of recurrence risk in prostate cancer. BMC Cancer 2019, 19, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Roberts, D.; Takhar, M.; Erho, N.; Bibby, B.A.; Thiruthaneeswaran, N.; Bhandari, V.; Cheng, W.-C.; Haider, S.; McCorry, A.M.; et al. Development and Validation of a 28-gene Hypoxia-related Prognostic Signature for Localized Prostate Cancer. EBioMedicine 2018, 31, 182–189. [Google Scholar] [CrossRef]
- Urbanucci, A.; Barfeld, S.J.; Kytölä, V.; Itkonen, H.M.; Coleman, I.M.; Vodák, D.; Sjöblom, L.; Sheng, X.; Tolonen, T.; Minner, S.; et al. Androgen Receptor Deregulation Drives Bromodomain-Mediated Chromatin Alterations in Prostate Cancer. Cell Rep. 2017, 19, 2045–2059. [Google Scholar] [CrossRef]
- Hsu, C.L.; Liu, J.S.; Wu, P.L.; Guan, H.H.; Chen, Y.L.; Lin, A.C.; Ting, H.-J.; Pang, S.-T.; Yeh, S.-D.; Ma, W.-L.; et al. Identification of a new androgen receptor (AR) co-regulator BUD31 and related peptides to suppress wild-type and mutated AR-mediated prostate cancer growth via peptide screening and X-ray structure analysis. Mol. Oncol. 2014, 8, 1575–1587. [Google Scholar] [CrossRef]
- Baek, J.-H.; Yun, H.S.; Kwon, G.T.; Lee, J.; Kim, J.-Y.; Jo, Y.; Cho, J.-M.; Lee, C.-W.; Song, J.-Y.; Ahn, J.; et al. PLOD3 suppression exerts an anti-tumor effect on human lung cancer cells by modulating the PKC-delta signaling pathway. Cell Death Dis. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- MSigDb. SCHAEFFER_PROSTATE_DEVELOPMENT_48HR_UP. Available online: https://www.gsea-msigdb.org/gsea/msigdb/cards/SCHAEFFER_PROSTATE_DEVELOPMENT_48HR_UP (accessed on 1 August 2020).
- Suyama, T.; Shiraishi, T.; Zeng, Y.; Yu, W.; Parekh, N.; Vessella, R.L.; Luo, J.; Getzenberg, R.H.; Kulkarni, P. Expression of cancer/testis antigens in prostate cancer is associated with disease progression. Prostate 2010, 70, 1778–1787. [Google Scholar] [CrossRef] [PubMed]
- Goel, M.; Agrawal, D.; Natu, S.; Goel, A. Hepsin immunohistochemical expression in prostate cancer in relation to Gleason′s grade and serum prostate specific antigen. Indian J. Pathol. Microbiol. 2011, 54, 476–481. [Google Scholar] [CrossRef]
- Ashikari, D.; Takayama, K.-I.; Obinata, D.; Takahashi, S.; Inoue, S. CLDN8, an androgen-regulated gene, promotes prostate cancer cell proliferation and migration. Cancer Sci. 2017, 108, 1386–1393. [Google Scholar] [CrossRef]
- Rotinen, M.; You, S.; Yang, J.; Coetzee, S.; Huang, W.-C.; Huang, F.; Pan, X.; Yyyez, A.; Hazelett, D.; Chu, C.-Y.; et al. ONECUT2 Is a Targetable Master Regulator of Lethal Prostate Cancer That Suppresses the Androgen Axis. SSRN Electron. J. 2018, 24, 1887. [Google Scholar] [CrossRef]
- Geistlinger, L.; Csaba, G.; Küffner, R.; Mulder, N.; Zimmer, R. From sets to graphs: Towards a realistic enrichment analysis of transcriptomic systems. Bioinformatics 2011, 27, i366–i373. [Google Scholar] [CrossRef]
- Shajari, N.; Davudian, S.; Kazemi, T.; Mansoori, B.; Salehi, S.; Shahgoli, V.K.; Shanehbandi, D.; Mohammadi, A.; Duijf, P.H.G.; Baradaran, B. Silencing of BACH1 inhibits invasion and migration of prostate cancer cells by altering metastasis-related gene expression. Artif. Cells Nanomed. Biotechnol. 2017, 46, 1495–1504. [Google Scholar] [CrossRef]
- Shin, S.H.; Lee, G.Y.; Lee, M.; Kang, J.; Shin, H.W.; Chun, Y.S.; Park, J.W. Aberrant expression of CITED2 promotes prostate cancer metastasis by activating the nucleolin-AKT pathway. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Lee, E.; Wang, J.; Yumoto, K.; Jung, Y.; Cackowski, F.C.; Decker, A.M.; Li, Y.; Franceschi, R.T.; Pienta, K.J.; Taichman, R.S. DNMT1 Regulates Epithelial-Mesenchymal Transition and Cancer Stem Cells, Which Promotes Prostate Cancer Metastasis. Neoplasia 2016, 18, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Alonso, L.M.; Iorio, F.; Matchan, A.; Fonseca, N.A.; Jaaks, P.; Peat, G.; Pignatelli, M.; Falcone, F.; Benes, C.H.; Dunham, I.; et al. Transcription Factor Activities Enhance Markers of Drug Sensitivity in Cancer. Cancer Res. 2018, 78, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.A.A.; De Santiago, I.; Campbell, T.M.; Vaughn, C.; Hickey, T.E.; Ross, E.; Tilley, W.D.; Markowetz, F.; Ponder, B.A.; Meyer, K.B. Regulators of genetic risk of breast cancer identified by integrative network analysis. Nat. Genet. 2016, 48, 12–21. [Google Scholar] [CrossRef]
- Lee, C.-R.; Lee, S.H.; Rigas, N.K.; Kim, R.H.; Kang, M.K.; Park, N.-H.; Shin, K.-H. Elevated expression of JMJD6 is associated with oral carcinogenesis and maintains cancer stemness properties. Carcinogenesis 2015, 37, 119–128. [Google Scholar] [CrossRef]
- Gao, W.-W.; Xiao, R.-Q.; Zhang, W.-J.; Hu, Y.-R.; Peng, B.-L.; Li, W.-J.; He, Y.-H.; Shen, H.-F.; Ding, J.-C.; Huang, Q.-X.; et al. JMJD6 Licenses ERα-Dependent Enhancer and Coding Gene Activation by Modulating the Recruitment of the CARM1/MED12 Co-activator Complex. Mol. Cell 2018, 70, 340–357.e8. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Sun, Y.; Xi, Z.; Milazzo, G.; Poulos, R.C.; Bartenhagen, C.; Bell, J.L.; Mayoh, C.; Ho, N.; Tee, A.E.; et al. JMJD6 is a tumorigenic factor and therapeutic target in neuroblastoma. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Liu, X.; Si, W.; Liu, X.; He, L.; Ren, J.; Yang, Z.; Yang, G.; Li, Y.; Liu, S.; Pei, F.; et al. JMJD6 promotes melanoma carcinogenesis through regulation of the alternative splicing of PAK1, a key MAPK signaling component. Mol. Cancer 2017, 16, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Tie, Y.; Fang, Z.; Wu, X.; Yi, T.; Huang, S.; Liang, X.; Qian, Y.; Wang, X.; Pi, R.; et al. Jumonji domain-containing 6 (JMJD6) identified as a potential therapeutic target in ovarian cancer. Signal Transduct. Target. Ther. 2019, 4, 1–14. [Google Scholar] [CrossRef]
- Mitra, A.; Jameson, C.; Barbachano, Y.; Sanchez, L.; Kote-Jarai, Z.; Peock, S.; Sodha, N.; Bancroft, E.; Fletcher, A.; Cooper, C.; et al. Overexpression of RAD51 occurs in aggressive prostatic cancer. Histopathology 2009, 55, 696–704. [Google Scholar] [CrossRef]
- Na, R.; Wu, Y.; Ding, Q.; Xu, J. Clinically available RNA profiling tests of prostate tumors: Utility and comparison. Asian J. Androl. 2016, 18, 575–579. [Google Scholar] [CrossRef]
- Testa, U.; Castelli, G.; Pelosi, E. Cellular and Molecular Mechanisms Underlying Prostate Cancer Development: Therapeutic Implications. Medicines 2019, 6, 82. [Google Scholar] [CrossRef]
- Mills, I.G. Maintaining and reprogramming genomic androgen receptor activity in prostate cancer. Nat. Rev. Cancer 2014, 14, 187–198. [Google Scholar] [CrossRef]
- Paschalis, A.; Welti, J.; Neeb, A.J.; Yuan, W.; Figueiredo, I.; Pereira, R.; Ferreira, A.; Riisnaes, R.; Rodrigues, D.N.; Jiménez-Vacas, J.M.; et al. JMJD6 Is a Druggable Oxygenase That Regulates AR-V7 Expression in Prostate Cancer. Cancer Res. 2021, 81, 1087–1100. [Google Scholar] [CrossRef] [PubMed]
- Unoki, M.; Masuda, A.; Dohmae, N.; Arita, K.; Yoshimatsu, M.; Iwai, Y.; Fukui, Y.; Ueda, K.; Hamamoto, R.; Shirakawa, M.; et al. Lysyl 5-Hydroxylation, a Novel Histone Modification, by Jumonji Domain Containing 6 (JMJD6)*. J. Biol. Chem. 2013, 288, 6053–6062. [Google Scholar] [CrossRef] [PubMed]
- Poulard, C.; Rambaud, J.; Lavergne, E.; Jacquemetton, J.; Renoir, J.-M.; Trédan, O.; Chabaud, S.; Treilleux, I.; Corbo, L.; Le Romancer, M. Role of JMJD6 in Breast Tumourigenesis. PLoS ONE 2015, 10, e0126181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ni, S.-S.; Zhao, W.-L.; Dong, X.-C.; Wang, J.-L. High expression of JMJD6 predicts unfavorable survival in lung adenocarcinoma. Tumor Biol. 2013, 34, 2397–2401. [Google Scholar] [CrossRef]
- Aprelikova, O.; Chen, K.; El Touny, L.H.; Brignatz-Guittard, C.; Han, J.; Qiu, T.; Yang, H.H.; Lee, M.P.; Zhu, M.; Green, J.E. The epigenetic modifier JMJD6 is amplified in mammary tumors and cooperates with c-Myc to enhance cellular transformation, tumor progression, and metastasis. Clin. Epigenetics 2016, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Fard, A.T.; Ragan, M.A. Modeling the Attractor Landscape of Disease Progression: A Network-Based Approach. Front. Genet. 2017, 8, 48. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Cohorts | UTA | EMC | ICGC | |||
|---|---|---|---|---|---|---|
| Number (n) | n = 27 | % | n = 37 | % | n = 85 | % |
| age at diagnosis | ||||||
| range | 47–71 | NA | 32–52 | |||
| mean | 60 | NA | 47 | |||
| median | 61 | NA | 48 | |||
| na | 0 | |||||
| psa at diagnosis (ng/mL) | ||||||
| range | 3.5–48.1 | 0.3–36.2 | 3.1–743 | |||
| mean | 10.4 | 11.8 | 30.48 | |||
| median | 8.3 | 9.4 | 8.21 | |||
| na | 0 | 0 | 0 | |||
| tumour stage | ||||||
| t1 | 10 | 37.0 | 1 | 2.7 | 0 | 0.0 |
| t2 | 16 | 59.3 | 15 | 40.5 | 61 | 71.8 |
| t3 | 1 | 3.7 | 13 | 35.1 | 23 | 27.1 |
| t4 | 0 | 0.0 | 8 | 21.6 | 1 | 1.2 |
| na | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 |
| gleason score | ||||||
| <7 | 7 | 25.9 | 6 | 16.2 | 12 | 14.1 |
| 7 | 13 | 48.2 | 19 | 51.4 | 65 | 76.5 |
| >7 | 7 | 25.9 | 0 | 0.0 | 8 | 9.4 |
| na | 0 | 0.0 | 12 | 32.4 | 0 | 0.0 |
| therapy | ||||||
| Radical prostatectomy | 27 | 100 | 37 | 100 | 85 | 100 |
| (A) | ||
|---|---|---|
| Ensembl ID | Hugo Symbol | p Value |
| ENSG00000070495 | JMJD6 | 0.002 |
| ENSG00000196132 | MYT1 | 0.006 |
| ENSG00000100410 | PHF5A | 0.02 |
| ENSG00000065057 | NTHL1 | 0.02 |
| ENSG00000159210 | SNF8 | 0.02 |
| ENSG00000171222 | SCAND1 | 0.02 |
| ENSG00000123091 | RNF11 | 0.02 |
| ENSG00000120798 | NR2C1 | 0.02 |
| ENSG00000107882 | SUFU | 0.03 |
| ENSG00000146083 | RNF44 | 0.04 |
| (B) | ||
| Ensembl ID | Hugo Symbol | pValue |
| ENSG00000070495 | JMJD6 | 0.003 |
| ENSG00000095002 | MSH2 | 0.006 |
| ENSG00000107882 | SUFU | 0.007 |
| ENSG00000136826 | KLF4 | 0.01 |
| ENSG00000119969 | HELLS | 0.01 |
| ENSG00000151929 | BAG3 | 0.02 |
| ENSG00000105607 | GCDH | 0.02 |
| ENSG00000092607 | TBX15 | 0.02 |
| ENSG00000188486 | H2AFX | 0.02 |
| ENSG00000180596 | HIST1H2BC | 0.03 |
| (A) Univariate Analysis | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Clinical Cohorts | UTA | EMC | ICGC | ||||||
| Statistics | HR | 95% CI | p | HR | 95% CI | p | HR | 95% CI | p |
| Clinicopathological variables | |||||||||
| Gleason score | 2.7 | 1.6–4.7 | 0.0004 | 1.9 | 0.2–16 | 0.5 | 2 | 1.4–3 | 0.0004 |
| Tumor stage | 1.7 | 1–2.96 | 0.05 | 1.3 | 1–1.6 | 0.02 | 2.5 | 1.7–3.8 | <0.0001 |
| Signatures | |||||||||
| active JMJD6 regulon | 6 | 1.9–18 | 0.002 | 5.8 | 1.8–18.6 | 0.003 | 4.2 | 1.5–12 | 0.006 |
| TMCC11 | 4.5 | 1.1–17.8 | 0.03 | 1 | 0.3–3.7 | 1.0 | 4 | 1.6–10.5 | 0.004 |
| BROMO-10 | 0.06 | 0.0069–0.52 | 0.01 | 1.2 | 0.3–4.2 | 0.8 | 2.6 | 0.7–9.3 | 0.2 |
| HYPOXIA-28 | 2.1 | 0.7–6.24 | 0.2 | 1.1 | 0.4–3.5 | 0.8 | 3.4 | 1.3–9.2 | 0.01 |
| (B) Multivariate analysis | |||||||||
| UTA | EMC | ICGC | |||||||
| HR | 95% CI | P | HR | 95% CI | P | HR | 95% CI | P | |
| JMJD6 regulon | 6.5 | 1.3–32 | 0.02 | 4.4 | 1.3–14.6 | 0.01 | 1.2 | 0.3–4.8 | 0.7 |
| Gleason score | 1.6 | 0.8–3.1 | 0.2 | 1.2 | 0.6–2.4 | 0.6 | |||
| Tumor stage | 2.3 | 1.1–4.9 | 0.03 | 1.2 | 1–1.5 | 0.05 | 2.6 | 1.3–4.9 | 0.004 |
| TMCC11 | 3.4 | 0.8–14.4 | 0.1 | 1.8 | 0.6–5.6 | 0.3 | |||
| Gleason score | 2.5 | 1.4–4.4 | 0.002 | 1.3 | 0.7–2.4 | 0.5 | |||
| Tumor stage | 1.58 | 0.8–3.2 | 0.2 | 2.2 | 1.1–4.4 | 0.02 | |||
| BROMO-10 | 0.3 | 0.03–4.2 | 0.4 | ||||||
| Gleason score | 2.15 | 1.08–4.27 | 0.03 | ||||||
| Tumor stage | 1.5 | 0.8–2.8 | 0.2 | ||||||
| HYPOXIA-28 | 2.1 | 0.7–6.1 | 0.2 | ||||||
| Gleason score | 1.3 | 0.7–2.4 | 0.4 | ||||||
| Tumor stage | 2.2 | 1.13–4.24 | 0.02 | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cangiano, M.; Grudniewska, M.; Salji, M.J.; Nykter, M.; Jenster, G.; Urbanucci, A.; Granchi, Z.; Janssen, B.; Hamilton, G.; Leung, H.Y.; et al. Gene Regulation Network Analysis on Human Prostate Orthografts Highlights a Potential Role for the JMJD6 Regulon in Clinical Prostate Cancer. Cancers 2021, 13, 2094. https://doi.org/10.3390/cancers13092094
Cangiano M, Grudniewska M, Salji MJ, Nykter M, Jenster G, Urbanucci A, Granchi Z, Janssen B, Hamilton G, Leung HY, et al. Gene Regulation Network Analysis on Human Prostate Orthografts Highlights a Potential Role for the JMJD6 Regulon in Clinical Prostate Cancer. Cancers. 2021; 13(9):2094. https://doi.org/10.3390/cancers13092094
Chicago/Turabian StyleCangiano, Mario, Magda Grudniewska, Mark J. Salji, Matti Nykter, Guido Jenster, Alfonso Urbanucci, Zoraide Granchi, Bart Janssen, Graham Hamilton, Hing Y. Leung, and et al. 2021. "Gene Regulation Network Analysis on Human Prostate Orthografts Highlights a Potential Role for the JMJD6 Regulon in Clinical Prostate Cancer" Cancers 13, no. 9: 2094. https://doi.org/10.3390/cancers13092094
APA StyleCangiano, M., Grudniewska, M., Salji, M. J., Nykter, M., Jenster, G., Urbanucci, A., Granchi, Z., Janssen, B., Hamilton, G., Leung, H. Y., & Beumer, I. J. (2021). Gene Regulation Network Analysis on Human Prostate Orthografts Highlights a Potential Role for the JMJD6 Regulon in Clinical Prostate Cancer. Cancers, 13(9), 2094. https://doi.org/10.3390/cancers13092094