
Novel Target Opportunities in Non-Metastatic Castrate Resistant Prostate Cancer


 ,
,
Abstract
Simple Summary
Abstract
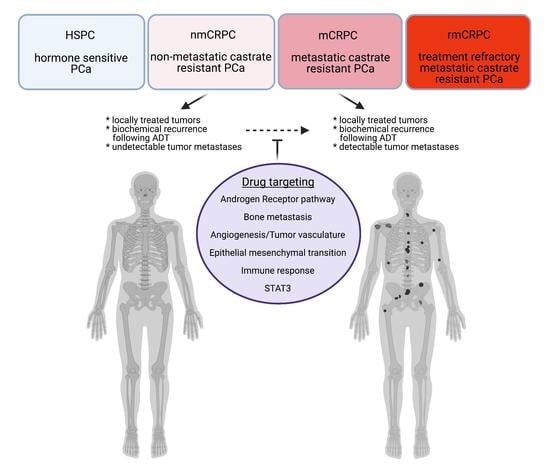
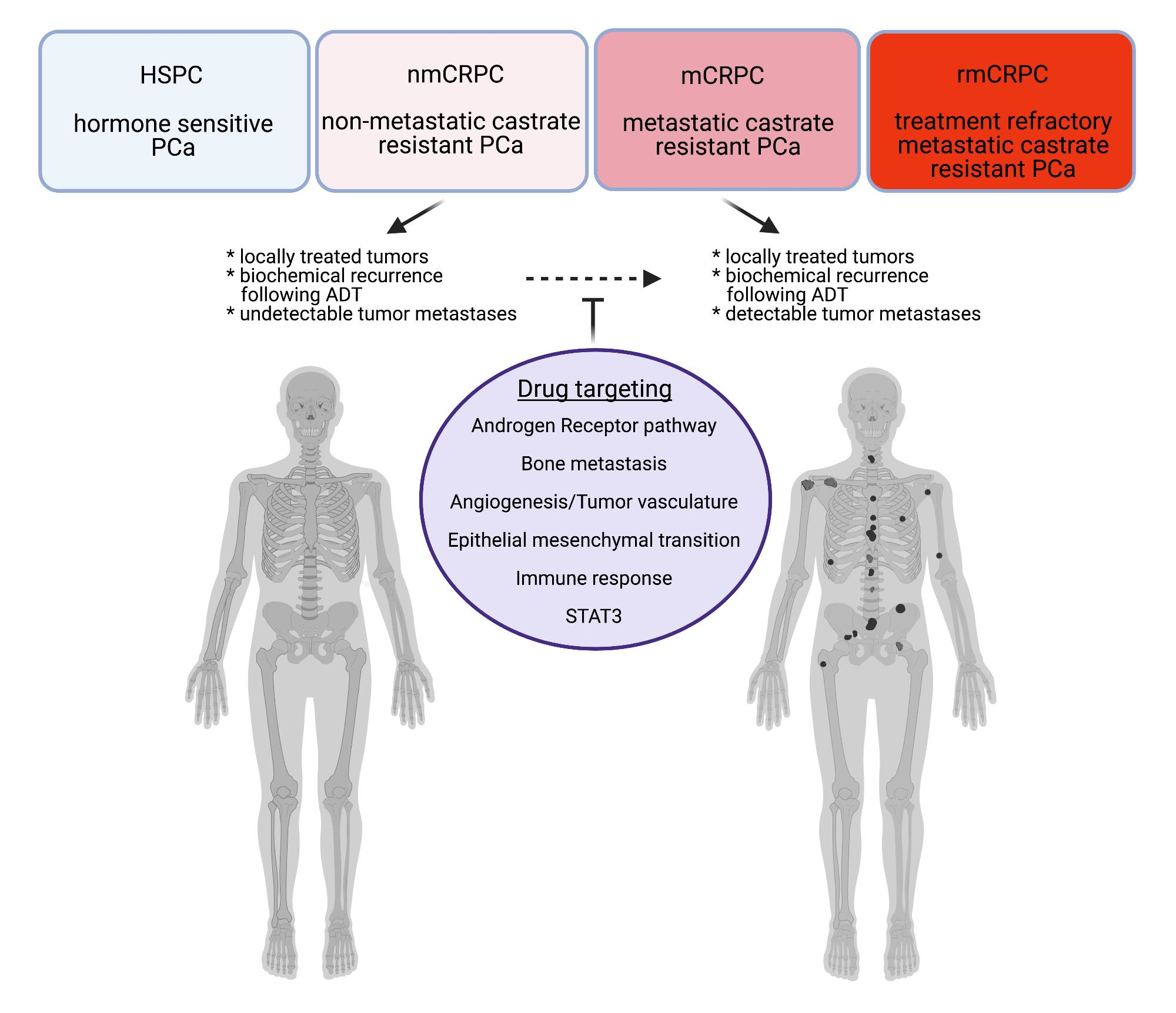
1. Introduction
2. Current Treatment Paradigm for nmCRPC


{kind=link}
{kind=link}
{kind=link}
| Trial Identifier | Trial Name/Details | Name | Drug Target | Sample Size (n) | Conclusions/Metastasis Free Survival (MFS) |
|---|---|---|---|---|---|
| NCT01946204 | Selective Prostate Androgen Receptor Targeting with ARN-509 (SPARTAN), Phase 3 | Apalutamide | AR antagonist | n = 806 ADT/ apalutamide n = 401 ADT/placebo | 40.5 months versus 16.2 months with placebo (HR 0.28, 95% CI 0.23–0.35, p < 0.001) [10] |
| NCT02003924 | PROSPER, Phase 3 | Enzalutamide | AR antagonist | n = 933 ADT/ enzalutamide n = 468 ADT/placebo | 36.6 months versus 14.7 months with placebo (HR 0.29, 95% CI 0.24–0.35, p < 0.001) [11] |
| NCT02200614 | ARAMIS, Phase 3 | Darolutamide | AR antagonist | n = 955 ADT/ darolutamide n = 554 ADT/placebo | 40.4 months versus 18.4 months with placebo (HR 0.41, 95% CI 0.34–0.50, p < 0.001) [12] |
| NCT01314118 | IMAAGEN trial Phase 2 | Abiraterone acetate + prednisone | Cytochrome C17 enzyme (CYP17), androgen synthesis | n = 131 ADT/ abiraterone and prednisone | Median PSA progression at 28.7 months, radiographic progression not reached (95% CI 21.2–38.2) [16] |
| NCT00121238 | Phase 2 | Cilengitide | selective antagonist of αv β3 and αv β5 integrins | n = 16 cilengitide | No detectable clinical activity [17] |
| NCT00036556 | Atrasentan, Phase 3 | Atrasentan | selective endothelin -A receptor antagonist | n = 467 Atrasenten n = 474 placebo | No significant delay in time to disease progression, but did show a prolongation of TTP among patients outside the US only [18] |
| NCT00626548 | ENTHUSE M0, Phase 3 | Zibotentan | ETA receptor antagonist | n = 705 Zibotentan n = 716 placebo | No difference in overall survival or progression free survival resulting in trial termination [19] |
| NCT00510224 | Sandostatin, Phase 2 | Octreotide Acetate | Insulin-like growth factor (IGF) signaling pathway inhibition, somatostatin analogue that inhibits growth hormone release from the pituitary | n = 13 Octreotide | No decline in PSA levels despite three cycles of treatment and a decline in IGF levels [20] |
| NCT00286091 | Phase 3 | Denosumab | anti-RANKL monoclonal antibody | n = 716 Denosumab n = 716 placebo | 29.5 months versus 25.2 months with placebo to delay bone MFS specifically (HR 0.85, 95% CI 0.73–0.98, p = 0.028) [21] |
| NCT01046916 | TAK-700, Phase 2 | Orteronel (TAK-700) | CYP17A selective inhibitor | n = 39 Orteronel | Median time to PSA progression and metastases to be 14 months and 25 months [22] |
| NCT00849121 | DNA vaccine, Phase 2 | DNA Vaccine: pTVG-HP with rhGM-CSF | Sipuleucel-T +/− DNA-based vaccine booster (pTVG-HP) for prostatic acid phosphatase (PAP) | n = 9 pTVG-HP n = 9 No booster | Repetitive immunization with pTVG-HP maintained antigen-specific T-cells that target prostate cells [23] |
| N/A | Randomized control trial | Poxvirus-based PSA vaccine | Vaccine with transgenes for PSA and human t-cell costimulatory molecule B7.1; priming vaccine followed by monthly boosts with GM-CSF | n = 21 Vaccine n = 21 Nilutamide Crossover design with PSA progression | Noted trend toward survival benefit for patients randomized to vaccine arm (median 5.1 years in vaccine vs. 3.4 years in ADT, p = 0.13) [24] |
| NCT01656304 | Pilot phase 2 trial | Bevacizumab | Humanized monoclonal antibody against vascular endothelial growth factor (VEGF) | n = 15 Bevacizumab | No benefit noted [25] |
3. Targets beyond Androgen Receptor for the nmCRPC Therapy
4. Novel Epithelial Mesenchymal Transition (EMT) Process to Delay nmCRPC
5. Targeting STAT3 as Master Regulator in nmCRPC
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Freedland, S.J.; Humphreys, E.B.; Mangold, L.A.; Eisenberger, M.; Dorey, F.J.; Walsh, P.C.; Partin, A.W. Death in patients with recurrent prostate cancer after radical prostatectomy: Prostate-specific antigen doubling time subgroups and their associated contributions to all-cause mortality. J. Clin. Oncol. 2007, 25, 1765–1771. [Google Scholar] [CrossRef] [PubMed]
- Huggins, C.; Hodges, C.V. Studies on prostatic cancer: I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. 1941. J. Urol. 2002, 168, 9–12. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network: Prostate Cancer 2021 [updated 2/2021]. Available online: https://www.nccn.org/professionals/physician_gls/pdf/prostate.pdf (accessed on 8 April 2021).
- Shore, N.D.; Saad, F.; Cookson, M.S.; George, D.J.; Saltzstein, D.R.; Tutrone, R.; Akaza, H.; Bossi, A.; van Veenhuyzen, D.F.; Selby, B.; et al. Oral Relugolix for Androgen-Deprivation Therapy in Advanced Prostate Cancer. N. Engl. J. Med. 2020, 382, 2187–2196. [Google Scholar] [CrossRef]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.E.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Bhattacharya, S.; Carles, J.; Chowdhury, S.; et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N. Engl. J. Med. 2014, 371, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Afshar, M.; Evison, F.; James, N.D.; Patel, P. Shifting paradigms in the estimation of survival for castration-resistant prostate cancer: A tertiary academic center experience. Urol. Oncol. 2015, 33, 338.e1–338.e7. [Google Scholar] [CrossRef] [PubMed]
- Aly, M.; Hashim, M.; Heeg, B.; Liwing, J.; Leval, A.; Mehra, M.; Lawson, J.; Brookman-May, S.D.; Akre, O. Time-to-event Outcomes in Men with Nonmetastatic Castrate-resistant Prostate Cancer-A Systematic Literature Review and Pooling of Individual Participant Data. Eur. Urol. Focus 2019, 5, 788–798. [Google Scholar] [CrossRef]
- Wallitt, K.L.; Khan, S.R.; Dubash, S.; Tam, H.H.; Khan, S.; Barwick, T.D. Clinical PET Imaging in Prostate Cancer. Radiographics 2017, 37, 1512–1536. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Saad, F.; Chowdhury, S.; Oudard, S.; Hadaschik, B.A.; Graff, J.N.; Olmos, D.; Mainwaring, P.N.; Lee, J.Y.; Uemura, H.; et al. Apalutamide Treatment and Metastasis-free Survival in Prostate Cancer. N. Engl. J. Med. 2018, 378, 1408–1418. [Google Scholar] [CrossRef]
- Hussain, M.; Fizazi, K.; Saad, F.; Rathenborg, P.; Shore, N.; Ferreira, U.; Ivashchenko, P.; Demirhan, E.; Modelska, K.; De Phung, B.S.; et al. Enzalutamide in Men with Nonmetastatic, Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2018, 378, 2465–2474. [Google Scholar] [CrossRef]
- Fizazi, K.; Shore, N.; Tammela, T.L.; Ulys, A.; Vjaters, E.; Polyakov, S.; Jievaltas, M.; Luz, M.; Alekseev, B.; Kuss, I.; et al. Darolutamide in Nonmetastatic, Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2019, 380, 1235–1246. [Google Scholar] [CrossRef]
- El-Amm, J.; Aragon-Ching, J.B. The Current Landscape of Treatment in Non-Metastatic Castration-Resistant Prostate Cancer. Clin. Med. Insights Oncol. 2019, 13, 1179554919833927. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.A.; Malhotra, S.V.; Stoyanova, T. Second-Generation Antiandrogens: From Discovery to Standard of Care in Castration Resistant Prostate Cancer. Front. Oncol. 2019, 9, 801. [Google Scholar] [CrossRef]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.-E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased Survival with Enzalutamide in Prostate Cancer after Chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef]
- Ryan, C.J.; Crawford, E.D.; Shore, N.D.; Underwood, W., 3rd; Taplin, M.E.; Londhe, A.; Francis, P.S.J.; Phillips, J.; McGowan, T.; Kantoff, P.W. The IMAAGEN Study: Effect of Abiraterone Acetate and Prednisone on Prostate Specific Antigen and Radiographic Disease Progression in Patients with Nonmetastatic Castration Resistant Prostate Cancer. J. Urol. 2018, 200, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Alva, A.; Slovin, S.; Daignault, S.; Carducci, M.; Dipaola, R.; Pienta, K.; Agus, D.; Cooney, K.; Chen, A.; Smith, D.C.; et al. Phase II study of cilengitide (EMD 121974, NSC 707544) in patients with non-metastatic castration resistant prostate cancer, NCI-6735. A study by the DOD/PCF prostate cancer clinical trials consortium. Investig. New Drugs 2012, 30, 749–757. [Google Scholar] [CrossRef]
- Nelson, J.B.; Love, W.; Chin, J.L.; Saad, F.; Schulman, C.C.; Sleep, D.J.; Qian, J.; Steinberg, J.; Carducci, M. Phase 3, randomized, controlled trial of atrasentan in patients with nonmetastatic, hormone-refractory prostate cancer. Cancer 2008, 113, 2478–2487. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.; Moul, J.W.; Gleave, M.; Fizazi, K.; Nelson, J.B.; Morris, T.; Nathan, F.E.; McIntosh, S.; Pemberton, K.; Higano, C.S. Phase III, randomized, placebo-controlled study of once-daily oral zibotentan (ZD4054) in patients with non-metastatic castration-resistant prostate cancer. Prostate Cancer Prostatic Dis. 2013, 16, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, T.W.; Weinberg, V.K.; Small, E.J.; Sharib, J.; Harzstark, A.L.; Lin, A.M.; Fong, L.; Ryan, C.J. Effect of the somatostatin analog octreotide acetate on circulating insulin-like growth factor-1 and related peptides in patients with non-metastatic castration-resistant prostate cancer: Results of a phase II study. Urol. Oncol. 2012, 30, 408–414. [Google Scholar] [CrossRef]
- Smith, M.R.; Saad, F.; Coleman, R.; Shore, N.; Fizazi, K.; Tombal, B.; Miller, K.; Sieber, P.; Karsh, L.; Damiao, R.; et al. Denosumab and bone-metastasis-free survival in men with castration-resistant prostate cancer: Results of a phase 3, randomised, placebo-controlled trial. Lancet 2012, 379, 39–46. [Google Scholar] [CrossRef]
- Hussain, M.; Corn, P.G.; Michaelson, M.D.; Hammers, H.J.; Alumkal, J.J.; Ryan, C.J.; Bruce, J.Y.; Moran, S.; Lee, S.Y.; Lin, H.M.; et al. Phase II study of single-agent orteronel (TAK-700) in patients with nonmetastatic castration-resistant prostate cancer and rising prostate-specific antigen. Clin. Cancer Res. 2014, 20, 4218–4227. [Google Scholar] [CrossRef]
- Wargowski, E.; Johnson, L.E.; Eickhoff, J.C.; Delmastro, L.; Staab, M.J.; Liu, G.; McNeel, D.G. Prime-boost vaccination targeting prostatic acid phosphatase (PAP) in patients with metastatic castration-resistant prostate cancer (mCRPC) using Sipuleucel-T and a DNA vaccine. J. Immunother. Cancer 2018, 6, 21. [Google Scholar] [CrossRef]
- Madan, R.A.; Gulley, J.L.; Schlom, J.; Steinberg, S.M.; Liewehr, D.J.; Dahut, W.L.; Arlen, P.M. Analysis of overall survival in patients with nonmetastatic castration-resistant prostate cancer treated with vaccine, nilutamide, and combination therapy. Clin. Cancer Res. 2008, 14, 4526–4531. [Google Scholar] [CrossRef] [PubMed]
- Ogita, S.; Tejwani, S.; Heilbrun, L.; Fontana, J.; Heath, E.; Freeman, S.; Smith, D.; Baranowski, K.; Vaishampayan, U. Pilot Phase II Trial of Bevacizumab Monotherapy in Nonmetastatic Castrate-Resistant Prostate Cancer. ISRN Oncol. 2012, 2012, 242850. [Google Scholar] [CrossRef]
- Denmeade, S.R.; Isaacs, J.T. A history of prostate cancer treatment. Nat. Rev. Cancer 2002, 2, 389–396. [Google Scholar] [CrossRef]
- Yuan, X.; Cai, C.; Chen, S.; Chen, S.; Yu, Z.; Balk, S.P. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene 2014, 33, 2815–2825. [Google Scholar] [CrossRef] [PubMed]
- Attard, G.; Reid, A.H.; Yap, T.A.; Raynaud, F.; Dowsett, M.; Settatree, S.; Barrett, M.; Parker, C.; Martins, V.; Folkerd, E.; et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J. Clin. Oncol. 2008, 26, 4563–4571. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Dong, X.; Gleave, M. Molecular model for neuroendocrine prostate cancer progression. BJU Int. 2018, 122, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Imamura, Y.; Sadar, M.D. Androgen receptor targeted therapies in castration-resistant prostate cancer: Bench to clinic. Int. J. Urol. 2016, 23, 654–665. [Google Scholar] [CrossRef]
- Vlachostergios, P.J.; Puca, L.; Beltran, H. Emerging Variants of Castration-Resistant Prostate Cancer. Curr. Oncol. Rep. 2017, 19, 32. [Google Scholar] [CrossRef]
- Hormone Therapy, Radiation Therapy, and Steroid 17alpha-monooxygenase TAK-700 in Treating Patients With High-Risk Prostate Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT01546987?term=NCT01546987&draw=2&rank=1 (accessed on 20 March 2019).
- Study Comparing Orteronel Plus Prednisone in Participants With Chemotherapy-Naive Metastatic Castration-Resistant Prostate Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT01193244?term=NCT01193244&draw=2&rank=1 (accessed on 20 December 2019).
- McNeel, D.G.; Becker, J.T.; Eickhoff, J.C.; Johnson, L.E.; Bradley, E.; Pohlkamp, I.; Staab, M.J.; Liu, G.; Wilding, G.; Olson, B.M. Real-time immune monitoring to guide plasmid DNA vaccination schedule targeting prostatic acid phosphatase in patients with castration-resistant prostate cancer. Clin. Cancer Res. 2014, 20, 3692–3704. [Google Scholar] [CrossRef] [PubMed]
- Arlen, P.M.; Gulley, J.L.; Todd, N.; Lieberman, R.; Steinberg, S.M.; Morin, S.; Bastian, A.; Marte, J.; Tsang, K.Y.; Beetham, P.; et al. Antiandrogen, vaccine and combination therapy in patients with nonmetastatic hormone refractory prostate cancer. J. Urol. 2005, 174, 539–546. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Grant, C.M.; Kyprianou, N. Epithelial mesenchymal transition (EMT) in prostate growth and tumor progression. Transl. Androl. Urol. 2013, 2, 202–211. [Google Scholar]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Meng, W.; Takeichi, M. Adherens junction: Molecular architecture and regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a002899. [Google Scholar] [CrossRef] [PubMed]
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [PubMed]
- Berx, G.; van Roy, F. Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb. Perspect. Biol. 2009, 1, a003129. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, U.; Christofori, G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat. Rev. Cancer 2004, 4, 118–132. [Google Scholar] [CrossRef]
- Cakouros, D.; Raices, R.M.; Gronthos, S.; Glackin, C.A. Twist-ing cell fate: Mechanistic insights into the role of twist in lineage specification/differentiation and tumorigenesis. J. Cell. Biochem. 2010, 110, 1288–1298. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Yang, L.; Li, R.; Rodrigues, D.N.; Crespo, M.; Hsieh, J.T.; Tilley, W.D.; de Bono, J.; Selth, L.A.; Raj, G.V. Disrupting Androgen Receptor Signaling Induces Snail-Mediated Epithelial-Mesenchymal Plasticity in Prostate Cancer. Cancer Res. 2017, 77, 3101–3112. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Itsumi, M.; Takeuchi, A.; Imada, K.; Yokomizo, A.; Kuruma, H.; Inokuchi, J.; Tatsugami, K.; Uchiumi, T.; Oda, Y.; et al. Crosstalk between epithelial-mesenchymal transition and castration resistance mediated by Twist1/AR signaling in prostate cancer. Endocr. Relat. Cancer 2015, 22, 889–900. [Google Scholar] [CrossRef]
- Yochum, Z.A.; Cades, J.; Mazzacurati, L.; Neumann, N.M.; Khetarpal, S.K.; Chatterjee, S.; Wang, H.; Attar, M.A.; Huang, E.H.; Chatley, S.N.; et al. A First-in-Class TWIST1 Inhibitor with Activity in Oncogene-Driven Lung Cancer. Mol. Cancer Res. 2017, 15, 1764–1776. [Google Scholar] [CrossRef]
- Yochum, Z.A.; Cades, J.; Wang, H.; Chatterjee, S.; Simons, B.W.; O’Brien, J.P.; Khetarpal, S.K.; Lemtiri-Chlieh, G.; Myers, K.V.; Huang, E.H.; et al. Targeting the EMT transcription factor TWIST1 overcomes resistance to EGFR inhibitors in EGFR-mutant non-small-cell lung cancer. Oncogene 2019, 38, 656–670. [Google Scholar] [CrossRef]
- Martin, S.K.; Pu, H.; Penticuff, J.C.; Cao, Z.; Horbinski, C.; Kyprianou, N. Multinucleation and Mesenchymal-to-Epithelial Transition Alleviate Resistance to Combined Cabazitaxel and Antiandrogen Therapy in Advanced Prostate Cancer. Cancer Res. 2016, 76, 912–926. [Google Scholar] [CrossRef]
- Nath, D.; Li, X.; Mondragon, C.; Post, D.; Chen, M.; White, J.R.; Hryniewicz-Jankowska, A.; Caza, T.; Kuznetsov, V.A.; Hehnly, H.; et al. Abi1 loss drives prostate tumorigenesis through activation of EMT and non-canonical WNT signaling. Cell Commun. Signal. 2019, 17, 120. [Google Scholar] [CrossRef] [PubMed]
- Lau, Y.K.; Ramaiyer, M.; Johnson, D.E.; Grandis, J.R. Targeting STAT3 in Cancer with Nucleotide Therapeutics. Cancers 2019, 11, 1681. [Google Scholar] [CrossRef]
- Matsuda, T.; Nakamura, T.; Nakao, K.; Arai, T.; Katsuki, M.; Heike, T.; Yokota, T. STAT3 activation is sufficient to maintain an undifferentiated state of mouse embryonic stem cells. EMBO J. 1999, 18, 4261–4269. [Google Scholar] [CrossRef]
- Gujral, T.S.; Chan, M.; Peshkin, L.; Sorger, P.K.; Kirschner, M.W.; MacBeath, G. A noncanonical Frizzled2 pathway regulates epithelial-mesenchymal transition and metastasis. Cell 2014, 159, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Sandsmark, E.; Hansen, A.F.; Selnaes, K.M.; Bertilsson, H.; Bofin, A.M.; Wright, A.J.; Viset, T.; Richardsen, E.; Drablos, F.; Bathen, T.F.; et al. A novel non-canonical Wnt signature for prostate cancer aggressiveness. Oncotarget 2017, 8, 9572–9586. [Google Scholar] [CrossRef] [PubMed]
- Leibold, J.; Ruscetti, M.; Cao, Z.; Ho, Y.J.; Baslan, T.; Zou, M.; Abida, W.; Feucht, J.; Han, T.; Barriga, F.M.; et al. Somatic Tissue Engineering in Mouse Models Reveals an Actionable Role for WNT Pathway Alterations in Prostate Cancer Metastasis. Cancer Discov. 2020, 10, 1038–1057. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, D.T.; Zheng, Y.; Wittner, B.S.; Lee, R.J.; Zhu, H.; Broderick, K.T.; Desai, R.; Fox, D.B.; Brannigan, B.W.; Trautwein, J.; et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science 2015, 349, 1351–1356. [Google Scholar] [CrossRef] [PubMed]
- Virgo, K.S.; Rumble, R.B.; de Wit, R.; Mendelson, D.S.; Smith, T.J.; Taplin, M.-E.; Wade, J.L., 3rd; Bennett, C.L.; Scher, H.I.; Nguyen, P.L.; et al. Initial Management of Noncastrate Advanced, Recurrent, or Metastatic Prostate Cancer: ASCO Guideline Update. J. Clin. Oncol. 2021, 39, 1274–1305. [Google Scholar] [CrossRef]
- Moreira, D.; Adamus, T.; Zhao, X.; Su, Y.L.; Zhang, Z.; White, S.V.; Swiderski, P.; Lu, X.; DePinho, R.A.; Pal, S.K.; et al. STAT3 Inhibition Combined with CpG Immunostimulation Activates Antitumor Immunity to Eradicate Genetically Distinct Castration-Resistant Prostate Cancers. Clin. Cancer Res. 2018, 24, 5948–5962. [Google Scholar] [CrossRef] [PubMed]
- Stylianou, N.; Lehman, M.L.; Wang, C.; Fard, A.T.; Rockstroh, A.; Fazli, L.; Jovanovic, L.; Ward, M.; Sadowski, M.C.; Kashyap, A.S.; et al. A molecular portrait of epithelial-mesenchymal plasticity in prostate cancer associated with clinical outcome. Oncogene 2019, 38, 913–934. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Beltran, H. Clinical and Biological Features of Neuroendocrine Prostate Cancer. Curr. Oncol. Rep. 2021, 23, 15. [Google Scholar] [CrossRef] [PubMed]
- Ku, S.Y.; Gleave, M.E.; Beltran, H. Towards precision oncology in advanced prostate cancer. Nat. Rev. Urol. 2019, 16, 645–654. [Google Scholar] [CrossRef]
- Schmid, S.; Omlin, A.; Higano, C.; Sweeney, C.; Chanza, N.M.; Mehra, N.; Kuppen, M.C.P.; Beltran, H.; Conteduca, V.; Vargas Pivato de Almeida, D.; et al. Activity of Platinum-Based Chemotherapy in Patients With Advanced Prostate Cancer With and Without DNA Repair Gene Aberrations. JAMA Netw. Open 2020, 3, e2021692. [Google Scholar] [CrossRef] [PubMed]
- Pencik, J.; Wiebringhaus, R.; Susani, M.; Culig, Z.; Kenner, L. IL-6/STAT3/ARF: The guardians of senescence, cancer progression and metastasis in prostate cancer. Swiss Med. Wkly. 2015, 145, w14215. [Google Scholar] [PubMed]
- Pencik, J.; Schlederer, M.; Gruber, W.; Unger, C.; Walker, S.M.; Chalaris, A.; Marie, I.J.; Hassler, M.R.; Javaheri, T.; Aksoy, O.; et al. STAT3 regulated ARF expression suppresses prostate cancer metastasis. Nat. Commun. 2015, 6, 7736. [Google Scholar] [CrossRef]
- Moreira, D.; Sampath, S.; Won, H.; White, S.V.; Su, Y.L.; Alcantara, M.; Wang, C.; Lee, P.; Maghami, E.; Massarelli, E.; et al. Myeloid cell-targeted STAT3 inhibition sensitizes head and neck cancers to radiotherapy and T cell-mediated immunity. J. Clin. Investig. 2021, 131, e137001. [Google Scholar] [CrossRef] [PubMed]
- Alme, A.K.; Karir, B.S.; Faltas, B.M.; Drake, C.G. Blocking immune checkpoints in prostate, kidney, and urothelial cancer: An overview. Urol. Oncol. 2016, 34, 171–181. [Google Scholar] [CrossRef]
- Su, Y.L.; Banerjee, S.; White, S.V.; Kortylewski, M. STAT3 in Tumor-Associated Myeloid Cells: Multitasking to Disrupt Immunity. Int. J. Mol. Sci. 2018, 19, 1803. [Google Scholar] [CrossRef]
- Kortylewski, M.; Yu, H. Role of Stat3 in suppressing anti-tumor immunity. Curr. Opin. Immunol. 2008, 20, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Furtek, S.L.; Backos, D.S.; Matheson, C.J.; Reigan, P. Strategies and Approaches of Targeting STAT3 for Cancer Treatment. ACS Chem. Biol. 2016, 11, 308–318. [Google Scholar] [CrossRef]
- Zou, S.; Tong, Q.; Liu, B.; Huang, W.; Tian, Y.; Fu, X. Targeting STAT3 in Cancer Immunotherapy. Mol. Cancer 2020, 19, 145. [Google Scholar] [CrossRef]
- Huynh, J.; Chand, A.; Gough, D.; Ernst, M. Therapeutically exploiting STAT3 activity in cancer - using tissue repair as a road map. Nat. Rev. Cancer 2019, 19, 82–96. [Google Scholar] [CrossRef]
- Zhou, H.; Bai, L.; Xu, R.; Zhao, Y.; Chen, J.; McEachern, D.; Chinnaswamy, K.; Wen, B.; Dai, L.; Kumar, P.; et al. Structure-Based Discovery of SD-36 as a Potent, Selective, and Efficacious PROTAC Degrader of STAT3 Protein. J. Med. Chem. 2019, 62, 11280–11300. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhu, Y.; Lou, W.; Cui, Y.; Evans, C.P.; Gao, A.C. Inhibition of constitutively active Stat3 reverses enzalutamide resistance in LNCaP derivative prostate cancer cells. Prostate 2014, 74, 201–209. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gleicher, S.; Porter, B.A.; Nath, D.; Li, G.; Khanna, R.; Goldberg, H.; Kortylewski, M.; Bratslavsky, G.; Kotula, L. Novel Target Opportunities in Non-Metastatic Castrate Resistant Prostate Cancer. Cancers 2021, 13, 2426. https://doi.org/10.3390/cancers13102426
Gleicher S, Porter BA, Nath D, Li G, Khanna R, Goldberg H, Kortylewski M, Bratslavsky G, Kotula L. Novel Target Opportunities in Non-Metastatic Castrate Resistant Prostate Cancer. Cancers. 2021; 13(10):2426. https://doi.org/10.3390/cancers13102426
Chicago/Turabian StyleGleicher, Stephanie, Baylee A. Porter, Disharee Nath, Guanqun Li, Rakesh Khanna, Hanan Goldberg, Marcin Kortylewski, Gennady Bratslavsky, and Leszek Kotula. 2021. "Novel Target Opportunities in Non-Metastatic Castrate Resistant Prostate Cancer" Cancers 13, no. 10: 2426. https://doi.org/10.3390/cancers13102426
APA StyleGleicher, S., Porter, B. A., Nath, D., Li, G., Khanna, R., Goldberg, H., Kortylewski, M., Bratslavsky, G., & Kotula, L. (2021). Novel Target Opportunities in Non-Metastatic Castrate Resistant Prostate Cancer. Cancers, 13(10), 2426. https://doi.org/10.3390/cancers13102426