
B-Cell Regeneration Profile and Minimal Residual Disease Status in Bone Marrow of Treated Multiple Myeloma Patients

 ,
,  , ,
, , 
 ,
,  , , , , , and
on behalf of the EuroFlow Consortiumadd
Show full author list
, , , , , and
on behalf of the EuroFlow Consortiumadd
Show full author list
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
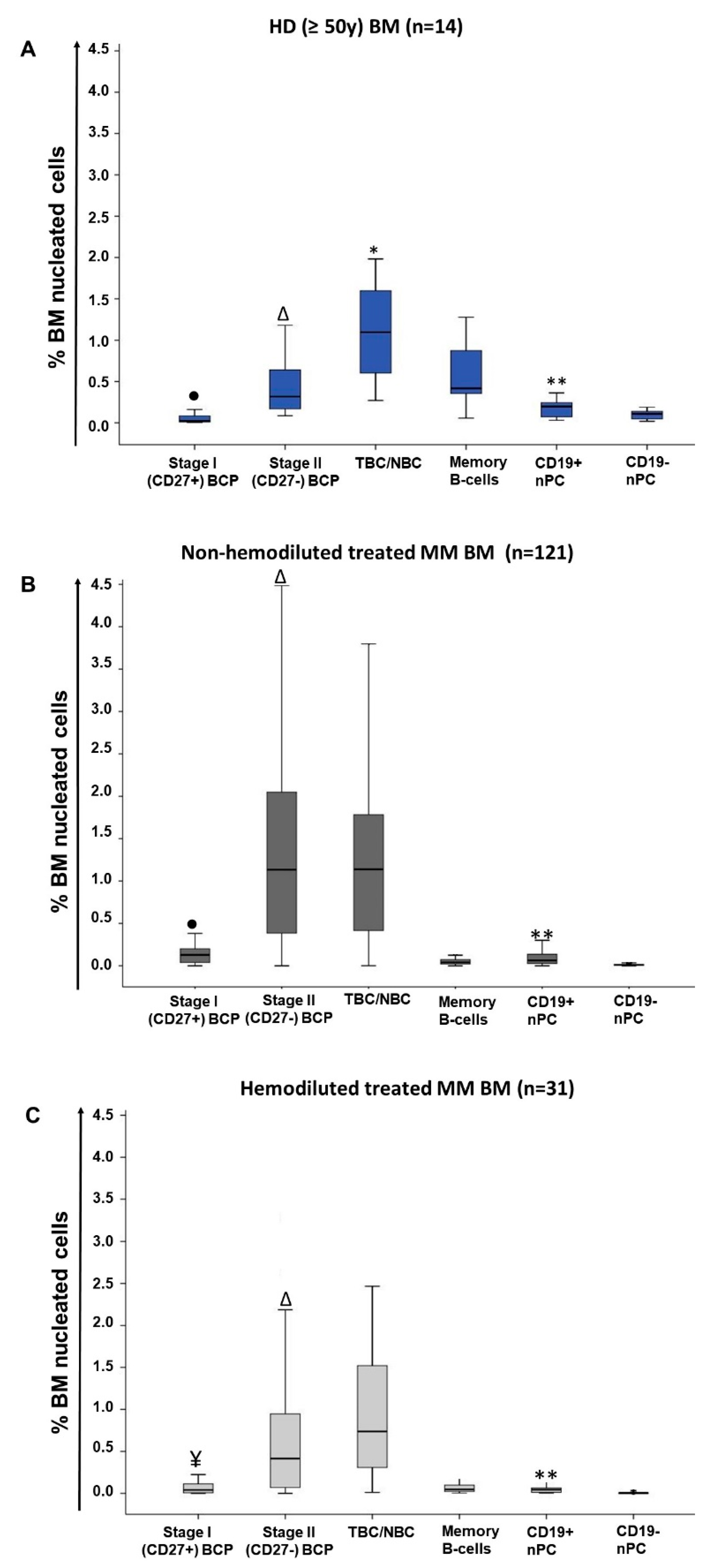
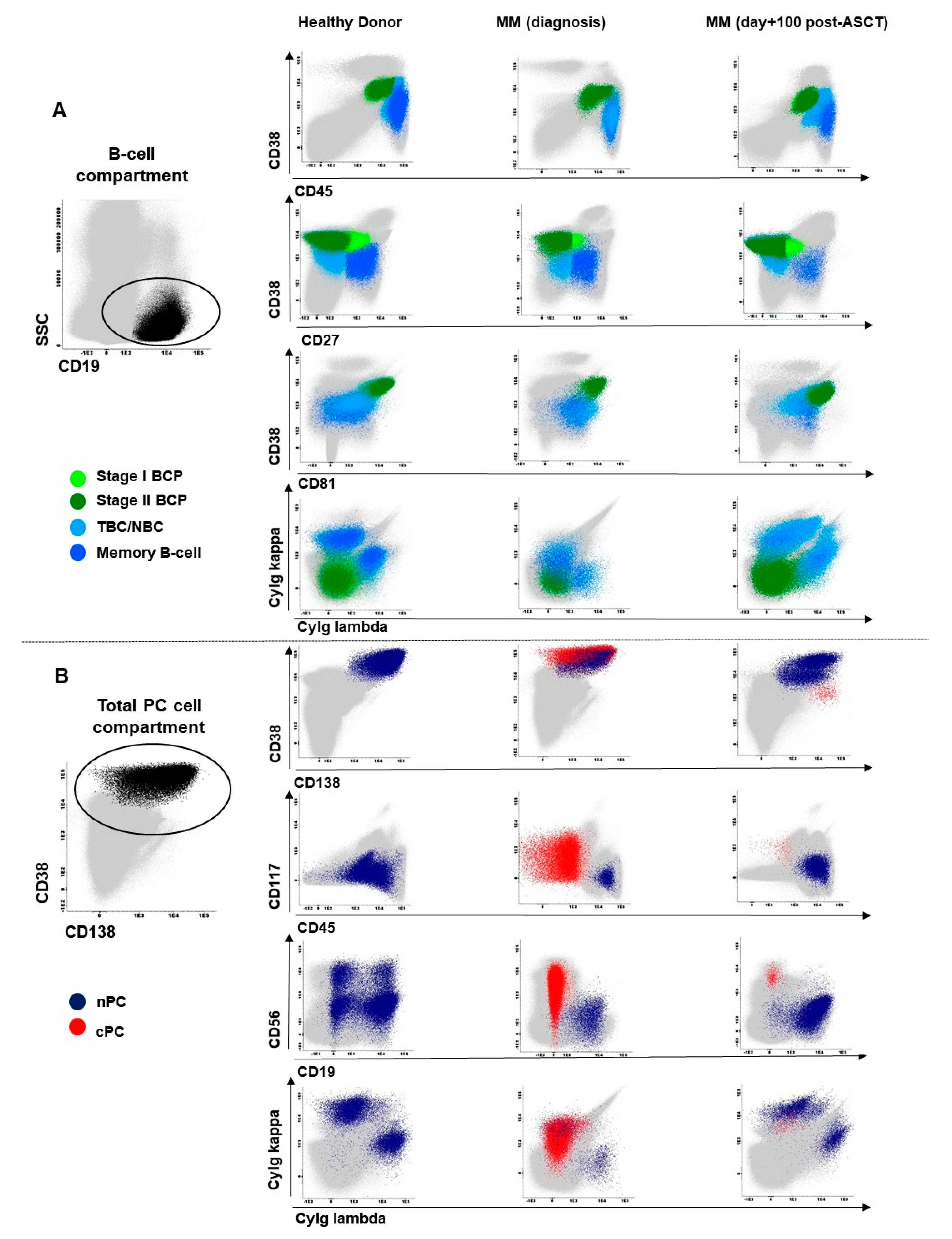
2.1. Distribution of Maturation-Associated B-Cell and PC Populations in Hemodiluted vs. Nonhemodiluted BM
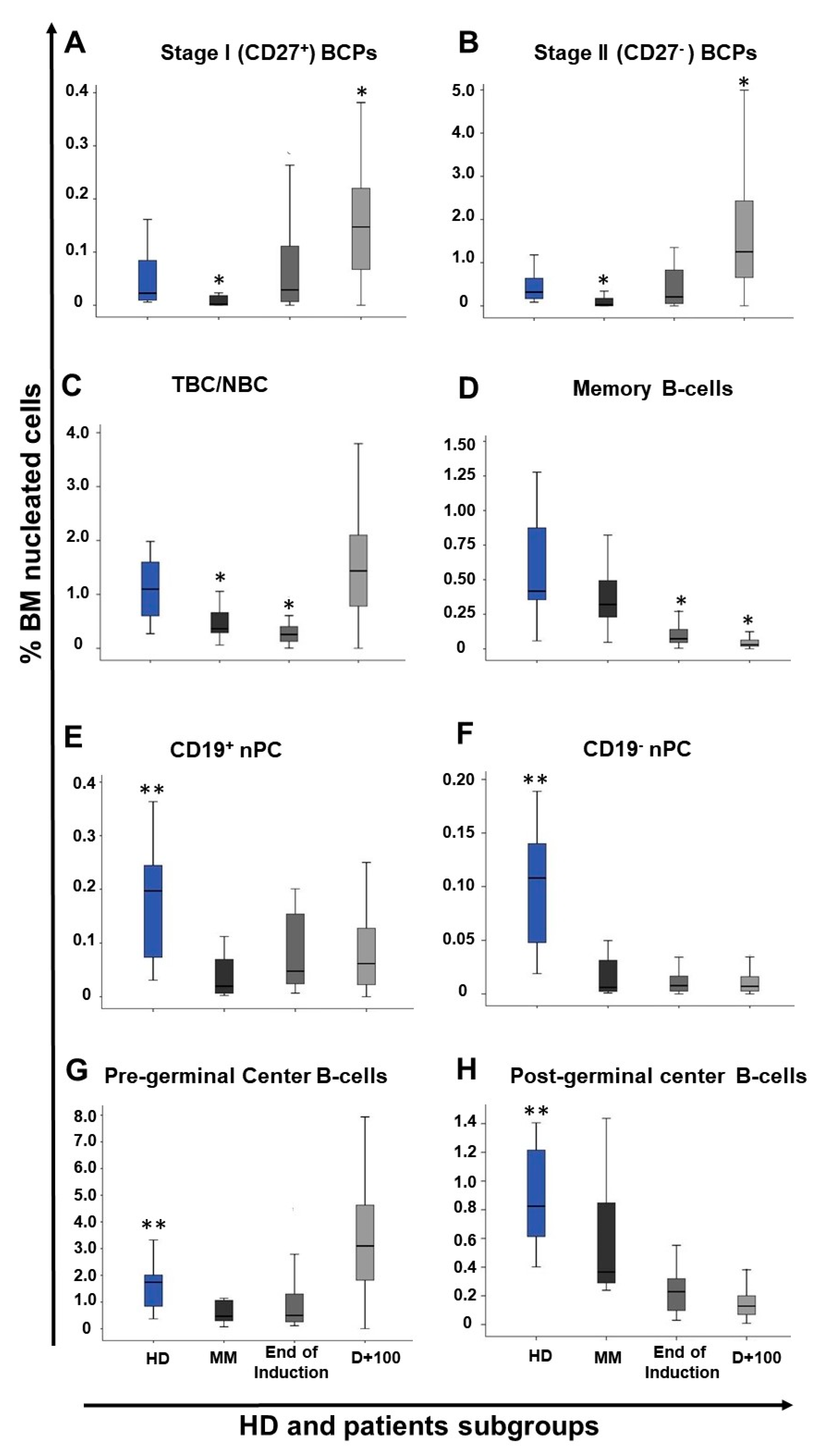
2.2. Distribution of Maturation-Associated B-Cell and nPC Populations in MM BM at Diagnosis and During Follow-Up
2.3. Impact of Response to Therapy and Type of Induction Therapy on the Distribution of Normal Residual BM B-Cell and nPC Populations in MM
2.4. Distribution of Maturation-Associated B-Cell and PC Populations in Follow-Up BM from MM Patients According to the Cytogenetic Risk at Diagnosis
3. Discussion
4. Materials and Methods
4.1. Patients, Samples, and Controls
4.2. Treatment Regimens
4.3. Immunophenotypic Studies
4.4. Molecular Cytogenetic Studies
4.5. Statistical Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kyle, R.A.; Rajkumar, S.V. Multiple myeloma. N. Engl. J. Med. 2004, 351, 1860–1873. [Google Scholar] [CrossRef]
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; Van Duin, M.; Sonneveld, P.; Mateos, M.V.; Gay, F.; Anderson, K.C. Multiple myeloma. Nat. Rev. Dis. Prim. 2017, 3, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Rajkumar, S.V. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia 2009, 23, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Borrello, I. Can we change the disease biology of multiple myeloma? Leuk. Res. 2012, 36, S3–S12. [Google Scholar] [CrossRef][Green Version]
- Kumar, S.K.; Anderson, K.C. Immune therapies in multiple myeloma. Clin. Cancer Res. 2016, 22, 5453–5460. [Google Scholar] [CrossRef]
- Lonial, S.; Durie, B.; Palumbo, A.; San-Miguel, J. Monoclonal antibodies in the treatment of multiple myeloma: Current status and future perspectives. Leukemia 2016, 30, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.D. CAR T cells and other cellular therapies for multiple myeloma: 2018 update. Am. Soc. Clin. Oncol. Educ. B 2018, e6–e15. [Google Scholar] [CrossRef]
- Bonello, F.; Mina, R.; Boccadoro, M.; Gay, F. Therapeutic monoclonal antibodies and antibody products: Current practices and development in multiple myeloma. Cancers 2020, 12, 15. [Google Scholar] [CrossRef]
- Mateos, M.-V.; Ocio, E.M.; Paiva, B.; Rosiñol, L.; Martínez-López, J.; Bladé, J.; Lahuerta, J.-J.; García-Sanz, R.; San Miguel, J.F. Treatment for patients with newly diagnosed multiple myeloma in 2015. Blood Rev. 2015, 29, 387–403. [Google Scholar] [CrossRef]
- Lahuerta, J.J.; Paiva, B.; Vidriales, M.B.; Cordón, L.; Cedena, M.T.; Puig, N.; Martinez-Lopez, J.; Rosiñol, L.; Gutierrez, N.C.; Martín-Ramos, M.L.; et al. Depth of response in multiple myeloma: A pooled analysis of three PETHEMA/GEM clinical trials. J. Clin. Oncol. 2017, 35, 2900–2910. [Google Scholar] [CrossRef] [PubMed]
- Paiva, B.; Vidriales, M.; Cervero, J.; Mateo, G.; Perez, J.; Montalban, M.A.; Sureda, A.; Montejano, L.; Gutierrez, N.; de Coca, A.G.; et al. The presence of residual normal plasma cells by multiparameter flow cytometry in the bone marrow of symptomatic multiple myeloma patients at diagnosis identifies a good prognostic subgroup. Haematol. Hematol. J. 2009, 94, 154–155. [Google Scholar] [CrossRef]
- Paiva, B.; Pérez-Andrés, M.; Vídriales, M.B.; Almeida, J.; De Las Heras, N.; Mateos, M.V.; López-Corral, L.; Gutiérrez, N.C.; Blanco, J.; Oriol, A.; et al. Competition between clonal plasma cells and normal cells for potentially overlapping bone marrow niches is associated with a progressively altered cellular distribution in MGUS vs myeloma. Leukemia 2011, 25, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Zent, C.S.; Wilson, C.S.; Tricot, G.; Jagannath, S.; Siegel, D.; Desikan, K.R.; Munshi, N.; Bracy, D.; Barlogie, B.; Butch, A.W. Oligoclonal protein bands and Ig isotype switching in multiple myeloma treated with high-dose therapy and hematopoietic cell transplantation. Blood 1998, 91, 3518–3523. [Google Scholar] [CrossRef]
- Rawstron, A.C.; Davies, F.E.; Owkn, R.G.; English, A.; Pratt, G.; Child, J.A.; Jack, A.S.; Morgan, G.J. B-lymphocyte suppression in multiple myeloma is a reversible phenomenon specific to normal B-cell progenitors and plasma cell precursors. Br. J. Haematol. 1998, 100, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Byrne, E.; Naresh, K.N.; Giles, C.; Rahemtulla, A. Excess bone marrow B-cells in patients with multiple myeloma achieving complete remission following autologous stem cell transplantation is a biomarker for improved survival. Br. J. Haematol. 2011, 155, 509–511. [Google Scholar] [CrossRef]
- González-Calle, V.; Cerdá, S.; Labrador, J.; Sobejano, E.; González-Mena, B.; Aguilera, C.; Ocio, E.M.; Vidriales, M.B.; Puig, N.; Gutiérrez, N.C.; et al. Recovery of polyclonal immunoglobulins one year after autologous stem cell transplantation as a long-term predictor marker of progression and survival in multiple myeloma. Haematologica 2017, 102, 922–931. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Kumar, S. Multiple myeloma: Diagnosis and treatment. Mayo Clin. Proc. 2016, 91, 101–119. [Google Scholar] [CrossRef]
- Nucci, M.; Anaissie, E. Infections in patients with multiple myeloma in the era of high-dose therapy and novel agents. Clin. Infect. Dis. 2009, 49, 1211–1225. [Google Scholar] [CrossRef]
- Teh, B.W.; Harrison, S.J.; Worth, L.J.; Thursky, K.A.; Slavin, M.A. Infection risk with immunomodulatory and proteasome inhibitor–based therapies across treatment phases for multiple myeloma: A systematic review and meta-analysis. Eur. J. Cancer 2016, 67, 21–37. [Google Scholar] [CrossRef]
- Teh, B.W.; Harrison, S.J.; Pellegrini, M.; Thursky, K.A.; Worth, L.J.; Slavin, M.A. Changing treatment paradigms for patients with plasma cell myeloma: Impact upon immune determinants of infection. Blood Rev. 2014, 28, 75–86. [Google Scholar] [CrossRef]
- De Magalhães, R.J.P.; Vidriales, M.B.; Paiva, B.; Fernandez-Gimenez, C.; García-Sanz, R.; Mateos, M.V.; Gutierrez, N.C.; Lecrevisse, Q.; Blanco, J.F.; Hernández, J.; et al. Analysis of the immune system of multiple myeloma patients achieving long-term disease control by multidimensional flow cytometry. Haematologica 2013, 98, 79–86. [Google Scholar] [CrossRef]
- Tovar, N.; de Larrea, C.F.; Aróstegui, J.I.; Cibeira, M.T.; Rosiñol, L.; Rovira, M.; Elena, M.; Filella, X.; Yagüe, J.; Bladé, J. Natural history and prognostic impact of oligoclonal humoral response in patients with multiple myeloma after autologous stem cell transplantation: Long-term results from a single institution. Haematologica 2013, 98, 1142–1146. [Google Scholar] [CrossRef] [PubMed]
- Paiva, B.; Cedena, M.-T.; Puig, N.; Arana, P.; Vidriales, M.-B.; Cordon, L.; Flores-Montero, J.; Gutierrez, N.C.; Martín-Ramos, M.-L.; Martinez-Lopez, J.; et al. Minimal residual disease monitoring and immune profiling using second generation flow cytometry in elderly multiple myeloma. Blood 2016, 127. [Google Scholar] [CrossRef] [PubMed]
- Arteche-López, A.; Kreutzman, A.; Alegre, A.; Sanz Martín, P.; Aguado, B.; González-Pardo, M.; Espiño, M.; Villar, L.M.; García Belmonte, D.; De La Cámara, R.; et al. Multiple myeloma patients in long-term complete response after autologous stem cell transplantation express a particular immune signature with potential prognostic implication. Bone Marrow Transpl. 2017, 52, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Tsou, P.; Katayama, H.; Ostrin, E.J.; Hanash, S.M. The emerging role of b cells in tumor immunity. Cancer Res. 2016, 76, 5591–5601. [Google Scholar] [CrossRef] [PubMed]
- Kroeger, D.R.; Milne, K.; Nelson, B.H. Tumor-infiltrating plasma cells are associated with tertiary lymphoid structures, cytolytic T-cell responses, and superior prognosis in ovarian cancer. Clin. Cancer Res. 2016, 22, 3005–3015. [Google Scholar] [CrossRef]
- Sharonov, G.V.; Serebrovskaya, E.O.; Yuzhakova, D.V.; Britanova, O.V.; Chudakov, D.M. B cells, plasma cells and antibody repertoires in the tumour microenvironment. Nat. Rev. Immunol. 2020, 20, 294–307. [Google Scholar] [CrossRef]
- San Miguel, J.F.; Almeida, J.; Mateo, G.; Blade, J.; Lopez-Berges, C.; Caballero, D.; Hernandez, J.; Moro, M.J.; Fernandez-Calvo, J.; Diaz-Mediavilla, J.; et al. Immunophenotypic evaluation of the plasma cell compartment in multiple myeloma: A tool for comparing the efficacy of different treatment strategies and predicting outcome. Blood 2002, 99, 1853–1856. [Google Scholar] [CrossRef]
- Rueff, J.; Medinger, M.; Heim, D.; Passweg, J.; Stern, M. Lymphocyte subset recovery and outcome after autologous hematopoietic stem cell transplantation for plasma cell myeloma. Biol. Blood Marrow Transpl. 2014, 20, 896–899. [Google Scholar] [CrossRef]
- Ho, C.M.; Mccarthy, P.L.; Wallace, P.K.; Zhang, Y.; Fora, A.; Mellors, P.; Tario, J.D.; Mccarthy, B.L.S.; Chen, G.L.; Holstein, S.A.; et al. Immune signatures associated with improved progression-free and overall survival for myeloma patients treated with AHSCT. Blood Adv. 2017, 1, 1056–1066. [Google Scholar] [CrossRef] [PubMed]
- Flores-Montero, J.; Sanoja-Flores, L.; Paiva, B.; Puig, N.; García-Sánchez, O.; Böttcher, S.; Van Der Velden, V.H.J.; Pérez-Morán, J.J.; Vidriales, M.B.; García-Sanz, R.; et al. Next generation flow for highly sensitive and standardized detection of minimal residual disease in multiple myeloma. Leukemia 2017, 31, 2094–2103. [Google Scholar] [CrossRef] [PubMed]
- Ciudad, J.; Orfao, A.; Vidriales, B.; Macedo, A.; Martínez, A.; González, M.; López-Berges, M.C.; Valverde, B.; San Miguel, J.F. Immunophenotypic analysis of CD19+ precursors in normal human adult bone marrow: Implications for minimal residual disease detection. Haematologica 1998, 83, 1069–1075. [Google Scholar]
- Delgado, J.A.; Guillén-Grima, F.; Moreno, C.; Panizo, C.; Pérez-Robles, C.; Mata, J.J.; Moreno, L.; Arana, P.; Chocarro, S.; Merino, J. A simple flow-cytometry method to evaluate peripheral blood contamination of bone marrow aspirates. J. Immunol. Methods 2017, 442, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Rawstron, A.C.; Orfao, A.; Beksac, M.; Bezdickova, L.; Brooimans, R.A.; Bumbea, H.; Dalva, K.; Fuhler, G.; Gratama, J.; Hose, D.; et al. Report of the European Myeloma Network on multiparametric flow cytometry in multiple myeloma and related disorders. Haematologica 2008, 93, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Bhaskar, A.; Kumar, L.; Sharma, A.; Jain, P. Flow cytometric immunophenotyping and minimal residual disease analysis in multiple myeloma. Am. J. Clin. Pathol. 2009, 132, 728–732. [Google Scholar] [CrossRef]
- Arroz, M.; Came, N.; Lin, P.; Chen, W.; Yuan, C.; Lagoo, A.; Monreal, M.; de Tute, R.; Vergilio, J.A.; Rawstron, A.C.; et al. Consensus guidelines on plasma cell myeloma minimal residual disease analysis and reporting. Cytom. Part B Clin. Cytom. 2016, 90, 31–39. [Google Scholar] [CrossRef]
- Orfao, A.; Escribano, L.; Villarrubia, J.; Velasco, J.L.; Cerveró, C.; Ciudad, J.; Navarro, J.L.; San Miguel, J.F. Flow cytometric analysis of mast cells from normal and pathological human bone marrow samples: Identification and enumeration. Am. J. Pathol. 1996, 149, 1493–1499. [Google Scholar] [PubMed]
- Escribano, L.; Orfao, A.; Díaz-Agustin, B.; Villarrubia, J.; Cerveró, C.; López, A.; Marcos, M.A.; Bellas, C.; Fernández-Cañadas, S.; Cuevas, M.; et al. Indolent systemic mast cell disease in adults: Immunophenotypic characterization of bone marrow mast cells and its diagnostic implications. Blood 1998, 91, 2731–2736. [Google Scholar] [CrossRef]
- Escribano, L.; Díaz-Agustín, B.; Bellas, C.; Navalón, R.; Nuñez, R.; Sperr, W.R.; Schernthaner, G.H.; Valent, P.; Orfao, A. Utility of flow cytometric analysis of mast cells in the diagnosis and classification of adult mastocytosis. Leuk. Res. 2001, 25, 563–570. [Google Scholar] [CrossRef]
- Brooimans, R.A.; Kraan, J.; van Putten, W.; Cornelissen, J.J.; Löwenberg, B.; Gratama, J.W. Flow cytometric differential of leukocyte populations in normal bone marrow: Influence of peripheral blood contamination. Cytom. Part B Clin. Cytom. 2009, 76, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Gener, G.; Espasa, A.; Raya, M.; Vergara, S.; Juncà, J.; Sorigue, M. Brief communication: Distribution of bone marrow cell subsets and hemodilution in patients with acute leukemia. Int. J. Lab. Hematol. 2020, 42, e192–e195. [Google Scholar] [CrossRef] [PubMed]
- Linton, P.J.; Dorshkind, K. Age-related changes in lymphocyte development and function. Nat. Immunol. 2004, 5, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Radbruch, A.; Muehlinghaus, G.; Luger, E.O.; Inamine, A.; Smith, K.G.; Dorner, T.; Hiepe, F. Competence and competition: The challenge of becoming a long-lived plasma cell. Nat. Rev. Immunol. 2006, 6, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Tokoyoda, K.; Egawa, T.; Sugiyama, T.; Choi, B.; Nagasawa, T. Cellular niches controlling B lymphocyte behavior within bone marrow during development. Immunity 2004, 20, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Mayack, S.R.; Shadrach, J.L.; Kim, F.S.; Wagers, A.J. Systemic signals regulate ageing and rejuvenation of blood stem cell niches. Nature 2010, 463, 495–500. [Google Scholar] [CrossRef]
- Perez-Andres, M.; Paiva, B.; Nieto, W.G.; Caraux, A.; Schmitz, A.; Almeida, J.; Vogt, R.F.; Marti, G.E.; Rawstron, A.C.; Van Zelm, M.C.; et al. Human peripheral blood B-Cell compartments: A crossroad in B-cell traffic. Cytom. Part B Clin. Cytom. 2010, 78, 47–60. [Google Scholar] [CrossRef]
- Tsujimoto, T.; Lisukov, A.; Huang, N.; Mahmoud, M.S.; Kawano, M.M. Plasma cells induce apoptosis. Blood 1996, 87, 3375–3383. [Google Scholar] [CrossRef]
- Theunissen, P.M.J.; van den Branden, A.; Van Der Sluijs-Gelling, A.; De Haas, V.; Beishuizen, A.; van Dongen, J.J.M.; Van Der Velden, V.H.J. Understanding the reconstitution of the B-cell compartment in bone marrow and blood after treatment for B-cell precursor acute lymphoblastic leukaemia. Br. J. Haematol. 2017, 178, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Zepeda, V.; Duggan, P.; Neri, P.; Chaudhry, A.; Tay, J.; Bahlis, N.J. Immunoglobulin recovery after ASCT for patients with multiple myeloma: Impact on overall survival and progression-free survival. Blood 2016, 128, 2263. [Google Scholar] [CrossRef]
- Tete, S.M.; Bijl, M.; Sahota, S.S.; Bos, N.A. Immune defects in the risk of infection and response to vaccination in monoclonal gammopathy of undetermined significance and multiple myeloma. Front. Immunol. 2014, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Paiva, B.; Anderson, K.C.; Durie, B.; Landgren, O.; Moreau, P.; Munshi, N.; Lonial, S.; Bladé, J.; Kyle, R.A.; et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016, 17, e328–e346. [Google Scholar] [CrossRef]
- Kalina, T.; Flores-Montero, J.; Van Der Velden, V.H.J.; Martin-Ayuso, M.; Böttcher, S.; Ritgen, M.; Almeida, J.; Lhermitte, L.; Asnafi, V.; Mendonça, A.; et al. EuroFlow standardization of flow cytometer instrument settings and immunophenotyping protocols. Leukemia 2012, 26, 1986–2010. [Google Scholar] [CrossRef]
- Nilsson, A.; De Milito, A.; Mowafi, F.; Winberg, G.; Björk, O.; Wolpert, E.Z.; Chiodi, F. Expression of CD27-CD70 on early B cell progenitors in the bone marrow: Implication for diagnosis and therapy of childhood ALL. Exp. Hematol. 2005, 33, 1500–1507. [Google Scholar] [CrossRef] [PubMed]
- Vaskova, M.; Fronkova, E.; Starkova, J.; Kalina, T.; Mejstrikova, E.; Hrusak, O. CD44 and CD27 delineate B-precursor stages with different recombination status and with an uneven distribution in nonmalignant and malignant hematopoiesis. Tissue Antigens 2008, 71, 57–66. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Population (%) | Treated MM (n = 152) | |||
|---|---|---|---|---|
| HD (n = 14) | MM at Diagnosis (n = 25) | Non-hemodiluted BM (n = 121) | Hemodiluted BM (n = 31) | |
| Total B-cells | 2.6 (1–4.6) | 1.2 a (0.4–3.3) | 2.5 b (0.05 -11.4) | 1.5 c (0.04–7.9) |
| Pre-germinal center B-cells | 1.7 (0.4–3.3) | 0.5 a (0.07–2.2) | 2.4 a,b (0.003–11.2) | 1.3 b,c (0.03–7.8) |
| BCP | 0.3 (0.09–1.7) | 0.01 a (<0.0002–0.8) | 1.3 a,b (<0.0002–10.3) | 0.5 b, c (<0.0002–6.1) |
| Stage I BCP | 0.02 (0.006–0.2) | 0.0005 a (<0.0002–0.1) | 0.1a,b (<0.0002–0.9) | 0.04 b,c (<0.0002–0.6) |
| Stage II BCP | 0.3 (0.09–1.5) | 0.01 a (<0.0002–0.7) | 1.1 a,b (<0.0002–9.4) | 0.4 b,c (<0.0002–5.5) |
| Stage I/stage II BCP ratio | 0.1 (0.04–0.2) | 0.02 a (0–0.2) | 0.09 b (<0.0002 -1.6) | 0.1 b (<0.0002–1.2) |
| Transitional/naive B-cells | 1.1 (0.3–2) | 0.4 a (0.05–1.9) | 1.1 b (0.0008–5.7) | 0.7 (0.01–4,4) |
| Post-germinal center B-cells | 0.8 0.4–1.4) | 0.4 a (0.1–1.6) | 0.1 a,b (0.0008–0.9) | 0.1 a,b (0.01–1) |
| Memory B-cells | 0.4 (0.06–1.3) | 0.3 (0.05–1.5) | 0.04 a,b (0.0005–0.5) | 0.05 a (0.003–1) |
| nPC | 0.3 (0.08–0.9) | 0.04 a (0.005–0.5) | 0.08 a,b (0.002–0.8) | 0.05 a,c (0.003–0.4) |
| CD19+ nPC | 0.2 (0.03–0.6) | 0.03 a (0.002–0.4) | 0.06 a,b (<0.0002–0.8) | 0.04 a,c (0.002–0.4) |
| CD19− nPC | 0.1 (0.02–0.3) | 0.006 a (<0.0002–0.08) | 0.007 a (<0.0002–0.2) | 0.003 a,b,c (<0.0002–0.08) |
| CD19+/CD19− nPC ratio | 2 (0.7–8.6) | 2.2 (0–18.6) | 7.5 a,b (0–146) | 9.4 a,b (0–45) |
| Mature B-cells ¥ | 1.5 (0.4–3) | 0.8 a (0.1–2.8) | 1.2 (0.004–5.9) | 0.9 a (0.01–4.5) |
| BCP/mature B-cell ratio | 0.2 (0.08–1.6) | 0.005 a (0–2.5) | 1 a,b (0–30.2) | 0.4 b,c (0–5.9) |
| Cell population (%) | End of Induction (n = 27) | Post ASCT (day+100) (n = 94) | ||||
|---|---|---|---|---|---|---|
| Non-CR (n = 7) | sCR/CR MRD+ (n = 13) | sCR/CR MRD- (n = 7) | Non-CR (n = 38) | sCR/CR MRD+ (n = 27) | sCR/CR MRD- (n = 29) | |
| Total B-cells | 1.3 (0.3–2.9) | 0.7 (0.3–2.1) | 0.9 (0.5–5) | 3.7 (0.06–11.4) | 4.1 (1.1–10.7) | 2.6 (0.05–9.3) |
| Pre-germinal center B-cells | 0.8 a (0.2–2.9) | 0.3 (0.1–1.9) | 0.7 (0.4–4.5) | 3.6 (0.05–11.2) | 3.9 (0.5–10.5) | 2.6 (0.003–9.1) |
| BCP | 0.5 (0.05–2.3) | 0.1 (<0.0002–1.4) | 0.3 (0.01–2) | 1.7 (0.04–10.3) | 1.9 b (0.06–9.5) | 1.3 (0.002–4.1) |
| Stage I BCP | 0.1 (0.005–0.3) | 0.008 (<0.0002–0.2) | 0.02 (0.001–0.7) | 0.2 (0.01–0.9) | 0.2 (0.002–0.8) | 0.1 (<0.0002–0.4) |
| Stage II BCP | 0.2 (0.05–2.1) | 0.1 (<0.0002–1.2) | 0.3 (0.01–1.4) | 1.5 (0.03–9.4) | 1.7 b (0.06–8.6) | 1.1 (0.002–3.8) |
| Stage I/stage II BCP ratio | 0.1 (0.07–1.6) | 0.1 (<0.0002–0.8) | 0.09 (0.03–0.5) | 0.09 (0.02–0.6) | 0.09 (0.01–0.5) | 0.08 (<0.0002–1.2) |
| Transitional/naive B-cells | 0.3 a (0.2–0.6) | 0.1 (0.004–0.4) | 0.3 (0.1–2.5) | 1.5 (0.09–5.7) | 1.3 (0.3–4.9) | 1.5 (0.0008–5.7) |
| Post-germinal center B-cells | 0.2 (0.08–0.5) | 0.2 (0.03–0.6) | 0.2 (0.08–0.6) | 0.1 (0.03–0.9) | 0.2 a,b (0.02–0.6) | 0.1 (0.009–0.5) |
| Memory B-cells | 0.08 (0.05–0.5) | 0.07 (0.005–0.5) | 0.08 (0.05–0.3) | 0.03 (0.005–0.2) | 0.03 (0.006–0.5) | 0.03 (0.0005–0.2) |
| nPC | 0.03 (0.01–0.2) | 0.06 (0.008–0.4) | 0.1 (0.04–0.4) | 0.07 (0.004–0.8) | 0.1 a,b (0.002–0.6) | 0.07 (0.008–0.3) |
| CD19+ nPC | 0.03 (0.01–0.2) | 0.05 (0.007–0.4) | 0.09 (0.03–0.4) | 0.05 (<0.0002–0.8) | 0.09 a,b (0.002–0.4) | 0.06 (0.0008–0.3) |
| CD19− nPC | 0.003 (≤0.0002–0.02) | 0.01 (0.006–0.04) | 0.01 (0.003–0.07) | 0.006 (<0.0002–0.1) | 0.009 (<0.0002–0.2) | 0.01 (0.001–0.06) |
| CD19+/CD19− nPC ratio | 8.4 (0–72.2) | 5.4 (2.7–19.3) | 4.8 (1.3–147) | 11.3 (0–42) | 9.5 (0–39) | 5 (0–21.5) |
| Mature B-cells ¥ | 0.5 a (0.3–0.8) | 0.2 (0.009–0.7) | 0.4 (0.3–2.6) | 1.5 (0.01–5.8) | 1.4 (0.3–5) | 1.5 (0.004–5.9) |
| BCP/Mature B-cell ratio | 0.6 (0.1–5.9) | 0.6 (0–30.2) | 0.8 (0.03–3.4) | 1 (0–13.3) | 1.7 (0–8.5) | 0.9 (0.08–3.5) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendonça de Pontes, R.; Flores-Montero, J.; Sanoja-Flores, L.; Puig, N.; Pessoa de Magalhães, R.J.; Corral-Mateos, A.; Salgado, A.B.; García-Sánchez, O.; Pérez-Morán, J.; Mateos, M.-V.; et al. B-Cell Regeneration Profile and Minimal Residual Disease Status in Bone Marrow of Treated Multiple Myeloma Patients. Cancers 2021, 13, 1704. https://doi.org/10.3390/cancers13071704
Mendonça de Pontes R, Flores-Montero J, Sanoja-Flores L, Puig N, Pessoa de Magalhães RJ, Corral-Mateos A, Salgado AB, García-Sánchez O, Pérez-Morán J, Mateos M-V, et al. B-Cell Regeneration Profile and Minimal Residual Disease Status in Bone Marrow of Treated Multiple Myeloma Patients. Cancers. 2021; 13(7):1704. https://doi.org/10.3390/cancers13071704
Chicago/Turabian StyleMendonça de Pontes, Robéria, Juan Flores-Montero, Luzalba Sanoja-Flores, Noemi Puig, Roberto J. Pessoa de Magalhães, Alba Corral-Mateos, Anna Beatriz Salgado, Omar García-Sánchez, José Pérez-Morán, Maria-Victoria Mateos, and et al. 2021. "B-Cell Regeneration Profile and Minimal Residual Disease Status in Bone Marrow of Treated Multiple Myeloma Patients" Cancers 13, no. 7: 1704. https://doi.org/10.3390/cancers13071704
APA StyleMendonça de Pontes, R., Flores-Montero, J., Sanoja-Flores, L., Puig, N., Pessoa de Magalhães, R. J., Corral-Mateos, A., Salgado, A. B., García-Sánchez, O., Pérez-Morán, J., Mateos, M.-V., Burgos, L., Paiva, B., te Marvelde, J., van der Velden, V. H. J., Aguilar, C., Bárez, A., García-Mateo, A., Labrador, J., Leoz, P., ... Orfao, A., on behalf of the EuroFlow Consortium. (2021). B-Cell Regeneration Profile and Minimal Residual Disease Status in Bone Marrow of Treated Multiple Myeloma Patients. Cancers, 13(7), 1704. https://doi.org/10.3390/cancers13071704