Simple Summary
Multiple myeloma is a deadly blood cancer, but fortunately drug development has substantially prolonged the lifespan of patients to average more than a decade after diagnosis with optimal therapy. As a result, the population of patients living with multiple myeloma has grown considerably. Through its course, patients suffer repeated relapses for which they require new lines of treatment. Currently, the key drug classes for treatment are immunomodulatory drugs, proteasome inhibitors, and monoclonal antibodies. The goal of this review is to summarize the understanding of the problem of resistance to these drugs, which is ultimately responsible for patient fatality. In addition, we will focus on how new agents that are promising in clinical trials overcome resistance.
Abstract
Multiple myeloma is a malignant plasma cell neoplasm that remains incurable and is ultimately fatal when patients acquire multi-drug resistance. Thus, advancing our understanding of the mechanisms behind drug resistance in multi-relapsed patients is critical for developing better strategies to extend their lifespan. Here, we review the understanding of resistance to the three key drug classes approved for multiple myeloma treatment: immunomodulatory drugs, proteasome inhibitors, and monoclonal antibodies. We consider how the complex, heterogenous biology of multiple myeloma may influence the acquisition of drug resistance and reflect on the gaps in knowledge where additional research is needed to improve our treatment approaches. Fortunately, many agents are currently being evaluated preclinically and in clinical trials that have the potential to overcome or delay drug resistance, including next-generation immunomodulatory drugs and proteasome inhibitors, novel small molecule drugs, chimeric antigen receptor T cells, antibody-drug conjugates, and bispecific antibodies. For each class, we discuss the potential of these strategies to overcome resistance through modifying agents within each class or new classes without cross-resistance to currently available drugs.
1. Introduction
Multiple myeloma (MM) is a malignant plasma cell (PC) neoplasm which is controllable initially with treatment, but ultimately the development of drug resistant disease leads to patient mortality. MM afflicts more than 30,000 Americans each year and is the second most prevalent hematologic malignancy [1]. Over the last decade the incidence of MM has continued to increase, with an estimated 12,830 deaths predicted in 2020 [1]. MM is characterized by the clonal proliferation of plasma cells, predominantly in the bone marrow, associated with overproduction of monoclonal immunoglobulin (Ig) known as M-protein. Complications of MM include bone lesions, hypercalcemia, renal failure, cytopenias, and immune suppression [2]. Treatment options for MM have significantly improved over the past two decades with the emergence of many new anti-myeloma drugs, increasing the average life expectancy from three-to-four years to seven-to-eight years for standard-risk disease [2]. However, patients still suffer repeated relapses and inevitably develop multi-drug resistance.
Multiple myeloma is a complex and heterogeneous disease. The underlying genetic abnormalities that initiate and promote MM are extraordinarily diverse. Cytogenetic abnormalities in multiple myeloma that confer poorer outcomes include translocations t (4;14), t (14;16), and t (14;20), deletion of chromosome 17p and gain of chromosome 1q. In addition, accumulation of genetic mutations increases over the disease course and occurs more rapidly in cytogenetically high-risk patients [3]. However, although these genetic abnormalities have prognostic value, they do not correlate with initial disease response. Recently, subclonal heterogeneity has become appreciated to occur in MM and tends to evolve with disease progression through selection of more drug-resistant or aggressive subclones [4,5]. Spatial heterogeneity adds further complexity, as subclones can vary in distribution across simultaneous biopsy sites [6]. Although clonal evolution parallels drug resistance development, how these are mechanistically linked remains unclear. Irrespective of genetic risk factors, multi-drug resistance development is a critical issue, as patients who are triple-class refractory to proteasome inhibitors (PIs), immunomodulatory drugs (IMiDs), and anti-CD38 monoclonal antibodies have only 5.6 months median overall survival [7]. Consequently, triple-refractory patients are in dire need of new therapies which can circumvent triple-class resistance.
Acquired drug resistance, leading to triple-class refractory MM, is the root cause of relapsed disease and remains the greatest obstacle to prolonging patients’ lives. With many drugs available, patients are cycled through combinations of two to four agents at a time. These regimens include at least sixteen individual agents in seven major drug classes. This diverse toolbox has created a complex situation where patients develop drug resistance to the available agents in a gradual manner that varies considerably from patient-to-patient. Advancing our understanding of multi-drug resistance is critical for the development of more effective strategies for the treatment of relapsed/refractory MM (RRMM). This starts with clear characterization of the underlying mechanisms of resistance to each drug class. In this review, we focus on the development of drug resistance to the three key therapeutic classes in MM treatment: Immunomodulatory Drugs (IMiDs), Proteasome Inhibitors (PIs), and immunotherapies (Table 1). We will also touch on the resistance to steroids and chemotherapies. We summarize the current knowledge of the genetic, molecular, and cellular mechanisms of resistance. To develop a complete picture of the current state of MM drug resistance, we will also consider lessons from ongoing clinical trials and examine how current drug development for MM may overcome drug resistance and improve the bleak outcome of triple-class refractory MM.

Table 1.
Resistance mechanisms to targeted therapies in multiple myeloma.
2. Immunomodulatory Drugs
IMiDs have revolutionized MM treatment in the past 20 years. The first IMiD, thalidomide, was initially used as an antiemetic for pregnant women, but unfortunately had teratogenic effects. Serendipitously, it was later discovered to be cytotoxic to MM cells and was repurposed for MM treatment [29]. Due to some undesirable side effects, thalidomide has largely been superseded by more efficacious and tolerable derivatives. Chemical modification led to second generation lenalidomide, and structural elements of both thalidomide and lenalidomide were combined to form pomalidomide, the most potent of the IMiDs to be FDA-approved [30]. Lenalidomide is a key part of standard of care drug combinations in the diagnosis setting and as maintenance [31]. In RRMM, multiple lenalidomide and pomalidomide-based regimens are FDA-approved.
2.1. IMiD Mechanism of Action
Only relatively recently did the unique anti-myeloma mechanism of the IMiDs come to light. IMiDs bind to Cereblon (CRBN), a protein which dictates the substrate specificity of the CRL4CRBN E3 ubiquitin ligase [32,33]. IMiD binding to CRBN alters its substrate affinity and leads CRL4CRBN to target a new set of substrates for proteasomal degradation [34]. The key neosubstrates in MM cells are Ikaros (IKZF1) and Aiolos (IKZF3) [35,36]. These transcription factors (TFs) regulate cell fate decisions in normal lymphopoiesis and PC development [37]. IKZF1 and IKZF3 upregulate the TFs interferon regulatory factor 4 (IRF4) and c-MYC (MYC), which form a positive autoregulatory loop necessary for MM survival and proliferation [38]. These four TFs can be collectively referred to as the Ikaros axis, and knockdown experiments support that IRF4 and MYC dependence is the means by which IMiDs ultimately exert their activity [38,39]. In summary, IMiDs act through promoting the rapid degradation of IKZF1 and IKZF3 via CRBN-dependent ubiquitination, leading to downregulation of IRF4 and MYC (Figure 1).
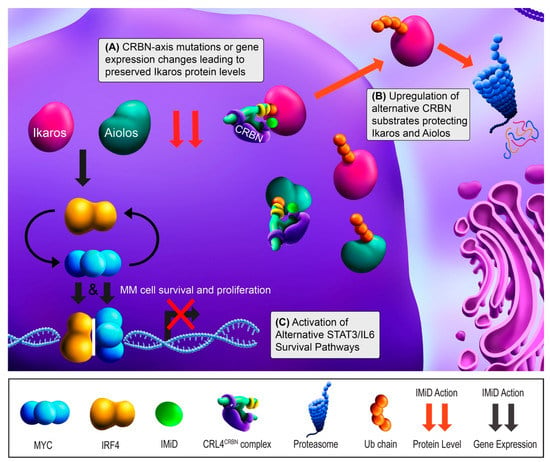
Figure 1.
Immunomodulatory drug (IMiD) mechanism of action and resistance mechanisms. Ikaros (IKZF1) and Aiolos (IKZF3) increase the gene expression of IRF4 (yellow) and MYC (cyan), leading to multiple myeloma (MM) cell survival and proliferation. IRF4 and MYC bind to their respective promoters independently, but also upregulate each other’s gene expression. With treatment, IMiD binding leads to the Cereblon (CRBN)-dependent proteasomal degradation of ubiquitin (Ub)-tagged Ikaros and Aiolos (red arrows). IKZF1/3 degradation leads to the downregulation of IRF4 and MYC gene expression (black arrows) and results in growth arrest and apoptosis (red “X”). (A) IMiD resistance can occur when components in the CRBN-Ikaros axis are mutated or undergo altered expression, most frequently downregulation of CRBN. (B) IMiD resistance can also occur through increased expression of alternative CRBN substrates that effectively prevent IKZF1/3 degradation by binding CRBN or by mediating the CRBN degradation. (C) Finally, IMiD resistance can occur via upregulation of alternative survival pathways to bypass Ikaros axis dependence, including IL-6/STAT3 activation.
2.2. IMiD Resistance
Most patients are initially sensitive to IMiDs. First tested in the RRMM setting, single-agent lenalidomide showed an overall response rate (ORR; ≥50% decrease in disease burden) of 26% [40]. Based on the IMiD mechanism of action, perturbations in the CRL4CRBN complex and Ikaros axis are logical candidate drivers of resistance (Figure 1A). This hypothesis was initially supported with MM cell lines, as targeted disruption of CRBN and IKZF1/3 binding yielded IMiD resistance [35,36,41]. Later, a study of primary samples showed CRBN mutations in 12% (6/50) of RRMM patients, 88% of which were clinically IMiD-refractory. These mutations were not present in matched pre-treatment samples, suggesting these were acquired in response to treatment [11]. Similar studies have also showed acquired CRBN mutations in RRMM patients [10,42]. With lower frequencies, mutations in IKZF1, IRF4, and the CUL4B component of CRL4CRBN have also been reported [10,11]. Although CRBN mutations are relative rare, separate studies have found CRBN downregulation after lenalidomide treatment in 89% (8/9) of patients at the mRNA level and 71% (5/7) of patients at the protein level [12,41]. Similarly, high CRBN mRNA expression has been associated with IMiD responsiveness [43,44,45,46], and IMiD-resistant MM cell lines reflect this at the protein level [8,9,32]. CRBN protein levels are affected by the SCFFbxo7 E3 ubiquitin ligase, which is regulated by the CSN signalosome [15]. Studies have also looked at IKZF1/3, but conflicting results have emerged as to whether their expression correlates with IMiD response [45,47,48]. Collectively, mutations in the CRBN-Ikaros axis exist in IMiD resistant patients, but the patients tested thus far suggest that CRBN downregulation may a more commonly acquired event.
Additional modulators of CRBN or IKZF1/3 activity may also contribute to resistance. This can occur if IKZF1/3 degradation is prevented through interference of binding with CRBN (Figure 1B). Zhou and colleagues found that RUNX1/3 compete with CRBN for binding and prevent IKZF1/3 degradation [14]. In MM cell lines and patient samples (n = 5), inhibiting RUNX1/3 reversed lenalidomide resistance. CRBN also has several substrates that if upregulated may outcompete IKZF1/3 for CRBN binding in the presence of IMiDs [8]. Interestingly, Eichner et al. found that CRBN also functions as a chaperone for MCT1 and CD147, and IMiD binding leads to mislocalization of those proteins [16]. This study also found that lenalidomide-resistant cell lines maintained CD147 and MCT1 levels in response to IMiDs. In aggregate, these studies clearly show the complexity of CRBN biology, but their IMiD resistance mechanisms have only been characterized in cell lines to this point and still need to be investigated in patient samples to fully assess clinical relevance. While IKZF1/3 and CRBN have been studied more extensively, investigating the downstream effects on IRF4 and MYC will also be important in determining the functional consequence of these mechanisms in clinical IMiD resistance.
Other IMiD resistance mechanisms could bypass CBRN and the Ikaros axis to promote MM cell survival (Figure 1C). Although low CRBN expression tends to correlate with IMiD resistance, some patients with similarly high CRBN levels have differential drug sensitivity and many IMiD-resistant MM cases do not show abnormalities in CRBN, IKZF1 or IKZF3 [11,44]. The activation of other signaling pathways may compensate, potentially through upregulating IRF4, MYC, or other unrelated pro-survival factors. Consistent with this, Zhu and colleagues found that lenalidomide-adapted MM cell lines lacking CRBN abnormalities showed impaired IRF4 downregulation and upregulation of IL-6/STAT3 signaling [9]. In patients, high IL-6 expression was associated with shorter responses [9]. Downstream, IL-6/STAT3 signaling may maintain IRF4 and/or MYC upregulation and induce pro-survival MAPK and PI3K pathway activation [9,13,17]. Based on these findings, investigating IL-6 antagonists and STAT3 inhibitors in the context of IMiD-refractory patients is warranted.
In sum, the evidence thus far supports the notion that IMiD resistance occurs mostly by mutation or gene expression changes in the CRBN-Ikaros axis or by compensatory growth pathways. Sequencing studies have found low mutation rates in the CRBN-Ikaros axis in the RRMM setting, whereas clonal evolution with mutations in other proliferative pathways have been comparatively common [4,10,11]. Currently, it remains unclear from to what degree RRMM patients’ disease remains Ikaros dependent [49]. The clinical trials discussed next have shown that IKZF1/3 remain valid targets in at least some IMiD-exposed patients.
2.3. Overcoming IMiD Resistance
Next-generation CRBN E3 Ligase Modulators (CELMoDs) show higher potency and may be able to overcome IMiD resistance in Ikaros axis-dependent MM. The CELMoDs iberdomide, avadomide, and CC-92480 are currently in clinical development for RRMM (Table 2). Iberdomide and avadomide have 20-fold higher binding affinity for CRBN compared to lenalidomide and increased potency of IKZF1/3 degradation [29,38]. CELMoDs overcome IMiD resistance to some degree in MM cell lines, likely through accelerating the degradation of IKZF1/3 [8,50,51]. CELMoDs are currently being evaluated as for treatment of RRMM (Table 2). In a Phase 1b/2a dose escalation trial of IMiD exposed patients, the ORR for iberdomide with dexamethasone was 31% [52]. The promising results with iberdomide show that there remains a significant portion of patients whose IMiD resistance can be overcome with more potent IKZF1/3 degradation. To this point, avadomide has only been reported in two patients with RRMM thus far, neither of which responded [53]. Clinical results are not yet available with CC-92480. In the cases where IMiD resistance results from CRL4CRBN component or Ikaros mutations, these agents may be ineffective and thus studying the relationship between mutational status and clinical response will be helpful in the development of CELMoDs. The direct ex vivo measurement of IMiD response may be a valuable way to determine when CELMoDs are likely to benefit an IMiD-refractory patient [54].
Table 2.
Key therapies currently in clinical trials for overcoming drug resistance in multiple myeloma.
On the other hand, targeting alternative pathways will likely be needed if patients’ disease become Ikaros axis independent. This could occur by MYC decoupling from IKZF1/3 downregulation by IMiDs, making other means of MYC inhibition attractive. Bromodomain extra-terminal (BET) inhibitors are one possibility, as they downregulate MYC transcription. The BET inhibitor JQ1 has shown efficacy in both IMiD sensitive and resistant MM cell lines [68]. The MYC-MAX dimerization inhibitor 10058-F4 also displayed anti-myeloma effects in MM cell lines [69]. Other MYC inhibitors have also shown promise as anti-cancer therapeutics, but these have yet to be evaluated in MM [70]. Further study of these approaches is merited in IMiD-resistant MM.
3. Proteasome Inhibitors
Proteasome inhibitors have been a standard-of-care in MM treatment over the past two decades. Bortezomib is a first-in class proteasome inhibitor (PI) that is used in both the newly diagnosed and RRMM setting. Newer-generation PIs carfilzomib and orally bioavailable ixazomib are FDA-approved for combination therapy in the RRMM setting.
3.1. PI Mechanism of Action
Approximately 90% of protein turnover occurs through the ubiquitin-proteasome system [71]. Since MM cells are highly secretory, they are exceptionally dependent on their ability to dispose of misfolded and aggregated proteins via proteasomal degradation. Consistent with this, normal PCs and malignant MM cells are heavily reliant on the unfolded protein response (UPR) to ensure proper protein folding and turnover [71]. Proteasomes are proteolytic complexes that degrade ubiquitylated proteins and are composed of a 20S core catalytic particle and 19S regulatory particle. There are three distinct catalytic sites within the 20S particle: the chymotrypsin-like site in the β5 subunit, trypsin-like site in the β2 subunit, and caspase-like site in the β1 subunit [72]. PIs are short peptide-based structures that bind to the catalytic sites. Bortezomib and ixazomib bind reversibly to the chymotrypsin-like site. In contrast, carfilzomib binds irreversibly and with much higher affinity to the β5 active site and also binds with lower affinity to the trypsin-like and caspase-like active sites [72,73]. Proteasome inhibition results in an excessive accumulation of proteins within MM cells, resulting in prolonged, irresolvable ER stress, and apoptosis (Figure 2) [74,75].
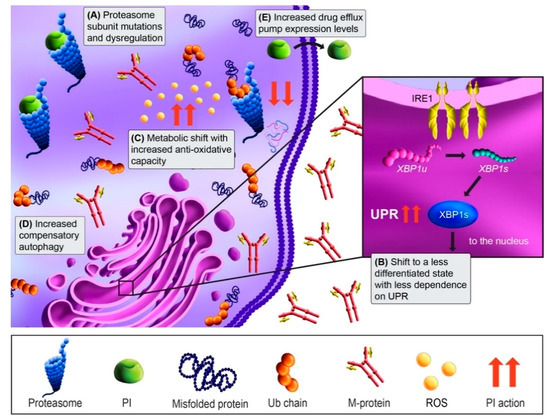
Figure 2.
Proteasome inhibitor (PI) mechanism of action and resistance mechanisms. MM cells are dependent on the proteasome to degrade and recycle the high level of misfolded proteins that result from the production of M-protein. Treatment with PIs (green) blocks the degradation of ubiquitinated (Ub) proteins by binding either reversibly (bortezomib) or irreversibly (carfilzomib) to the proteasome. This blocks protein degradation, leading to the accumulation of misfolded proteins and activation of the unfolded protein response (UPR) and apoptosis (red arrows). (A) PI resistance can occur through mutations in proteasome catalytic subunits, or via dysregulation of subunit expression. (B) PI resistance may also occur in MM cells through a shift to a less differentiated phenotype that produces less M-protein and thus exhibit less dependence on the IRE1-Xbp1s UPR arm. (C) In addition, a shift in metabolic state can occur with PI resistance that more effectively neutralizes the cytotoxic reactive oxygen species (ROS) that are generated by UPR. (D) Autophagy upregulation is another compensatory adaptation that can maintain proteostasis when the proteasome is inhibited. (E) PI-resistant MM cells may adapt to PI treatment via upregulation of drug efflux pumps that extrude the drugs from the cells.
3.2. PI Resistance
The proteasome, as the direct drug class target, is a well-studied candidate in acquired PI resistance (Figure 2A). Based on the binding site shared between bortezomib and carfilzomib, PSMB5 mutations in the β5 subunit were predicted be a hotspot for PI resistance, and bortezomib-adapted MM cell lines have shown mutations there [76,77]. In MM patient specimens, proteasome mutations have been extensively characterized by Barrio and colleagues. Mutations in PSMB5 occurred at a frequency of 1.1% (4/355) in relapsed patients, compared to 0.08% (1/1, 241) in newly diagnosed patients [19]. Among those characterized, bortezomib resistance was observed and carfilzomib resistance was a variable. Mutations outside the catalytic core also occur at low frequencies in 20S subunit genes PSMA1, PSMB8, and PSMB9, the 19S subunit gene PSMD1, and the proteasome assembly chaperone PSMG2, but their functional consequences are unknown [11,19]. While it was postulated that proteasome overexpression may mediate bortezomib resistance, a study of bortezomib-resistant primary MM found that most proteasome subunits, including PSMB5, were downregulated [18]. In addition, low 19S regulatory subunit gene expression appears to correlate with PI non-responsiveness [20,78]. Conversely, the 20S subunit PSMA3 was upregulated in bortezomib-resistant patients and conferred resistance when overexpressed in MM cell lines [79]. Thus, proteasome subunit expression changes in may contribute to PI resistance, but the frequency whereby this occurs in patients are not clear and warrant further investigation.
Resistance mechanisms independent of the proteasome itself have also been supported in MM. Since PIs rely on disrupting proteostasis for their cytotoxic effects, resistance may occur through shifting to a less secretory state that can withstand UPS disruption. X-box-protein 1 (XBP1) is critical for PC differentiation and is a master UPR regulator. XBP1 is spliced into its active form (Xbp1s) by Inositol-requiring enzyme 1α (Ire1α), which allows PCs and MM cells to maintain proteostasis [80]. Leung-Hagesteijn and colleagues showed IRE1α-Xbp1 pathway downregulation in PI-resistant patient samples [18]. In line with this, populations of bortezomib-resistant primary MM cells possessed a pre-plasmablast phenotype, and increased fractions of less differentiated cells have been observed following bortezomib-based regimens [18,81,82]. This switch to a less differentiated and secretory, state suggests that proteostasis disruption may no longer be catastrophic to resistant MM cells (Figure 2B). Another consequence of PI-induced ER stress is the overproduction of reactive oxygen species (ROS) and oxidative stress [83]. However, PI-resistant MM cells may rewire their metabolism to better handle ROS (Figure 2C) [84,85]. In addition to UPR, PCs also rely on autophagy for protein disposal [56,86]. Not surprisingly, increased autophagy has been observed in MM cell line acquired PI resistance (Figure 2D) [22,56,87,88]. Overall, it appears MM cells may evade PI-induced cytotoxicity through changes in cell state or compensatory pathways, suggesting novel therapeutic strategies that target PI-resistant cellular features or further exacerbate proteotoxic stress may be required.
A more generic pharmacokinetic mechanism of PI resistance may be increased drug efflux (Figure 2E). Drug efflux pumps, or ATP-binding cassette (ABC) transporters, shuttle drugs and their metabolites from the intracellular to the extracellular space [89]. Increased drug efflux leads to resistance by preventing drugs from interacting with their intracellular targets. Carfilzomib and bortezomib are substrates of the multidrug transporter P-glycoprotein (MDR1) [89,90]. MDR1 is upregulated in response to PI treatment and was also identified in PI-adapted MM cell lines [23,91]. MDR1 inhibition re-sensitizes MM cells to PIs in vitro, but clinically showed too many side effects and no benefit [92]. Thus, a different approach is needed. Nima-related protein kinase 2 (Nek2) controls many ABC pathways and has been implicated in PI resistance through several studies [24,91,93]. Pharmacologic inhibition of Nek2 reversed bortezomib resistance in MM xenograft models [21], suggesting this strategy may impede increased drug efflux in PI-refractory MM patients.
3.3. Overcoming PI Resistance
Many drugs are being investigated for MM treatment that may overcome PI resistance. Within the same drug class, the novel pan-proteasome inhibitor marizomib (NPI-0052) underwent clinical trials for RRMM. Marizomib irreversibly binds to all three proteolytic sites of the 20S proteasome (β5, β2, β1) and binds to the β5 site with two times higher potency compared to bortezomib [55,94]. Marizomib has activity in MM cells with acquired bortezomib resistance, but it has not been tested in carfilzomib resistant cells [94]. While Phase I and II clinical trials were completed for marizomib for RRMM in 2017, at this point there has been no further clinical development [55].
Several proteasome-independent strategies for inducing proteotoxic stress are being pursued for the PI refractory setting. Kinase inhibitors of IRE1α have shown cytotoxicity in MM cell lines, xenograft models and patient samples [80]. While not yet directly assessed with PI resistance, inhibiting IRE1α similarly triggers ER stress. Attacking another node of proteostasis, the E1 ubiquitin-activating enzyme inhibitor TAK-243 (MLN7243) overcame PI resistance in MM cell line models and RRMM primary samples, inducing all three ER stress pathways [95]. TAK-243 is currently in Phase I trials for myeloid malignancies and hopefully will be assessed in MM soon. Another potential target in PI-refractory disease is the Vasolin-containing protein (p97), which delivers ubiquitin-tagged proteins to the proteasome. The p97 inhibitor CB-5083 induces UPR in MM cells and demonstrated efficacy in PI-resistant patient samples and preclinical models [96,97]. Unfortunately, clinical trials for CB-5083 were terminated due to off-target effects, so this strategy may require development of less toxic inhibitors [97]. The pan-histone deacetylase (HDAC) inhibitor panobinostat is approved for RRMM treatment, and preclinical studies supported its synergy with bortezomib through inhibiting the aggresome pathway [98]. However, panobinostat is infrequently utilized in MM due to associated toxicities [99]. The new HDAC inhibitor ricolinostat is more selective and possesses less off-target effects. Ricolinostat showed activity in carfilzomib-refractory patients and is currently in a Phase I/II trial for RRMM [65]. Autophagy inhibitors are also being explored. The lysosomal inhibitor hydroxychloroquine enhanced carfilzomib effects in cell lines and is being evaluated in a Phase I trial for RRMM treatment in combination with bortezomib (Table 2) [56]. Lastly, protein translation inhibition may be another approach to disrupt proteostasis of MM cells. Recently, we found that the translation inhibitor omacetaxine is potent and efficacious in PI/IMiD refractory MM patient samples [100]. These strategies are still in early development for RRMM treatment, so further clinical trial results are needed to assess their potential to overcome PI resistance.
4. Immunotherapies
Immunotherapies have recently become a mainstay of MM treatment. Currently, three monoclonal antibodies (mAbs) are approved for MM treatment, and multiple next-generation immunotherapies are at various stages of clinical testing. mAbs are unique among anti-myeloma agents, acting to prime the immune system to specifically target malignant cells. In 2015, the FDA approved daratumumab monotherapy and elotuzumab-based combination treatment for RRMM. Daratumumab has since been approved in several combination regimens for newly diagnosed and RRMM, and elotuzumab in combination with dexamethasone and either lenalidomide or pomalidomide in RRMM. Daratumumab-based combinations have become highly important in treating RRMM, with the response rates being the highest in the relapsed setting to date [101]. Another anti-CD38 mAb, isatuximab, has also been approved for RRMM treatment in combination with pomalidomide and dexamethasone. Recently, the antibody-drug conjugate (ADC) targeting BCMA, belantamab mafodotin, received accelerated approval for RRMM. This appears to be the first of several new immunotherapies likely to be approved, as multiple chimeric antigen receptor (CAR) T-cell products, bispecific antibodies and antibody drug conjugates (ADCs) look promising in clinical trials.
4.1. Monoclonal Antibody Mechanism of Action
Daratumumab is a fully human mAb that binds to the surface ectoenzyme CD38, a PC marker highly expressed on MM cells [102]. It acts through multiple cytotoxic mechanisms, including antibody-dependent cell-mediated cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), antibody-dependent cellular phagocytosis (ADCP), and direct induction of cell death (Figure 3) [103,104]. Isatuximab targets a distinctly different CD38 epitope [105]. Elotuzumab targets the anti-signaling lymphocyte activation marker F7 (SLAMF7), which is expressed on more than 95% of MM cells [106]. SLAMF7 signaling also activates NK cell proliferation, so the elotuzumab effect may be accentuated through activation of NK cells.
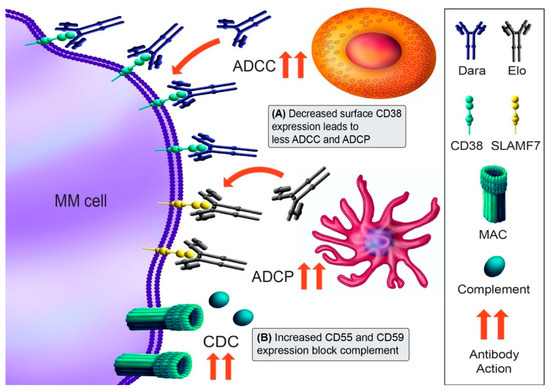
Figure 3.
Immunotherapy mechanisms of action and resistance mechanism. The monoclonal antibody (mAb) daratumumab (Dara) binds to CD38 and induces cytotoxicity through antibody-dependent cellular cytotoxicity (ADCC; NK cell [orange], macrophage [pink]), complement-dependent cytotoxicity (CDC; membrane attack complex [MAC]), antibody-dependent cellular phagocytosis (ADCP; macrophage), and direct blocking of CD38 function. Elotuzumab (Elo) recognizes SLAMF7 and results in direct activation of NK cells and ADCC. (A) Downregulation of CD38 mediates Dara resistance, in part by trogocytosis of CD38 from MM cells. (B) A second mechanism of Dara resistance may be the upregulation of complement-inhibitory proteins CD55 and CD59. Antibody action is denoted by red arrows. The mechanisms shown are for Dara resistance mechanisms, as no Elo resistance mechanisms have been described.
4.2. Immunotherapy Resistance
Despite the efficacy of daratumumab, responders eventually develop resistance over time [102,107]. Intrinsic resistance appears due to low CD38 expression in some patients [25]. Acquired resistance is also expression-dependent, as daratumumab decreases CD38 levels even following the first infusion, and this decrease correlates with reduced CDC and ADCC at progression (Figure 3A) [25]. Interestingly, CD38 mutations in daratumumab-refractory patients have not been reported, although a limited number of daratumumab-treated patients have been sequenced [11]. Aside from CD38, upregulation of the complement inhibitors CD55 and CD59 also occurs in some patients at progression (Figure 3B), implying that may be a second event leading to daratumumab resistance [25]. CD38 loss appears to be transient, as patient’s MM cells recover their CD38 approximately six months after daratumumab discontinuation [25]. In agreement with this, ex vivo daratumumab sensitivity appears to be partially restored greater than one year after daratumumab discontinuation [108]. Clinically, recovering anti-CD38 mAb sensitivity could be of substantial benefit to patients. As elotuzumab has not shown single-agent activity, there are no resistance mechanisms currently known.
4.3. Overcoming Immunotherapy Resistance
Various next-generation immunotherapies are being investigated for MM, mostly targeting antigens other than CD38 or SLAMF7. These therapies feature formats with enhanced cytotoxicity and include chimeric antigen receptor (CAR)-T cells, antibody-drug conjugates (ADCs), and bispecific antibodies. The most frequently targeted alternative antigen for these immunotherapies has been B-cell maturation antigen (BCMA), which is expressed on MM cells in the vast majority of patients [109]. Prominent among the anti-BCMA therapies are CAR-T cells, engineered to express a chimeric T-cell receptor to recognize and facilitate the elimination of MM cells [57]. In a Phase II trial, the anti-BCMA CAR-T cell therapy idecabtagene vicleucel (ide-cel, formerly bb2121) showed a 73% (94/128) ORR in heavily-pretreated RRMM patients, including daratumumab-refractory patients [110]. Phase III results for ide-cel are not yet public, but have led to the recent FDA approval for this to be the first commercially available CAR-T cell product for MM. In addition, ciltacabtagene autoleucel (cilta-cel, formerly LCAR) has shown very promising results in a phase I/IIb single arm study, with an ORR of 95% (92/97) [58]. Bispecific antibodies are novel antibody therapeutics that have dual specificity for a target cancer cell antigen and CD3, by which they engage T cell mediated cytotoxicity [109]. The bispecific antibody CC-93269 showed favorable results in a Phase I trial for RRMM with an 83% (10/19) ORR [59]. In support of overcoming mAb resistance, 89% of patients were daratumumab-refractory. Other promising anti-BCMA CAR-T cells and bispecific antibodies for MM are also in development and are showing similarly high response rates (Table 2) [60,110,111,112]. ADCs are antibodies that deliver a cytotoxic payload directly to their target cancer cells. The anti-BCMA ADC belantamab mafodotin showed favorable results in a Phase I study that included 40% of patients that were daratumumab-refractory, and belantumab mafodotin was recently granted approval for RRMM [62]. In the near future, BCMA-targeted therapies are expected to drastically change the treatment landscape for RRMM.
Aside from BCMA-targeted immunotherapies, several other promising targeted immunotherapies are being pursued for MM treatment and may be especially useful for patients that relapse after BCMA-based therapy. CD46 is a complement inhibitor that is overexpressed in nearly all MM patients, and anti-CD46-ADC induced potent MM cell death in vitro and in vivo [113]. CD46-ADC is currently being evaluated in a Phase I trial for RRMM (Table 2). The bispecific antibody talquetamab is currently in a Phase I trial for RRMM. This bispecific targets the G protein-coupled receptor C5D (GPRC5D) and CD3 [114]. GPRC5D is expressed on myeloma cells but not regular PCs, making it an attractive therapeutic target. GPRC5D is also a potential target for CAR-T cell therapy [115]. The bispecific antibody cevostamab (BFCR4350A), targeting FCRH5 and CD3, is another bispecific antibody under clinical evaluation for MM [61]. The large repertoire of immunotherapies currently being evaluated in RRMM will offer an array of highly specific therapies to target MM cells, and in combination with other anti-MM agents may elicit deeper and more durable therapeutic responses.
5. Other Drug Classes
In addition to the three aforementioned drug classes, glucocorticoids and chemotherapeutics also remain vital components of myeloma treatment in both newly-diagnosed and RRMM. Dexamethasone is the most commonly used glucocorticoid in MM, and melphalan is a frequently utilized alkylating chemotherapeutic agent that also possesses anti-MM immunostimulatory effects [2,26].
5.1. Dexamethasone Resistance
All glucocorticoids target the glucocorticoid receptor (GR) and binding leads to nuclear translocation and changes in gene expression. GRs have many downstream targets, ultimately repressing anti-apoptotic and metabolic pathways that drive MM cell death [26]. In MM and other hematologic malignancies where steroids are commonly used, the most common resistance mechanism is decreased GR expression, and low levels correlate with poorer outcomes. Mutations and alternatively-spliced isoforms of GR have also been reported, but these appear to be rare [11]. Studies have also suggested that certain subgroups of MM respond better to dexamethasone [116]. Dexamethasone sensitivity in RRMM needs to be further investigated, as it is used in almost all lines of therapy.
5.2. Melphalan Resistance
As with chemotherapeutic agents in general, increased expression of drug efflux pumps, such as P-gp and other multidrug resistance (MDR) proteins, have been implicated in MM melphalan resistance [27]. Since alkylating agents primarily act through inducing irreparable DNA damage, it follows that increased expression of factors related to DNA repair or replication, such as RECQ1 (encoding the RecQ1 helicase), are associated with melphalan resistance in MM [28,117]. Additionally, high expression of anti-apoptotic genes, including BCL-xL, have also been associated with melphalan resistance [27]. While agents that target P-gp have been largely unsuccessful, agents that further exacerbate DNA damage, or prevent escape from apoptosis, may accentuate alkylator efficacy in RRMM.
5.3. Overcoming Melphalan Resistance
Melflufen (Melphalan-flufenamide) is a first-in-class peptide-drug conjugate that was recently FDA-approved for triple-class refractory MM. It was designed to selectively deliver melphalan to MM cells [118]. Due to its lipophilic nature, melflufen is rapidly taken up by MM cells, and once inside the cell it is rapidly cleaved by peptidases to release melphalan [66]. This accelerated uptake and release likely explain the increased efficacy of melflufen compared to melphalan. When evaluated in a Phase II trial, melflufen had an ORR of 26% (31/119) in triple-refractory patients [66]. Melflufen is also being assessed in combination with dexamethasone and either bortezomib or daratumumab in RRMM (Table 2).
6. Other Strategies for Circumventing Drug Resistance
In addition to improvements within the above classes, other drugs are under development for RRMM treatment. These agents have distinct mechanisms of action and may better avoid cross-resistance with routinely used drugs. Selinexor is the best example of this, having received accelerated approval in the context of triple-class refractory MM patients. Selinexor is a SINE compound that inhibits Exportin 1 (XPO1), a nuclear export protein that controls the localization of many proteins important for MM survival [119]. Selinexor showed an ORR of 26% (32/122) in PI/IMiD triple-refractory patients in a Phase I/II study in combination with dexamethasone (Table 2) [120]. In addition, selinexor with bortezomib and dexamethasone showed an ORR of 76% (149/195), with a progression free survival of 13.9 months in a Phase III trial of myeloma patients who had been treated with one to three prior lines of therapy [121].
A more recent approach in anti-cancer therapeutics is to inhibit the anti-apoptotic proteins cancer cells exploit. MM shows a heterogenous dependency on the anti-apoptotic B-cell lymphoma 2 (BCL-2) family members BCL-2, BCL-xL, and MCL-1 [122]. Venetoclax is a BH3 mimetic that selectively inhibits BCL-2 and is approved for use in chronic lymphoid leukemia (CLL) and acute myeloid leukemia (AML). Early clinical trials in MM suggested that venetoclax synergizes with PIs [123]. In a phase III clinical trial in RRMM, venetoclax addition to bortezomib and dexamethasone led increased progression free survival, but unfortunately also increased mortality, leading to a FDA-imposed halt of the study [67]. However, preclinical and clinical results strongly support that venetoclax activity in MM is associated with t (11;14), leading to the possibility those specific patients would have a better risk-benefit ratio [67,122,124]. Thus, clinical study of venetoclax in RRMM is currently focused on t (11;14)-positive disease (Table 2).
7. Conclusions and Future Directions
Although MM has seen a series of therapeutic revolutions over the past two decades that have drastically improved clinical outcomes, patients still inevitably develop multi-drug resistance and ultimately succumb to their disease. Thus, the triple-class refractory setting is currently the greatest area of clinical need in MM. Understanding drug resistance mechanisms is critical for identifying novel and effective drug targets and designing rational therapeutic combinations for triple-class refractory patients. New drug classes that target these mechanisms or drugs within the same class that do not show cross-resistance are fitting strategies. However, drug resistance in MM, like the disease itself, is complex and heterogenous, so it is unlikely one mechanism alone explains every case of resistance to that drug class. Pinpointing drug resistance mechanisms in patients is further convoluted by the fact that patients nearly always receive combination therapy, so it is difficult to say exactly which drug(s) a patient is actually resistant to in the combination. This is exacerbated furthermore by the widespread occurrence of multi-drug resistance, as some of these agents work synergistically or have overlapping pathways involved in their mechanisms of action. It is possible that the best way to evaluate drug resistance will be on a patient-to-patient basis.
One feasible approach to complement drug development may be personalized medicine (also known as precision medicine). This would involve identifying reliable predictive biomarkers of response and profiling individual patients for what agents are likely to elicit a successful therapeutic response, which would inform effective treatment regimens. Approaches to accomplishing this could include predicting patient drug response based on their genetic profile or to profile patient’s sensitivity to anti-myeloma agents ex vivo over their disease course [54,125,126]. Several of these approaches are currently being assessed in clinical trials for MM. Previous studies have suggested that resistance to one drug class reprograms resistant clones in a manner that yields them increased sensitivity to other drug classes or to drugs within the same class [13,127]. It is also possible that patients may become re-sensitized to an agent as they are exposed to other lines of treatment, so this could also help guide if agents can be reintroduced in patients with recaptured efficacy at a later point in their disease.
The observation of more progenitor-like MM cells in PI resistant cell lines and patient samples also raises the question of whether our standard approaches to characterizing MM are robust/conserved enough to capture the full disease, especially in the relapsed/refractory setting. MM is generally characterized by double-positivity for the PC markers CD38 and CD138; however, studies have suggested that MM differentially express CD138 or downregulate CD138 expression [128,129]. Additional cellular markers of MM that allow us to account more accurately for the disease heterogeneity, such as CD46 or BCMA, should be considered when studying MM, of which next-generation flow and mass cytometry that allow for a greater number of cellular markers will be instrumental.
Continuing to expand our understanding of the complexities of drug resistance mechanisms will identify novel targets and strategies that enable us to overcome resistance and offer reliable predictive biomarkers. These discoveries could guide clinical decisions for multiple myeloma treatment and may potentially even lead to a cure in some patients.
Author Contributions
L.N.D. wrote and edited the manuscript, D.W.S. edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
The authors received research support from the National Cancer Institute (K08CA222704 to D.W.S.).
Acknowledgments
The authors would like to thank Gwendolyn Tice for creating the figure illustrations for this manuscript. The authors would also like to thank Zachary Walker, Olivia Perez de Acha, and Lauren Reiman for helpful discussions.
Conflicts of Interest
D.W.S. has received research support from Oncopeptides AB and Fortis Therapeutics. The authors have no other potential conflict of interests.
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; van Duin, M.; Sonneveld, P.; Mateos, M.V.; Gay, F.; Anderson, K.C. Multiple myeloma. Nat. Rev. Dis. Prim. 2017, 3, 17046. [Google Scholar] [CrossRef] [PubMed]
- Nijhof, I.S.; Van De Donk, N.W.C.J.; Zweegman, S.; Lokhorst, H.M. Current and New Therapeutic Strategies for Relapsed and Refractory Multiple Myeloma: An Update. Drugs 2018, 78, 19–37. [Google Scholar] [CrossRef]
- Keats, J.J.; Chesi, M.; Egan, J.B.; Garbitt, V.M.; Palmer, S.E.; Braggio, E.; Van Wier, S.; Blackburn, P.R.; Baker, A.S.; Dispenzieri, A.; et al. Clonal competition with alternating dominance in multiple myeloma. Blood 2012, 120, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef]
- Rasche, L.; Chavan, S.S.; Stephens, O.W.; Patel, P.H.; Tytarenko, R.; Ashby, C.; Bauer, M.; Stein, C.; Deshpande, S.; Wardell, C.; et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat. Commun. 2017, 8, 268. [Google Scholar] [CrossRef]
- Gandhi, U.H.; Cornell, R.F.; Lakshman, A.; Gahvari, Z.J.; McGehee, E.; Jagosky, M.H.; Gupta, R.; Varnado, W.; Fiala, M.A.; Chhabra, S.; et al. Outcomes of patients with multiple myeloma refractory to CD38-targeted monoclonal antibody therapy. Leukemia 2019, 33, 2266–2275. [Google Scholar] [CrossRef]
- Sperling, A.S.; Burgess, M.; Keshishian, H.; Gasser, J.A.; Bhatt, S.; Jan, M.; Słabicki, M.; Sellar, R.S.; Fink, E.C.; Miller, P.G.; et al. Patterns of substrate affinity, competition, and degradation kinetics underlie biological activity of thalidomide analogs. Blood 2019, 134, 160–170. [Google Scholar] [CrossRef]
- Zhu, Y.X.; Shi, C.-X.; Bruins, L.A.; Wang, X.; Riggs, D.L.; Porter, B.; Ahmann, J.M.; De Campos, C.B.; Braggio, E.; Bergsagel, P.L.; et al. Identification of lenalidomide resistance pathways in myeloma and targeted resensitization using cereblon replacement, inhibition of STAT3 or targeting of IRF4. Blood Cancer J. 2019, 9, 19. [Google Scholar] [CrossRef]
- Barrio, S.; Munawar, U.; Zhu, Y.X.; Giesen, N.; Shi, C.X.; Da Via, M.; Sanchez, R.; Bruins, L.; Demler, T.; Muller, N.; et al. IKZF1/3 and CRL4-CRBN E3 ubiquitin ligase mutations and IMiD resistance in multiple myeloma. Haematologica 2019, 105, e237–e241. [Google Scholar] [CrossRef]
- Kortüm, K.M.; Mai, E.K.; Hanafiah, N.H.; Shi, C.-X.; Zhu, Y.-X.; Bruins, L.; Barrio, S.; Jedlowski, P.; Merz, M.; Xu, J.; et al. Targeted sequencing of refractory myeloma reveals a high incidence of mutations in CRBN and Ras pathway genes. Blood 2016, 128, 1226–1233. [Google Scholar] [CrossRef]
- Dimopoulos, K.; Munch-Petersen, H.F.; Eskelund, C.W.; Sjö, L.D.; Ralfkiaer, E.; Gimsing, P.; Grønbaek, K. Expression of CRBN, IKZF1, and IKZF3 does not predict lenalidomide sensitivity and mutations in the cereblon pathway are infrequent in multiple myeloma. Leuk. Lymphoma 2018, 60, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Ocio, E.M.; Fernández-Lázaro, D.; San-Segundo, L.; López-Corral, L.; Sánchez, L.A.C.; Gutiérrez, N.C.; Garayoa, M.; Paíno, T.; Garcia-Gomez, A.; Delgado, M.; et al. In vivo murine model of acquired resistance in myeloma reveals differential mechanisms for lenalidomide and pomalidomide in combination with dexamethasone. Leukemia 2014, 29, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Gutierrez-Uzquiza, A.; Zheng, X.Y.; Chang, R.; Vogl, D.T.; Garfall, A.L.; Bernabei, L.; Saraf, A.; Florens, L.; Washburn, M.P.; et al. RUNX proteins desensitize multiple myeloma to lenalidomide via protecting IKZFs from degradation. Leukemia 2019, 33, 2006–2021. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Song, T.; Zhou, W.; Xing, L.; Wang, S.; Ho, M.; Peng, Z.; Tai, Y.-T.; Hideshima, T.; Anderson, K.C.; et al. A genome-scale CRISPR-Cas9 screening in myeloma cells identifies regulators of immunomodulatory drug sensitivity. Leukemia 2019, 33, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Eichner, R.; Heider, M.; Fernández-Sáiz, V.; Van Bebber, F.; Garz, A.-K.; Lemeer, S.; Rudelius, M.; Targosz, B.-S.; Jacobs, L.; Knorn, A.-M.; et al. Immunomodulatory drugs disrupt the cereblon–CD147–MCT1 axis to exert antitumor activity and teratogenicity. Nat. Med. 2016, 22, 735–743. [Google Scholar] [CrossRef]
- Shi, L.; Wang, S.; Zangari, M.; Xu, H.; Cao, T.M.; Xu, C.; Wu, Y.; Xiao, F.; Liu, Y.; Yang, Y.; et al. Over-expression of CKS1B activates both MEK/ERK and JAK/STAT3 signaling pathways and promotes myeloma cell drug-resistance. Oncotarget 2010, 1, 22–33. [Google Scholar] [CrossRef]
- Leung-Hagesteijn, C.; Erdmann, N.; Cheung, G.; Keats, J.J.; Stewart, A.K.; Reece, D.E.; Chung, K.C.; Tiedemann, R.E. Xbp1s-Negative Tumor B Cells and Pre-Plasmablasts Mediate Therapeutic Proteasome Inhibitor Resistance in Multiple Myeloma. Cancer Cell 2013, 24, 289–304. [Google Scholar] [CrossRef]
- Barrio, S.; Stühmer, T.; Da-Viá, M.; Barrio-Garcia, C.; Lehners, N.; Besse, A.; Cuenca, I.; Garitano-Trojaola, A.; Fink, S.; Leich, E.; et al. Spectrum and functional validation of PSMB5 mutations in multiple myeloma. Leukemia 2019, 33, 447–456. [Google Scholar] [CrossRef]
- Acosta-Alvear, D.; Cho, M.Y.; Wild, T.; Buchholz, T.J.; Lerner, A.G.; Simakova, O.; Hahn, J.; Korde, N.; Landgren, O.; Maric, I.; et al. Paradoxical resistance of multiple myeloma to proteasome inhibitors by decreased levels of 19S proteasomal subunits. Elife 2015, 4, e08153. [Google Scholar] [CrossRef]
- Franqui-Machin, R.; Hao, M.; Bai, H.; Gu, Z.; Zhan, X.; Habelhah, H.; Jethava, Y.; Qiu, L.; Frech, I.; Tricot, G.; et al. Destabilizing NEK2 overcomes resistance to proteasome inhibition in multiple myeloma. J. Clin. Investig. 2018, 128, 2877–2893. [Google Scholar] [CrossRef] [PubMed]
- Milan, E.; Perini, T.; Resnati, M.; Orfanelli, U.; Oliva, L.; Raimondi, A.; Cascio, P.; Bachi, A.; Marcatti, M.; Ciceri, F.; et al. A plastic SQSTM1/p62-dependent autophagic reserve maintains proteostasis and determines proteasome inhibitor susceptibility in multiple myeloma cells. Autophagy 2015, 11, 1161–1178. [Google Scholar] [CrossRef]
- Soriano, G.P.; Besse, L.; Li, N.; Kraus, M.; Besse, A.; Meeuwenoord, N.; Bader, J.; Everts, B.; Dulk, H.D.; Overkleeft, H.S.; et al. Proteasome inhibitor-adapted myeloma cells are largely independent from proteasome activity and show complex proteomic changes, in particular in redox and energy metabolism. Leukemia 2016, 30, 2198–2207. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Jing, X.; Holman, C.; Sompallae, R.; Zhan, F.; Tricot, G.; Yang, Y.; Janz, S. Upregulation of FOXM1 leads to diminished drug sensitivity in myeloma. BMC Cancer 2018, 18, 1152. [Google Scholar] [CrossRef] [PubMed]
- Nijhof, I.S.; Casneuf, T.; Van Velzen, J.; Van Kessel, B.; Axel, A.E.; Syed, K.; Groen, R.W.J.; Van Duin, M.; Sonneveld, P.; Minnema, M.C.; et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood 2016, 128, 959–970. [Google Scholar] [CrossRef]
- Burwick, N.; Sharma, S. Glucocorticoids in multiple myeloma: Past, present, and future. Ann. Hematol. 2019, 98, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Robak, P.; Drozdz, I.; Szemraj, J.; Robak, T. Drug resistance in multiple myeloma. Cancer Treat. Rev. 2018, 70, 199–208. [Google Scholar] [CrossRef]
- Viziteu, E.; Klein, B.; Basbous, J.; Lin, Y.-L.; Hirtz, C.; Gourzones, C.; Tiers, L.; Bruyer, A.; Vincent, L.; Grandmougin, C.; et al. RECQ1 helicase is involved in replication stress survival and drug resistance in multiple myeloma. Leukemia 2017, 31, 2104–2113. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.; Hideshima, T.; Anderson, K. Thalidomide in multiple myeloma. Biomed. Pharmacother. 2002, 56, 115–128. [Google Scholar] [CrossRef]
- Sehgal, K.; Das, R.; Zhang, L.; Verma, R.; Deng, Y.; Kocoglu, M.; Vasquez, J.; Koduru, S.; Ren, Y.; Wang, M.; et al. Clinical and pharmacodynamic analysis of pomalidomide dosing strategies in myeloma: Impact of immune activation and cereblon targets. Blood 2015, 125, 4042–4051. [Google Scholar] [CrossRef] [PubMed]
- Holstein, S.A.; Suman, V.J.; McCarthy, P.L. Update on the role of lenalidomide in patients with multiple myeloma. Ther. Adv. Hematol. 2018, 9, 175–190. [Google Scholar] [CrossRef]
- Lopez-Girona, A.; Mendy, D.; Ito, T.A.; Miller, K.H.; Gandhi, A.K.; Kang, J.; Karasawa, S.; Carmel, G.; Jackson, P.E.; Abbasian, M.; et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia 2012, 26, 2326–2335. [Google Scholar] [CrossRef]
- Ito, T.; Handa, H. Cereblon and its downstream substrates as molecular targets of immunomodulatory drugs. Int. J. Hematol. 2016, 104, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Sievers, Q.L.; Petzold, G.; Bunker, R.D.; Renneville, A.; Słabicki, M.; Liddicoat, B.J.; Abdulrahman, W.; Mikkelsen, T.; Ebert, B.L.; Thomä, N.H. Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science 2018, 362, eaat0572. [Google Scholar] [CrossRef] [PubMed]
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.-K.; Bradner, J.E.; Jr, W.G.K. The Myeloma Drug Lenalidomide Promotes the Cereblon-Dependent Destruction of Ikaros Proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef] [PubMed]
- John, L.B.; Ward, A.C. The Ikaros gene family: Transcriptional regulators of hematopoiesis and immunity. Mol. Immunol. 2011, 48, 1272–1278. [Google Scholar] [CrossRef]
- Bjorklund, C.C.; Lu, L.; Kang, J.; Hagner, P.; Havens, C.G.; Amatangelo, M.; Wang, M.; Ren, Y.; Couto, S.S.; Breider, M.; et al. Rate of CRL4CRBN substrate Ikaros and Aiolos degradation underlies differential activity of lenalidomide and pomalidomide in multiple myeloma cells by regulation of c-Myc and IRF4. Blood Cancer J. 2015, 5, e354. [Google Scholar] [CrossRef]
- Shaffer, A.L.; Emre, N.C.T.; Lamy, L.; Ngo, V.N.; Wright, G.; Xiao, W.; Powell, J.; Dave, S.; Yu, X.; Zhao, H.; et al. IRF4 addiction in multiple myeloma. Nat. Cell Biol. 2008, 454, 226–231. [Google Scholar] [CrossRef]
- Richardson, P.; Jagannath, S.; Hussein, M.; Berenson, J.; Singhal, S.; Irwin, D.; Williams, S.F.; Bensinger, W.; Badros, A.Z.; Vescio, R.; et al. Safety and efficacy of single-agent lenalidomide in patients with relapsed and refractory multiple myeloma. Blood 2009, 114, 772–778. [Google Scholar] [CrossRef]
- Zhu, Y.X.; Braggio, E.; Shi, C.-X.; Bruins, L.A.; Schmidt, J.E.; Van Wier, S.; Chang, X.-B.; Bjorklund, C.C.; Fonseca, R.; Bergsagel, P.L.; et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood 2011, 118, 4771–4779. [Google Scholar] [CrossRef] [PubMed]
- Gooding, S.; Ansari-Pour, N.; Towfic, F.; Estévez, M.O.; Chamberlain, P.P.; Tsai, K.-T.; Flynt, E.; Hirst, M.; Rozelle, D.; Dhiman, P.; et al. Multiple cereblon genetic changes are associated with acquired resistance to lenalidomide or pomalidomide in multiple myeloma. Blood 2021, 137, 232–237. [Google Scholar] [CrossRef]
- Broyl, A.; Kuiper, R.; Van Duin, M.; Van Der Holt, B.; El Jarari, L.; Bertsch, U.; Zweegman, S.; Buijs, A.; Hose, D.; Lokhorst, H.M.; et al. High cereblon expression is associated with better survival in patients with newly diagnosed multiple myeloma treated with thalidomide maintenance. Blood 2013, 121, 624–627. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.R.; Kortuem, K.M.; Zhu, Y.X.; Braggio, E.; Shi, C.-X.; Bruins, L.A.; Schmidt, J.E.; Ahmann, G.; Kumar, S.; Rajkumar, S.V.; et al. The clinical significance of cereblon expression in multiple myeloma. Leuk. Res. 2014, 38, 23–28. [Google Scholar] [CrossRef]
- Zhu, Y.X.; Braggio, E.; Shi, C.-X.; Kortuem, K.M.; Bruins, L.A.; Schmidt, J.E.; Chang, X.-B.; Langlais, P.; Luo, M.; Jedlowski, P.; et al. Identification of cereblon-binding proteins and relationship with response and survival after IMiDs in multiple myeloma. Blood 2014, 124, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Franssen, L.E.; Nijhof, I.S.; Couto, S.; Levin, M.-D.; Bos, G.M.; Broijl, A.; Klein, S.K.; Ren, Y.; Wang, M.; Koene, H.R.; et al. Cereblon loss and up-regulation of c-Myc are associated with lenalidomide resistance in multiple myeloma patients. Haematology 2018, 103, e368–e371. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-Y.; Lin, C.-W.; Lin, H.-H.; Yao, M.; Tang, J.-L.; Wu, S.-J.; Chen, Y.-C.; Lu, H.-Y.; Hou, H.-A.; Chen, C.-Y.; et al. Expression of cereblon protein assessed by immunohistochemicalstaining in myeloma cells is associated with superior response of thalidomide- and lenalidomide-based treatment, but not bortezomib-based treatment, in patients with multiple myeloma. Ann. Hematol. 2014, 93, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Krönke, J.; Kuchenbauer, F.; Kull, M.; Teleanu, V.; Bullinger, L.; Bunjes, D.; Greiner, A.; Kolmus, S.; Köpff, S.; Schreder, M.; et al. IKZF1 expression is a prognostic marker in newly diagnosed standard-risk multiple myeloma treated with lenalidomide and intensive chemotherapy: A study of the German Myeloma Study Group (DSMM). Leukemia 2016, 31, 1363–1367. [Google Scholar] [CrossRef]
- Yamamoto, J.; Suwa, T.; Murase, Y.; Tateno, S.; Mizutome, H.; Asatsuma-Okumura, T.; Shimizu, N.; Kishi, T.; Momose, S.; Kizaki, M.; et al. ARID2 is a pomalidomide-dependent CRL4CRBN substrate in multiple myeloma cells. Nat. Chem. Biol. 2020, 16, 1208–1217. [Google Scholar] [CrossRef]
- Matyskiela, M.E.; Zhang, W.; Man, H.-W.; Muller, G.; Khambatta, G.; Baculi, F.; Hickman, M.; Lebrun, L.; Pagarigan, B.; Carmel, G.; et al. A Cereblon Modulator (CC-220) with Improved Degradation of Ikaros and Aiolos. J. Med. Chem. 2017, 61, 535–542. [Google Scholar] [CrossRef]
- Bjorklund, C.C.; Kang, J.; Amatangelo, M.; Polonskaia, A.; Katz, M.; Chiu, H.; Couto, S.; Wang, M.; Ren, Y.; Ortiz, M.; et al. Iberdomide (CC-220) is a potent cereblon E3 ligase modulator with antitumor and immunostimulatory activities in lenalidomide- and pomalidomide-resistant multiple myeloma cells with dysregulated CRBN. Leukemia 2020, 34, 1197–1201. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Van De Donk, N.W.; Popat, R.; Zonder, J.A.; Minnema, M.C.; Larsen, J.; Nguyen, T.V.; Chen, M.S.; Bensmaine, A.; Cota, M.; et al. First clinical (phase 1b/2a) study of iberdomide (CC-220; IBER), a CELMoD, in combination with dexamethasone (DEX) in patients (pts) with relapsed/refractory multiple myeloma (RRMM). J. Clin. Oncol. 2019, 37, 8006. [Google Scholar] [CrossRef]
- Rasco, D.W.; Papadopoulos, K.P.; Pourdehnad, M.; Gandhi, A.K.; Hagner, P.R.; Li, Y.; Wei, X.; Chopra, R.; Hege, K.; DiMartino, J.F.; et al. A First-in-Human Study of Novel Cereblon Modulator Avadomide (CC-122) in Advanced Malignancies. Clin. Cancer Res. 2018, 25, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Walker, Z.J.; VanWyngarden, M.J.; Stevens, B.M.; Abbott, D.; Hammes, A.; Langouët-Astrie, C.; Smith, C.A.; Palmer, B.E.; Forsberg, P.A.; Mark, T.M.; et al. Measurement of ex vivo resistance to proteasome inhibitors, IMiDs, and daratumumab during multiple myeloma progression. Blood Adv. 2020, 4, 1628–1639. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Zimmerman, T.M.; Hofmeister, C.C.; Talpaz, M.; Chanan-Khan, A.A.; Kaufman, J.L.; Laubach, J.P.; Chauhan, D.; Jakubowiak, A.J.; Reich, S.; et al. Phase 1 study of marizomib in relapsed or relapsed and refractory multiple myeloma: NPI-0052-101 Part 1. Blood 2016, 127, 2693–2700. [Google Scholar] [CrossRef] [PubMed]
- Baranowska, K.; Misund, K.; Starheim, K.K.; Holien, T.; Johansson, I.; Darvekar, S.; Buene, G.; Waage, A.; Bjørkøy, G.; Sundan, A. Hydroxychloroquine potentiates carfilzomib toxicity towards myeloma cells. Oncotarget 2016, 7, 70845–70856. [Google Scholar] [CrossRef] [PubMed]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef]
- Madduri, D.; Berdeja, J.G.; Usmani, M.S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, M.A.K.; Hari, M.P.; Htut, M.; O’Donnell, E.; et al. CARTITUDE-1: Phase 1b/2 Study of Ciltacabtagene Autoleucel, a B-Cell Maturation Antigen-Directed Chimeric Antigen Receptor T Cell Therapy, in Relapsed/Refractory Multiple Myeloma. Blood 2020, 136, 22–25. [Google Scholar] [CrossRef]
- Costa, L.J.; Wong, S.W.; Bermúdez, A.; De La Rubia, J.; Mateos, M.-V.; Ocio, E.M.; Rodríguez-Otero, P.; San-Miguel, J.; Li, S.; Sarmiento, R.; et al. First Clinical Study of the B-Cell Maturation Antigen (BCMA) 2+1 T Cell Engager (TCE) CC-93269 in Patients (Pts) with Relapsed/Refractory Multiple Myeloma (RRMM): Interim Results of a Phase 1 Multicenter Trial. Blood 2019, 134, 143. [Google Scholar] [CrossRef]
- Pillarisetti, K.; Powers, G.; Luistro, L.; Babich, A.; Baldwin, E.; Li, Y.; Zhang, X.; Mendonça, M.; Majewski, N.; Nanjunda, R.; et al. Teclistamab is an active T cell–redirecting bispecific antibody against B-cell maturation antigen for multiple myeloma. Blood Adv. 2020, 4, 4538–4549. [Google Scholar] [CrossRef]
- Cohen, A.D.; Trudel, S.; Forsberg, P.A.; Fonseca, R.; Krishnan, A.Y.; Spencer, A.; Berdeja, J.G.; Hamirani, S.M.; Li, M.; Cooper, J.; et al. GO39775: A multicenter phase I trial evaluating the safety, pharmacokinetics, and activity of BFCR4350A, a FcRH5/CD3 T-cell dependent bispecific antibody, in patients with relapsed or refractory multiple myeloma. J. Clin. Oncol. 2020, 38, TPS8551. [Google Scholar] [CrossRef]
- Trudel, S.; Lendvai, N.; Popat, R.; Voorhees, P.M.; Reeves, B.; Libby, E.N.; Richardson, P.G.; Hoos, A.; Gupta, I.; Bragulat, V.; et al. Antibody–drug conjugate, GSK2857916, in relapsed/refractory multiple myeloma: An update on safety and efficacy from dose expansion phase I study. Blood Cancer J. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- VanWyngarden, M.; Walker, Z.; Su, Y.; Bearrows, S.; Stevens, B.; Forsberg, P.; Mark, T.; Smith, C.; Matsui, W.; Jordan, C.; et al. CD46 Antibody Drug Conjugate Impedes Myeloma Engraftment in Patient-Derived Xenografts. Clin. Lymphoma Myeloma Leuk. 2019, 19, e151. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Delimpasi, S.; Simonova, M.; Spicka, I.; Pour, L.; Kryachok, I.; Gavriatopoulou, M.; Pylypenko, H.; Auner, H.W.; Leleu, X.; et al. Weekly selinexor, bortezomib, and dexamethasone (SVd) versus twice weekly bortezomib and dexamethasone (Vd) in patients with multiple myeloma (MM) after one to three prior therapies: Initial results of the phase III BOSTON study. J. Clin. Oncol. 2020, 38, 8501. [Google Scholar] [CrossRef]
- Vogl, D.T.; Raje, N.; Jagannath, S.; Richardson, P.; Hari, P.; Orlowski, R.; Supko, J.G.; Tamang, D.; Yang, M.; Jones, S.S.; et al. Ricolinostat, the First Selective Histone Deacetylase 6 Inhibitor, in Combination with Bortezomib and Dexamethasone for Relapsed or Refractory Multiple Myeloma. Clin. Cancer Res. 2017, 23, 3307–3315. [Google Scholar] [CrossRef]
- Richardson, P.G.; Oriol, A.; Larocca, A.; Bladé, J.; Cavo, M.; Rodriguez-Otero, P.; Leleu, X.; Nadeem, O.; Hiemenz, J.W.; Hassoun, H.; et al. Melflufen and Dexamethasone in Heavily Pretreated Relapsed and Refractory Multiple Myeloma. J. Clin. Oncol. 2021, 39, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Harrison, S.J.; Cavo, M.; de la Rubia, J.; Popat, R.; Gasparetto, C.; Hungria, V.; Salwender, H.; Suzuki, K.; Kim, I.; et al. Venetoclax or placebo in combination with bortezomib and dexamethasone in patients with relapsed or refractory multiple myeloma (BELLINI): A randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2020, 21, 1630–1642. [Google Scholar] [CrossRef]
- Mertz, J.A.; Conery, A.R.; Bryant, B.M.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Sims, R.J., 3rd. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar] [CrossRef] [PubMed]
- Holien, T.; Våtsveen, T.K.; Hella, H.; Waage, A.; Sundan, A. Addiction to c-MYC in multiple myeloma. Blood 2012, 120, 2450–2453. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Jain, A.D.; Truica, M.I.; Izquierdo-Ferrer, J.; Anker, J.F.; Lysy, B.; Sagar, V.; Luan, Y.; Chalmers, Z.R.; Unno, K.; et al. Small-Molecule MYC Inhibitors Suppress Tumor Growth and Enhance Immunotherapy. Cancer Cell 2019, 36, 483–497.e15. [Google Scholar] [CrossRef] [PubMed]
- Lecker, S.H.; Goldberg, A.L.; Mitch, W.E. Protein Degradation by the Ubiquitin–Proteasome Pathway in Normal and Disease States. J. Am. Soc. Nephrol. 2006, 17, 1807–1819. [Google Scholar] [CrossRef]
- Kubiczkova, L.; Pour, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. Proteasome inhibitors—Molecular basis and current perspectives in multiple myeloma. J. Cell. Mol. Med. 2014, 18, 947–961. [Google Scholar] [CrossRef]
- Besse, A.; Besse, L.; Kraus, M.; Mendez-Lopez, M.; Bader, J.; Xin, B.-T.; de Bruin, G.; Maurits, E.; Overkleeft, H.S.; Driessen, C. Proteasome Inhibition in Multiple Myeloma: Head-to-Head Comparison of Currently Available Proteasome Inhibitors. Cell Chem. Biol. 2019, 26, 340–351.e3. [Google Scholar] [CrossRef]
- Lee, A.-H.; Iwakoshi, N.N.; Anderson, K.C.; Glimcher, L.H. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc. Natl. Acad. Sci. USA 2003, 100, 9946–9951. [Google Scholar] [CrossRef]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef]
- Oerlemans, R.; Franke, N.E.; Assaraf, Y.G.; Cloos, J.; van Zantwijk, I.; Berkers, C.R.; Scheffer, G.L.; Debipersad, K.; Vojtekova, K.; Lemos, C.; et al. Molecular basis of bortezomib resistance: Proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 2008, 112, 2489–2499. [Google Scholar] [CrossRef]
- Ri, M.; Iida, S.; Nakashima, T.; Miyazaki, H.; Mori, F.; Ito, A.; Inagaki, A.; Kusumoto, S.; Ishida, T.; Komatsu, H.; et al. Bortezomib-resistant myeloma cell lines: A role for mutated PSMB5 in preventing the accumulation of unfolded proteins and fatal ER stress. Leukemia 2010, 24, 1506–1512. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Sokol, E.; Jin, D.; Brune, Z.; Thiru, P.; Ghandi, M.; Garraway, L.A.; Gupta, P.B.; Santagata, S.; Whitesell, L.; et al. Suppression of 19S proteasome subunits marks emergence of an altered cell state in diverse cancers. Proc. Natl. Acad. Sci. USA 2016, 114, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Han, H.; Song, S.; Yi, N.; Qian, C.; Qiu, Y.; Zhou, W.; Hong, Y.; Zhuang, W.; Li, Z.; et al. Exosome-Transmitted PSMA3 and PSMA3-AS1 Promote Proteasome Inhibitor Resistance in Multiple Myeloma. Clin. Cancer Res. 2019, 25, 1923–1935. [Google Scholar] [CrossRef]
- Harnoss, J.M.; Le Thomas, A.; Shemorry, A.; Marsters, S.A.; Lawrence, D.A.; Lu, M.; Chen, Y.-C.A.; Qing, J.; Totpal, K.; Kan, D.; et al. Disruption of IRE1α through its kinase domain attenuates multiple myeloma. Proc. Natl. Acad. Sci. USA 2019, 116, 16420–16429. [Google Scholar] [CrossRef] [PubMed]
- Chaidos, A.; Barnes, C.P.; Cowan, G.; May, P.C.; Melo, V.; Hatjiharissi, E.; Papaioannou, M.; Harrington, H.; Doolittle, H.; Terpos, E.; et al. Clinical drug resistance linked to interconvertible phenotypic and functional states of tumor-propagating cells in multiple myeloma. Blood 2013, 121, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Paiva, B.; Puig, N.; Cedena, M.T.; de Jong, B.G.; Ruiz, Y.; Rapado, I.; Martinez-Lopez, J.; Cordon, L.; Alignani, D.; Delgado, J.A.; et al. Differentiation stage of myeloma plasma cells: Biological and clinical significance. Leukemia 2017, 31, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Lipchick, B.C.; Fink, E.E.; Nikiforov, M.A. Oxidative stress and proteasome inhibitors in multiple myeloma. Pharmacol. Res. 2016, 105, 210–215. [Google Scholar] [CrossRef]
- Zaal, E.A.; Wu, W.; Jansen, G.; Zweegman, S.; Cloos, J.; Berkers, C.R. Bortezomib resistance in multiple myeloma is associated with increased serine synthesis. Cancer Metab. 2017, 5, 7. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Detappe, A.; Cai, K.; Keys, H.R.; Brune, Z.; Ying, W.; Thiru, P.; Reidy, M.; Kugener, G.; Rossen, J.; et al. Mitochondrial metabolism promotes adaptation to proteotoxic stress. Nat. Chem. Biol. 2019, 15, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Pengo, N.; Scolari, M.; Oliva, L.; Milan, E.; Mainoldi, F.; Raimondi, A.; Fagioli, C.; Merlini, A.; Mariani, E.; Pasqualetto, E.; et al. Plasma cells require autophagy for sustainable immunoglobulin production. Nat. Immunol. 2013, 14, 298–305. [Google Scholar] [CrossRef]
- Jaganathan, S.; Malek, E.; Vallabhapurapu, S.; Vallabhapurapu, S.; Driscoll, J.J. Bortezomib induces AMPK-dependent autophagosome formation uncoupled from apoptosis in drug resistant cells. Oncotarget 2014, 5, 12358–12370. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Riz, I.; Hawley, T.S.; Hawley, R.G. KLF4-SQSTM1/p62-associated prosurvival autophagy contributes to carfilzomib resistance in multiple myeloma models. Oncotarget 2015, 6, 14814–14831. [Google Scholar] [CrossRef]
- Besse, A.; Besse, A.; Stolze, S.C.; Stolze, S.C.; Rasche, L.; Rasche, L.; Weinhold, N.; Weinhold, N.; Morgan, G.J.; Morgan, G.J.; et al. Carfilzomib resistance due to ABCB1/MDR1 overexpression is overcome by nelfinavir and lopinavir in multiple myeloma. Leukemia 2018, 32, 391–401. [Google Scholar] [CrossRef]
- O’Connor, R.; Ooi, M.G.; Meiller, J.; Jakubikova, J.; Klippel, S.; Delmore, J.; Richardson, P.; Anderson, K.; Clynes, M.; Mitsiades, C.S.; et al. The interaction of bortezomib with multidrug transporters: Implications for therapeutic applications in advanced multiple myeloma and other neoplasias. Cancer Chemother. Pharmacol. 2013, 71, 1357–1368. [Google Scholar] [CrossRef]
- Zhou, W.; Yang, Y.; Xia, J.; Wang, H.; Salama, M.E.; Xiong, W.; Xu, H.; Shetty, S.; Chen, T.; Zeng, Z.; et al. NEK2 Induces Drug Resistance Mainly through Activation of Efflux Drug Pumps and Is Associated with Poor Prognosis in Myeloma and Other Cancers. Cancer Cell 2013, 23, 48–62. [Google Scholar] [CrossRef] [PubMed]
- Abraham, J.; Salama, N.N.; Azab, A.K. The role of P-glycoprotein in drug resistance in multiple myeloma. Leuk. Lymphoma 2014, 56, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhou, W.; Xia, J.; Gu, Z.; Wendlandt, E.; Zhan, X.; Janz, S.; Tricot, G.; Zhan, F. NEK2 mediates ALDH1A1-dependent drug resistance in multiple myeloma. Oncotarget 2014, 5, 11986–11997. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Catley, L.; Li, G.; Podar, K.; Hideshima, T.; Velankar, M.; Mitsiades, C.; Mitsiades, N.; Yasui, H.; Letai, A.; et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell 2005, 8, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Shirazi, F.; Singh, R.K.; Kuiatse, I.; Wang, H.; Lee, H.C.; Berkova, Z.; Berger, A.; Hyer, M.; Chattopadhyay, N.; et al. Ubiquitin-activating enzyme inhibition induces an unfolded protein response and overcomes drug resistance in myeloma. Blood 2019, 133, 1572–1584. [Google Scholar] [CrossRef] [PubMed]
- Le Moigne, R.; Aftab, B.T.; Djakovic, S.; Dhimolea, E.; Valle, E.; Murnane, M.; King, E.M.; Soriano, F.; Menon, M.-K.; Wu, Z.Y.; et al. The p97 Inhibitor CB-5083 Is a Unique Disrupter of Protein Homeostasis in Models of Multiple Myeloma. Mol. Cancer Ther. 2017, 16, 2375–2386. [Google Scholar] [CrossRef]
- Tang, W.K.; Odzorig, T.; Jin, W.; Xia, D. Structural Basis of p97 Inhibition by the Site-Selective Anticancer Compound Compound CB-5083. Mol. Pharmacol. 2019, 95, 286–293. [Google Scholar] [CrossRef]
- Catley, L.; Weisberg, E.; Kiziltepe, T.; Tai, Y.-T.; Hideshima, T.; Neri, P.; Tassone, P.; Atadja, P.; Chauhan, D.; Munshi, N.C.; et al. Aggresome induction by proteasome inhibitor bortezomib and α-tubulin hyperacetylation by tubulin deacetylase (TDAC) inhibitor LBH589 are synergistic in myeloma cells. Blood 2006, 108, 3441–3449. [Google Scholar] [CrossRef]
- Gao, X.; Shen, L.; Li, X.; Liu, J. Efficacy and toxicity of histone deacetylase inhibitors in relapsed/refractory multiple myeloma: Systematic review and meta-analysis of clinical trials. Exp. Ther. Med. 2019, 18, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Walker, Z.J.; Idler, B.M.; Davis, L.N.; Stevens, B.M.; VanWyngarden, M.J.; Ohlstrom, D.; Bearrows, S.C.; Hammes, A.; Smith, C.A.; Jordan, C.T.; et al. Exploiting Protein Translation Dependence in Multiple Myeloma with Omacetaxine-Based Therapy. Clin. Cancer Res. 2021, 27, 819–830. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; San-Miguel, J.; Belch, A.; White, D.; Benboubker, L.; Cook, G.; Leiba, M.; Morton, J.; Ho, P.J.; Kim, K.; et al. Daratumumab plus lenalidomide and dexamethasone versus lenalidomide and dexamethasone in relapsed or refractory multiple myeloma: Updated analysis of POLLUX. Haematology 2018, 103, 2088–2096. [Google Scholar] [CrossRef] [PubMed]
- Lokhorst, H.M.; Plesner, T.; Laubach, J.P.; Nahi, H.; Gimsing, P.; Hansson, M.; Minnema, M.C.; Lassen, U.; Krejcik, J.; Palumbo, A.; et al. Targeting CD38 with Daratumumab Monotherapy in Multiple Myeloma. N. Engl. J. Med. 2015, 373, 1207–1219. [Google Scholar] [CrossRef] [PubMed]
- De Weers, M.; Tai, Y.-T.; Van Der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.H.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a Novel Therapeutic Human CD38 Monoclonal Antibody, Induces Killing of Multiple Myeloma and Other Hematological Tumors. J. Immunol. 2010, 186, 1840–1848. [Google Scholar] [CrossRef] [PubMed]
- Overdijk, M.B.; Verploegen, S.; Bögels, M.; Van Egmond, M.; Van Bueren, J.J.L.; Mutis, T.; Groen, R.W.J.; Breij, E.; Martens, A.C.M.; Bleeker, W.K.; et al. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. mAbs 2015, 7, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Overdijk, M.B.; Jansen, J.H.; Nederend, M.; Lammerts van Bueren, J.J.; Groen, R.W.; Parren, P.W.; Leusen, J.H.; Boross, P. The Therapeutic CD38 Monoclonal Antibody Daratumumab Induces Programmed Cell Death via Fcgamma Receptor-Mediated Cross-Linking. J. Immunol. 2016, 197, 807–813. [Google Scholar] [CrossRef]
- Lonial, S.; Dimopoulos, M.A.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.V.; Magen, H.; et al. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Oriol, A.; Nahi, H.; San-Miguel, J.; Bahlis, N.J.; Usmani, S.Z.; Rabin, N.; Orlowski, R.Z.; Komarnicki, M.; Suzuki, K.; et al. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 1319–1331. [Google Scholar] [CrossRef]
- Acha, O.P.D.; Idler, B.M.; Walker, Z.J.; Forsberg, P.A.; Mark, T.M.; Sherbenou, D.W. Myeloma Drug Sensitivity Testing to Optimize Retreatment with Anti-CD38 Monoclonal Antibodies in Daratumumab-Refractory Patients. Blood 2020, 136, 36–37. [Google Scholar]
- Seckinger, A.; Delgado, J.A.; Moser, S.; Moreno, L.; Neuber, B.; Grab, A.; Lipp, S.; Merino, J.; Prosper, F.; Emde, M.; et al. Target Expression, Generation, Preclinical Activity, and Pharmacokinetics of the BCMA-T Cell Bispecific Antibody EM801 for Multiple Myeloma Treatment. Cancer Cell 2017, 31, 396–410. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, J.L.D.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Raje, N.S.; Jakubowiak, A.; Gasparetto, C.; Cornell, R.F.; Krupka, H.I.; Navarro, D.; Forgie, A.J.; Udata, C.; Basu, C.; Chou, J.; et al. Safety, Clinical Activity, Pharmacokinetics, and Pharmacodynamics from a Phase I Study of PF-06863135, a B-Cell Maturation Antigen (BCMA)-CD3 Bispecific Antibody, in Patients with Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2019, 134, 1869. [Google Scholar] [CrossRef]
- Topp, M.S.; Duell, J.; Zugmaier, G.; Attal, M.; Moreau, P.; Langer, C.; Kroenke, J.; Facon, T.; Salnikov, A.; Lesley, R.; et al. Evaluation of AMG 420, an anti-BCMA bispecific T-cell engager (BiTE) immunotherapy, in R/R multiple myeloma (MM) patients: Updated results of a first-in-human (FIH) phase I dose escalation study. J. Clin. Oncol. 2019, 37, 8007. [Google Scholar] [CrossRef]
- Sherbenou, D.W.; Aftab, B.T.; Su, Y.; Behrens, C.R.; Wiita, A.; Logan, A.C.; Acosta-Alvear, D.; Hann, B.C.; Walter, P.; Shuman, M.A.; et al. Antibody-drug conjugate targeting CD46 eliminates multiple myeloma cells. J. Clin. Investig. 2016, 126, 4640–4653. [Google Scholar] [CrossRef] [PubMed]
- Pillarisetti, K.; Edavettal, S.; Mendonça, M.; Li, Y.; Tornetta, M.; Babich, A.; Majewski, N.; Husovsky, M.; Reeves, D.; Walsh, E.; et al. A T-cell–redirecting bispecific G-protein–coupled receptor class 5 member D x CD3 antibody to treat multiple myeloma. Blood 2020, 135, 1232–1243. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.L.; Harrington, K.; Staehr, M.; Masakayan, R.; Jones, J.; Long, T.J.; Ng, K.Y.; Ghoddusi, M.; Purdon, T.J.; Wang, X.; et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci. Transl. Med. 2019, 11, eaau7746. [Google Scholar] [CrossRef]
- Kervoëlen, C.; Ménoret, E.; Gomez-Bougie, P.; Bataille, R.; Godon, C.; Marionneau-Lambot, S.; Moreau, P.; Pellat-Deceunynck, C.; Amiot, M. Dexamethasone-induced cell death is restricted to specific molecular subgroups of multiple myeloma. Oncotarget 2015, 6, 26922–26934. [Google Scholar] [CrossRef]
- Gkotzamanidou, M.; Terpos, E.; Bamia, C.; Munshi, N.C.; Dimopoulos, M.A.; Souliotis, V.L. DNA repair of myeloma plasma cells correlates with clinical outcome: The effect of the nonhomologous end-joining inhibitor SCR7. Blood 2016, 128, 1214–1225. [Google Scholar] [CrossRef] [PubMed]
- Wickström, M.; Nygren, P.; Larsson, R.; Harmenberg, J.; Lindberg, J.; Sjöberg, P.; Jerling, M.; Lehmann, F.; Richardson, P.; Anderson, K.; et al. Melflufen—A peptidase-potentiated alkylating agent in clinical trials. Oncotarget 2017, 8, 66641–66655. [Google Scholar] [CrossRef]
- Muz, B.; Azab, F.; De La Puente, P.; Landesman, Y.; Azab, A.K. Selinexor Overcomes Hypoxia-Induced Drug Resistance in Multiple Myeloma. Transl. Oncol. 2017, 10, 632–640. [Google Scholar] [CrossRef]
- Chari, A.; Vogl, D.T.; Gavriatopoulou, M.; Nooka, A.K.; Yee, A.J.; Huff, C.A.; Moreau, P.; Dingli, D.; Cole, C.; Lonial, S.; et al. Oral Selinexor–Dexamethasone for Triple-Class Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 381, 727–738. [Google Scholar] [CrossRef]
- Grosicki, S.; Simonova, M.; Spicka, I.; Pour, L.; Kriachok, I.; Gavriatopoulou, M.; Pylypenko, H.; Auner, H.W.; Leleu, X.; Doronin, V.; et al. Once-per-week selinexor, bortezomib, and dexamethasone versus twice-per-week bortezomib and dexamethasone in patients with multiple myeloma (BOSTON): A randomised, open-label, phase 3 trial. Lancet 2020, 396, 1563–1573. [Google Scholar] [CrossRef]
- Touzeau, C.; Ryan, J.; Guerriero, J.L.; Moreau, P.; Ni Chonghaile, T.; Le Gouill, S.; Richardson, P.G.; Anderson, K.C.; Amiot, M.; Letai, A. BH3 profiling identifies heterogeneous dependency on Bcl-2 family members in multiple myeloma and predicts sensitivity to BH3 mimetics. Leukemia 2016, 30, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Chanan-Khan, A.; Roberts, A.W.; Agarwal, A.B.; Facon, T.; Kumar, S.; Touzeau, C.; Punnoose, E.A.; Cordero, J.; Munasinghe, W.; et al. Promising efficacy and acceptable safety of venetoclax plus bortezomib and dexamethasone in relapsed/refractory MM. Blood 2017, 130, 2392–2400. [Google Scholar] [CrossRef] [PubMed]
- Punnoose, E.A.; Leverson, J.D.; Peale, F.; Boghaert, E.R.; Belmont, L.D.; Tan, N.; Young, A.; Mitten, M.; Ingalla, E.; Darbonne, W.C.; et al. Expression Profile of BCL-2, BCL-XL, and MCL-1 Predicts Pharmacological Response to the BCL-2 Selective Antagonist Venetoclax in Multiple Myeloma Models. Mol. Cancer Ther. 2016, 15, 1132–1144. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, B.; Han, H.; Song, S.; Xu, H.; Hong, Y.; Yi, N.; Zhuang, W. Predicting multi-level drug response with gene expression profile in multiple myeloma using hierarchical ordinal regression. BMC Cancer 2018, 18, 551. [Google Scholar] [CrossRef]
- Cetin, A.E.; Stevens, M.M.; Calistri, N.L.; Fulciniti, M.; Olcum, S.A.; Kimmerling, R.J.; Munshi, N.C.; Manalis, S.R. Determining therapeutic susceptibility in multiple myeloma by single-cell mass accumulation. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Stessman, H.A.; Baughn, L.B.; Sarver, A.; Xia, T.; Deshpande, R.; Mansoor, A.; Walsh, S.A.; Sunderland, J.J.; Dolloff, N.G.; Linden, M.A.; et al. Profiling Bortezomib Resistance Identifies Secondary Therapies in a Mouse Myeloma Model. Mol. Cancer Ther. 2013, 12, 1140–1150. [Google Scholar] [CrossRef]
- Kawano, Y.; Kikukawa, Y.; Fujiwara, S.; Wada, N.; Okuno, Y.; Mitsuya, H.; Hata, H. Hypoxia reduces CD138 expression and induces an immature and stem cell-like transcriptional program in myeloma cells. Int. J. Oncol. 2013, 43, 1809–1816. [Google Scholar] [CrossRef]
- Akhmetzyanova, I.; McCarron, M.J.; Parekh, S.; Chesi, M.; Bergsagel, P.L.; Fooksman, D.R. Dynamic CD138 surface expression regulates switch between myeloma growth and dissemination. Leukemia 2020, 34, 245–256. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).