
Therapeutic Targeting of Protein Disulfide Isomerase PDIA1 in Multiple Myeloma

 , , ,
, , , 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods

3. Results
3.1. Expression of PDIA1 Inversely Correlates with Survival in Patients with Relapsed or Refractory Myeloma
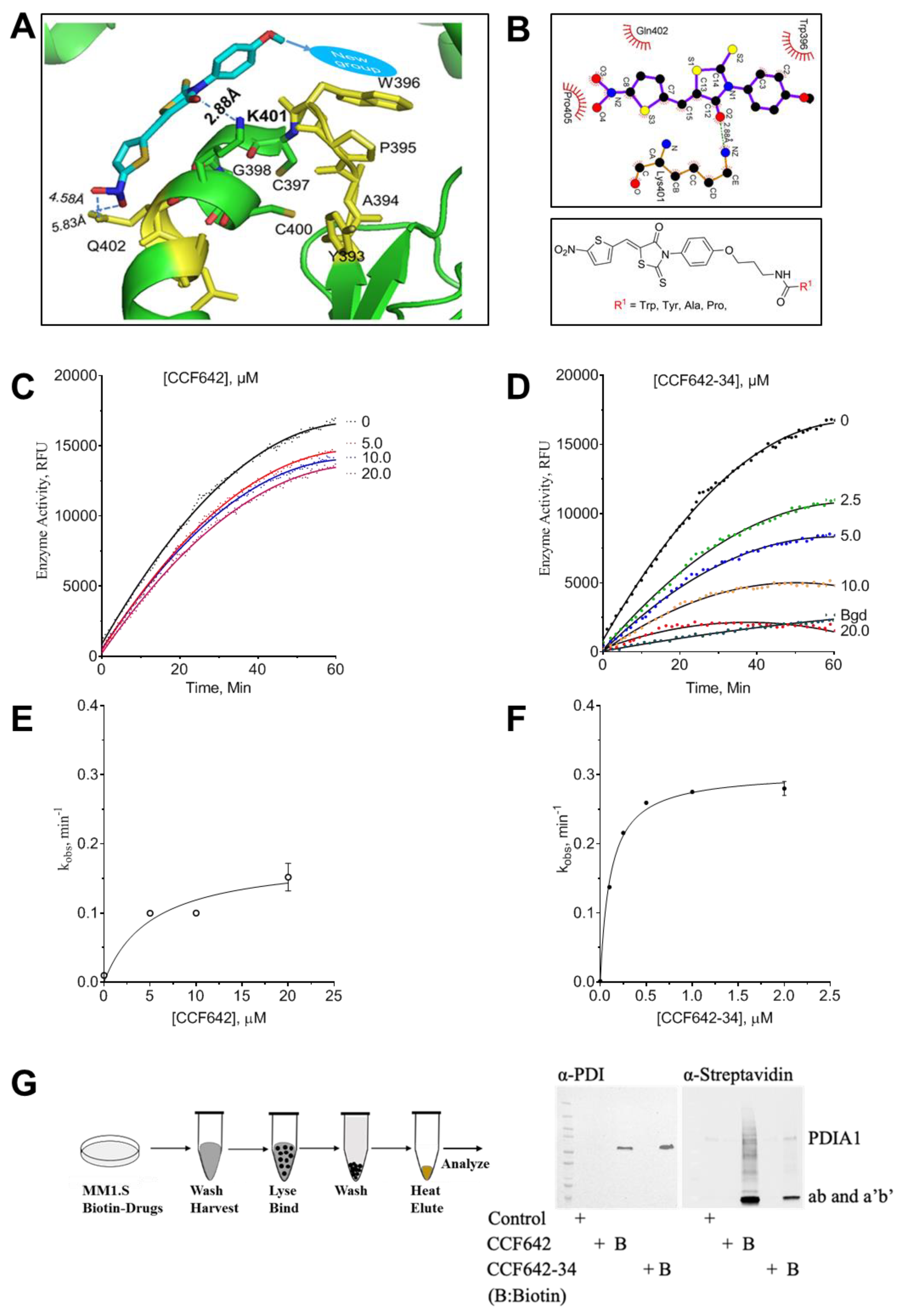
3.2. Development of a Potent PDIA1 Specific Inhibitor
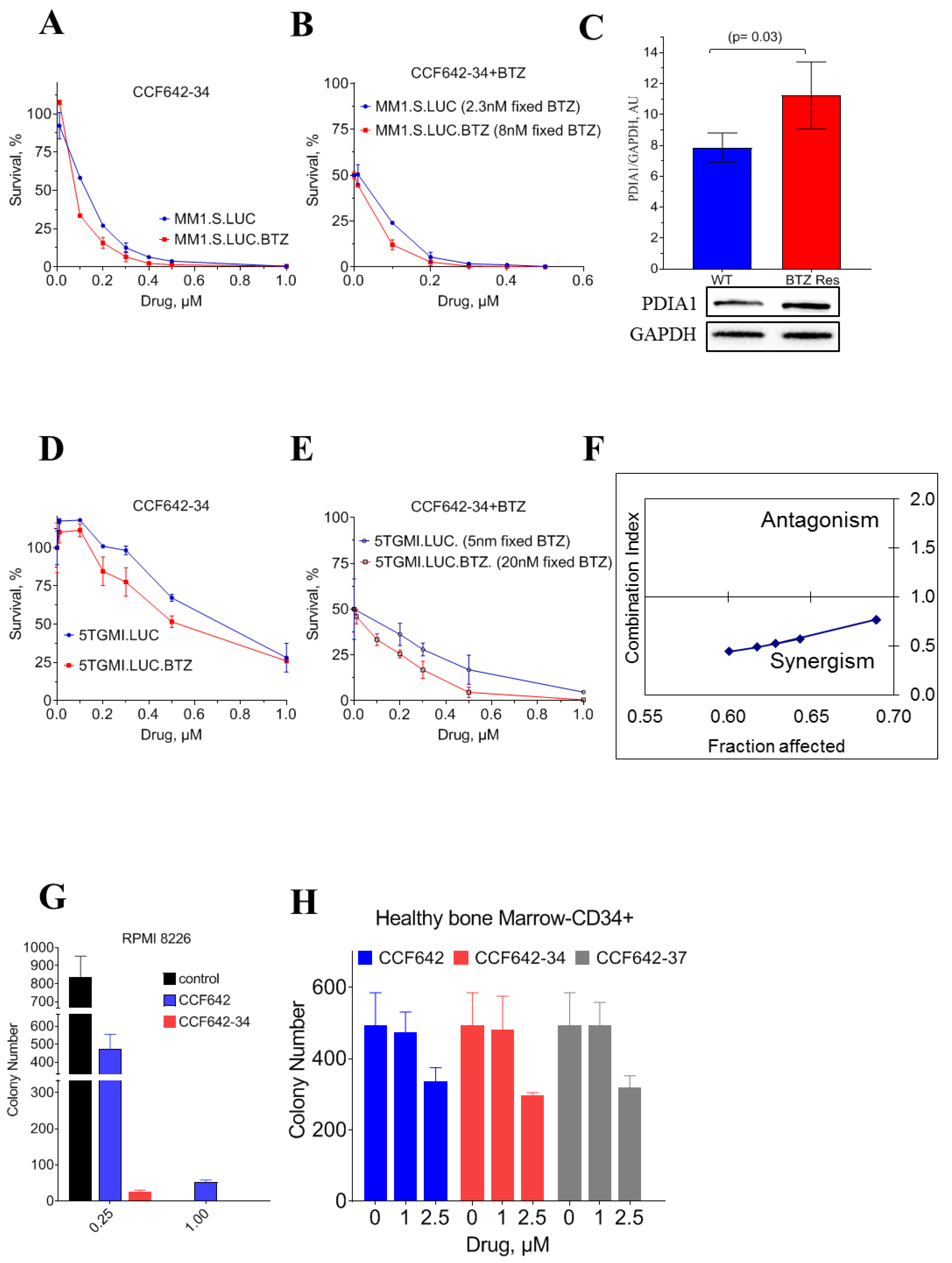
3.3. CCF642-34 Inhibits the Growth of Myeloma Cells without Any Significant Effects on Normal Bone Marrow-Derived CD34+ Cells
3.4. CCF642 Analogues Induce Acute ER Stress Response Followed by Apoptosis in MM1.S Cells
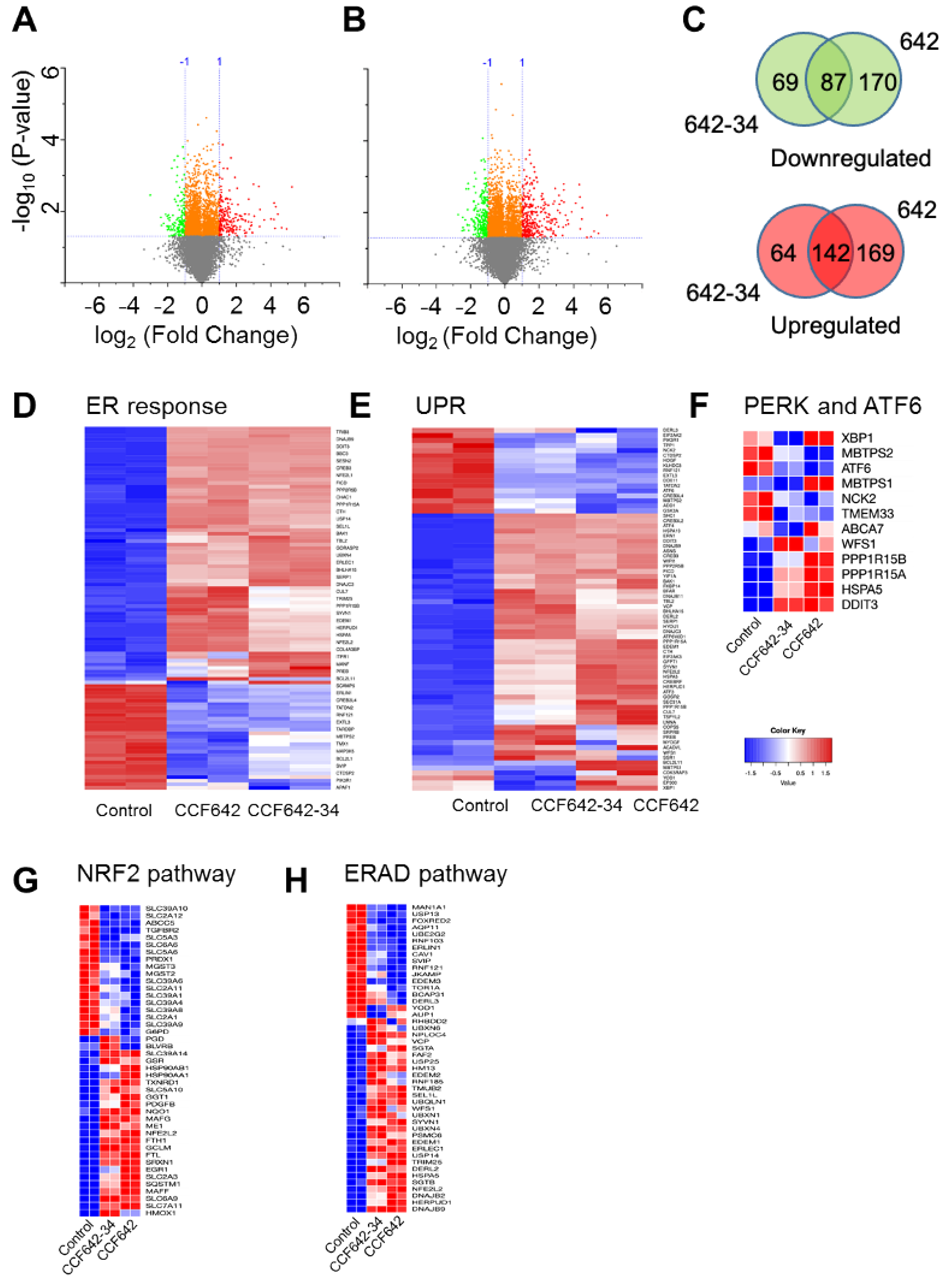
3.5. Greater Selectivity of CCF642-34 for PDIA1 Inhibition Translates into a Narrower Band Gene Expression Profile Than CCF642
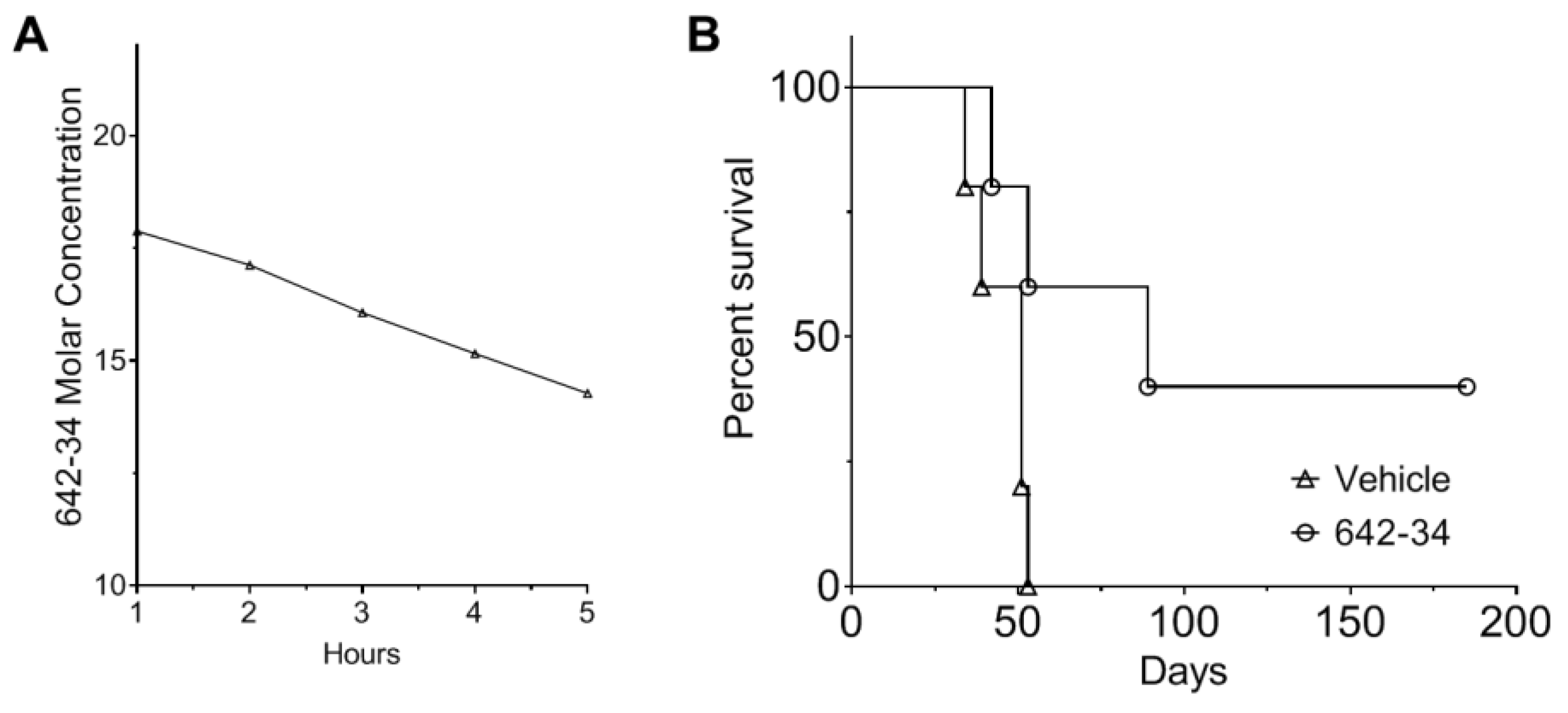
3.6. CCF642-34 Is Pharmacologically Stable to Acid Exposure and Does Not Undergo Rapid Hepatic Metabolism
3.7. CCF642-34 Prolonged Survival of Mice in the 5TGM1 Syngeneic Mouse Model of Myeloma
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement (ICF)
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Manier, S.; Salem, K.Z.; Park, J.; Landau, D.A.; Getz, G.; Ghobrial, I.M. Genomic complexity of multiple myeloma and its clinical im-plications. Nat. Rev. Clin. Oncol. 2017, 14, 100–113. [Google Scholar] [CrossRef]
- Weiss, B.M.; Abadie, J.; Verma, P.; Howard, R.S.; Kuehl, W.M. A monoclonal gammopathy precedes multiple myeloma in most pa-tients. Blood 2009, 113, 5418–5422. [Google Scholar] [CrossRef]
- Masciarelli, S.; Sitia, R. Building and operating an antibody factory: Redox control during B to plasma cell terminal differentia-tion. Biochim. Biophys. Acta 2008, 1783, 578–588. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef]
- Hideshima, T.; Richardson, P.; Chauhan, D.; Palombella, V.J.; Elliott, P.J.; Adams, J.; Anderson, K.C. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001, 61, 3071–3076. [Google Scholar]
- Gandhi, U.H.; Cornell, R.F.; Lakshman, A.; Gahvari, Z.J.; McGehee, E.; Jagosky, M.H.; Gupta, R.; Varnado, W.; Fiala, M.A.; Chhabra, S.; et al. Outcomes of patients with multiple myelo-ma refractory to CD38-targeted monoclonal antibody therapy. Leukemia 2019, 33, 2266–2275. [Google Scholar] [CrossRef]
- Fonseca, R.; Bergsagel, P.L.; Drach, J.; Shaughnessy, J.; Gutierrez, N.; Stewart, A.K.; Morgan, G.; Van Ness, B.; Chesi, M.; Minvielle, S.; et al. International Myeloma Working Group mo-lecular classification of multiple myeloma: Spotlight review. Leukemia 2009, 23, 2210–2221. [Google Scholar] [CrossRef]
- Ribatti, D. Edelman’s View on the Discovery of Antibodies. Immunol. Lett. 2015, 164, 59–68. [Google Scholar] [CrossRef]
- Freedman, R.B.; Hirst, T.R.; Tuite, M.F. Protein disulphide isomerase: Building bridges in protein folding. Trends Biochem. Sci. 1994, 19, 331–336. [Google Scholar] [CrossRef]
- Kersteen, E.A.; Raines, R.T. Catalysis of Protein Folding by Protein Disulfide Isomerase and Small-Molecule Mimics. Antioxid. Redox Signal. 2003, 5, 413–424. [Google Scholar] [CrossRef]
- Laurindo, F.R.; Pescatore, L.A.; Fernandes, D.D.C. Protein disulfide isomerase in redox cell signaling and homeostasis. Free. Radic. Biol. Med. 2012, 52, 1954–1969. [Google Scholar] [CrossRef]
- Turano, C.; Coppari, S.; Altieri, F.; Ferraro, A. Proteins of the PDI family: Unpredicted non-ER locations and functions. J. Cell. Physiol. 2002, 193, 154–163. [Google Scholar] [CrossRef]
- Beer, D.G.; Kardia, S.L.; Huang, C.C.; Giordano, T.J.; Levin, A.M.; Misek, D.E.; Lin, L.; Chen, G.; Gharib, T.G.; Thomas, D.G.; et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat. Med. 2002, 8, 816–824. [Google Scholar] [CrossRef]
- Rho, J.-H.; Roehrl, M.H.A.; Wang, J.Y. Glycoproteomic Analysis of Human Lung Adenocarcinomas Using Glycoarrays and Tandem Mass Spectrometry: Differential Expression and Glycosylation Patterns of Vimentin and Fetuin A Isoforms. Protein J. 2009, 28, 148–160. [Google Scholar] [CrossRef]
- Shin, B.K.; Wang, H.; Yim, A.M.; Le Naour, F.; Brichory, F.; Jang, J.H.; Zhao, R.; Puravs, E.; Tra, J.; Michael, C.W.; et al. Global Profiling of the Cell Surface Proteome of Cancer Cells Uncovers an Abundance of Proteins with Chaperone Function. J. Biol. Chem. 2003, 278, 7607–7616. [Google Scholar] [CrossRef]
- Xu, S.; Sankar, S.; Neamati, N. Protein disulfide isomerase: A promising target for cancer therapy. Drug Discov. Today 2014, 19, 222–240. [Google Scholar] [CrossRef]
- Yu, S.J.; Won, J.K.; Ryu, H.S.; Choi, W.M.; Cho, H.; Cho, E.J.; Lee, J.H.; Kim, Y.J.; Suh, K.S.; Jang, J.J.; et al. A novel prognostic factor for hepatocellular carcinoma: Protein di-sulfide isomerase. Korean J. Intern. Med. 2014, 29, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Vincenz-Donnelly, L.; Jäger, R.; O’Dwyer, M.; Samali, A. Endoplasmic Reticulum Stress and the Unfolded Protein Response: Targeting the Achilles Heel of Multiple Myeloma. Mol. Cancer Ther. 2013, 12, 831–843. [Google Scholar] [CrossRef]
- Vatolin, S.; Phillips, J.G.; Jha, B.K.; Govindgari, S.; Hu, J.; Grabowski, D.; Parker, Y.; Lindner, D.J.; Zhong, F.; Distelhorst, C.W.; et al. Novel Protein Disulfide Isomerase Inhibitor with An-ticancer Activity in Multiple Myeloma. Cancer Res. 2016, 76, 3340–3350. [Google Scholar] [CrossRef] [PubMed]
- Dallas, S.L.; Garrett, I.R.; Oyajobi, B.O.; Dallas, M.R.; Boyce, B.F.; Bauss, F.; Radl, J.; Mundy, G.R. Ibandronate reduces osteolytic lesions but not tumor burden in a murine model of myeloma bone disease. Blood 1999, 93, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R.M.; Reyes, L.; Duncan, R.M.; Bian, H.; Reitz, A.B.; Manevich, Y.; McClure, J.J.; Champion, M.M.; Chou, C.J.; Sharik, M.E.; et al. Inhibitors of the protein disulfide isomerase family for the treatment of multiple myeloma. Leukemia 2019, 33, 1011–1022. [Google Scholar] [CrossRef]
- Gu, X.; Ebrahem, Q.; Mahfouz, R.Z.; Hasipek, M.; Enane, F.; Radivoyevitch, T.; Rapin, N.; Przychodzen, B.; Hu, Z.; Balusu, R.; et al. Leukemogenic nucleophosmin mutation disrupts the transcription factor hub that regulates granulomonocytic fates. J. Clin. Investig. 2018, 128, 4260–4279. [Google Scholar] [CrossRef]
- Guan, Y.; Greenberg, E.F.; Hasipek, M.; Chen, S.; Liu, X.; Kerr, C.M.; Gackowski, D.; Zarakowska, E.; Radivoyevitch, T.; Gu, X.; et al. Context dependent effects of ascorbic acid treatment in TET2 mutant myeloid neoplasia. Commun. Biol. 2020, 3, 1–13. [Google Scholar] [CrossRef]
- Wang, C.; Li, W.; Ren, J.; Fang, J.; Ke, H.; Gong, W.; Feng, W.; Wang, C.C. Structural insights into the redox-regulated dynamic conformations of hu-man protein disulfide isomerase. Antioxid. Redox Signal. 2013, 19, 36–45. [Google Scholar] [CrossRef]
- Raturi, A.; Mutus, B. Characterization of redox state and reductase activity of protein disulfide isomerase under different redox environments using a sensitive fluorescent assay. Free. Radic. Biol. Med. 2007, 43, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Dobrusin, E.M.; Fry, D.W.; Haske, T.; Whitty, A.; McNamara, D.J. Structure-based design of a potent, selective, and irreversi-ble inhibitor of the catalytic domain of the erbB receptor subfamily of protein tyrosine kinases. J. Med. Chem. 1997, 40, 1130–1135. [Google Scholar] [CrossRef]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef]
- Leo, A.J.; Hoekman, D. Calculating log P(oct) with no missing fragments;The problem of estimating new interaction parame-ters. Perspect. Drug Discov. Des. 2000, 18, 19–38. [Google Scholar] [CrossRef]
- Mannhold, R.; Rekker, R.F. The hydrophobic fragmental constant approach forcalculating log P in octanol/water and aliphatic hydrocarbon/water systems. Perspect. Drug Discov. Des. 2000, 18, 1–18. [Google Scholar] [CrossRef]
- Robinson, R.M.; Reyes, L.; Duncan, R.M.; Bian, H.; Strobel, E.D.; Hyman, S.L.; Reitz, A.B.; Dolloff, N.G. Tuning isoform selectivity and bortezomib sensi-tivity with a new class of alkenyl indene PDI inhibitor. Eur. J. Med. Chem. 2020, 186, 111906. [Google Scholar] [CrossRef]
- Chou, T.-C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]
- Ellgaard, L.; Sevier, C.S.; Bulleid, N.J. How Are Proteins Reduced in the Endoplasmic Reticulum? Trends Biochem. Sci. 2018, 43, 32–43. [Google Scholar] [CrossRef]
- Eyers, P.A.; Keeshan, K.; Kannan, N. Tribbles in the 21st Century: The Evolving Roles of Tribbles Pseudokinases in Biology and Disease. Trends Cell Biol. 2017, 27, 284–298. [Google Scholar] [CrossRef]
- Du, K.; Herzig, S.; Kulkarni, R.N.; Montminy, M. TRB3: A tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science 2003, 300, 1574–1577. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, K.R.; Chaudhary, M.; Kim, H.-R.; Chae, H.-J. Endoplasmic Reticulum (ER) Stress Response Failure in Diseases. Trends Cell Biol. 2020, 30, 672–675. [Google Scholar] [CrossRef]
- Ron, D.; Harding, H.P. Protein-Folding Homeostasis in the Endoplasmic Reticulum and Nutritional Regulation. Cold Spring Harb. Perspect. Biol. 2012, 4, a013177. [Google Scholar] [CrossRef] [PubMed]
- Radl, J.; Croese, J.W.; Zurcher, C.; Van den Enden-Vieveen, M.H.; de Leeuw, A.M. Animal model of human disease. Multiple mye-loma. Am. J. Pathol. 1988, 132, 593–597. [Google Scholar] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Organism | Disease | Source | Media |
|---|---|---|---|---|
| MM1.S.luc | human | IgA lambda myeloma | ATCC | RPMI-1640 * |
| MM1.S.luc BtzR | human | Ig A lambda myeloma | CCF Lab | |
| RPMI-8226 | human | Plasmacytoma; myeloma | ATCC | |
| 5TGM1-luc | murine | Plasmacytoma; myeloma | Gift from Dr. Oyajobi [13] | IMDM * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasipek, M.; Grabowski, D.; Guan, Y.; Alugubelli, R.R.; Tiwari, A.D.; Gu, X.; DeAvila, G.A.; Silva, A.S.; Meads, M.B.; Parker, Y.; et al. Therapeutic Targeting of Protein Disulfide Isomerase PDIA1 in Multiple Myeloma. Cancers 2021, 13, 2649. https://doi.org/10.3390/cancers13112649
Hasipek M, Grabowski D, Guan Y, Alugubelli RR, Tiwari AD, Gu X, DeAvila GA, Silva AS, Meads MB, Parker Y, et al. Therapeutic Targeting of Protein Disulfide Isomerase PDIA1 in Multiple Myeloma. Cancers. 2021; 13(11):2649. https://doi.org/10.3390/cancers13112649
Chicago/Turabian StyleHasipek, Metis, Dale Grabowski, Yihong Guan, Raghunandan Reddy Alugubelli, Anand D. Tiwari, Xiaorong Gu, Gabriel A. DeAvila, Ariosto S. Silva, Mark B. Meads, Yvonne Parker, and et al. 2021. "Therapeutic Targeting of Protein Disulfide Isomerase PDIA1 in Multiple Myeloma" Cancers 13, no. 11: 2649. https://doi.org/10.3390/cancers13112649
APA StyleHasipek, M., Grabowski, D., Guan, Y., Alugubelli, R. R., Tiwari, A. D., Gu, X., DeAvila, G. A., Silva, A. S., Meads, M. B., Parker, Y., Lindner, D. J., Saunthararajah, Y., Shain, K. H., Maciejewski, J. P., Reu, F. J., Phillips, J. G., & Jha, B. K. (2021). Therapeutic Targeting of Protein Disulfide Isomerase PDIA1 in Multiple Myeloma. Cancers, 13(11), 2649. https://doi.org/10.3390/cancers13112649