
Residual Disease in Glioma Recurrence: A Dangerous Liaison with Senescence

Abstract
Simple Summary
Abstract
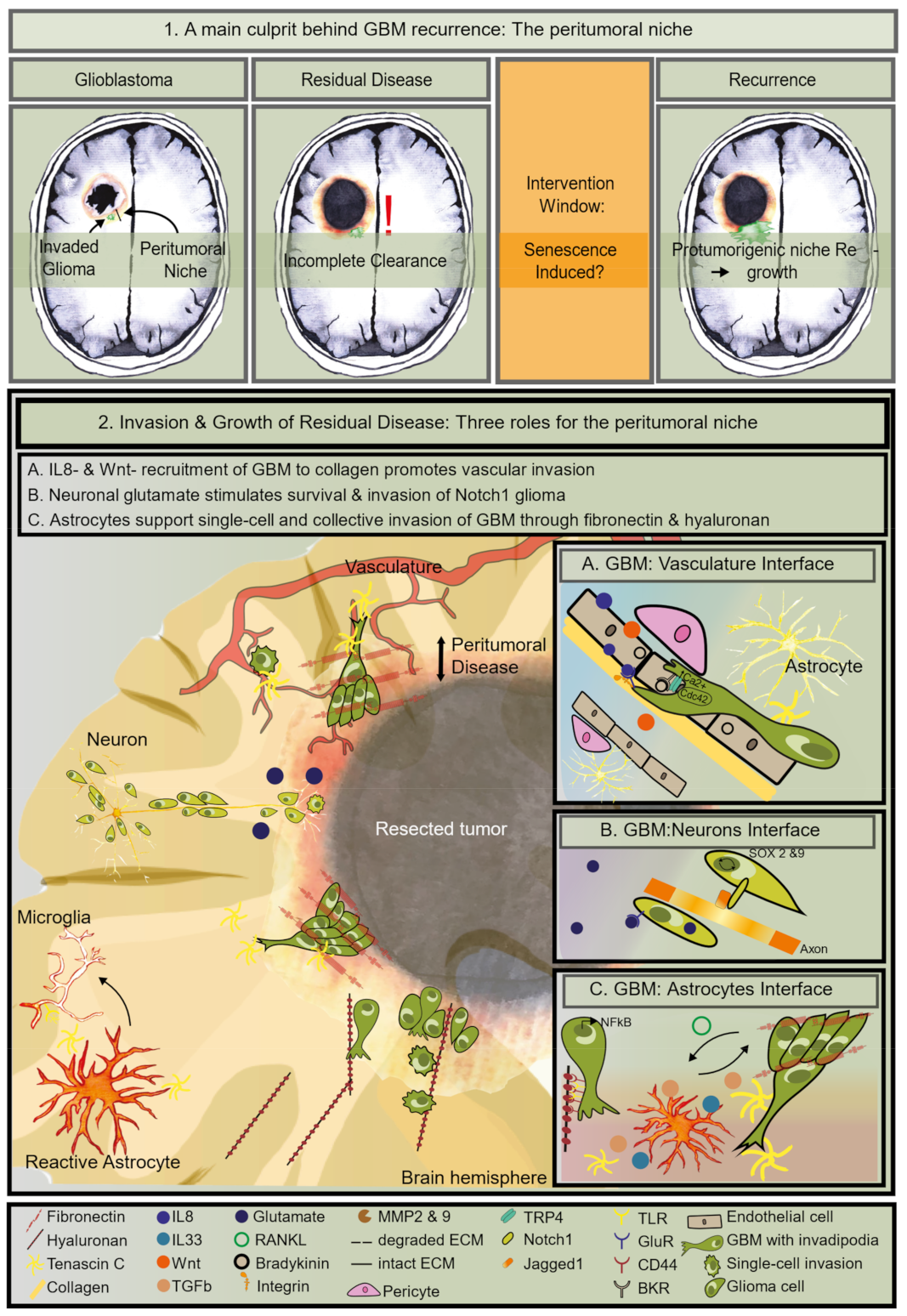
1. Residual Disease Is the Main Contributor to Treatment Failure of GBM
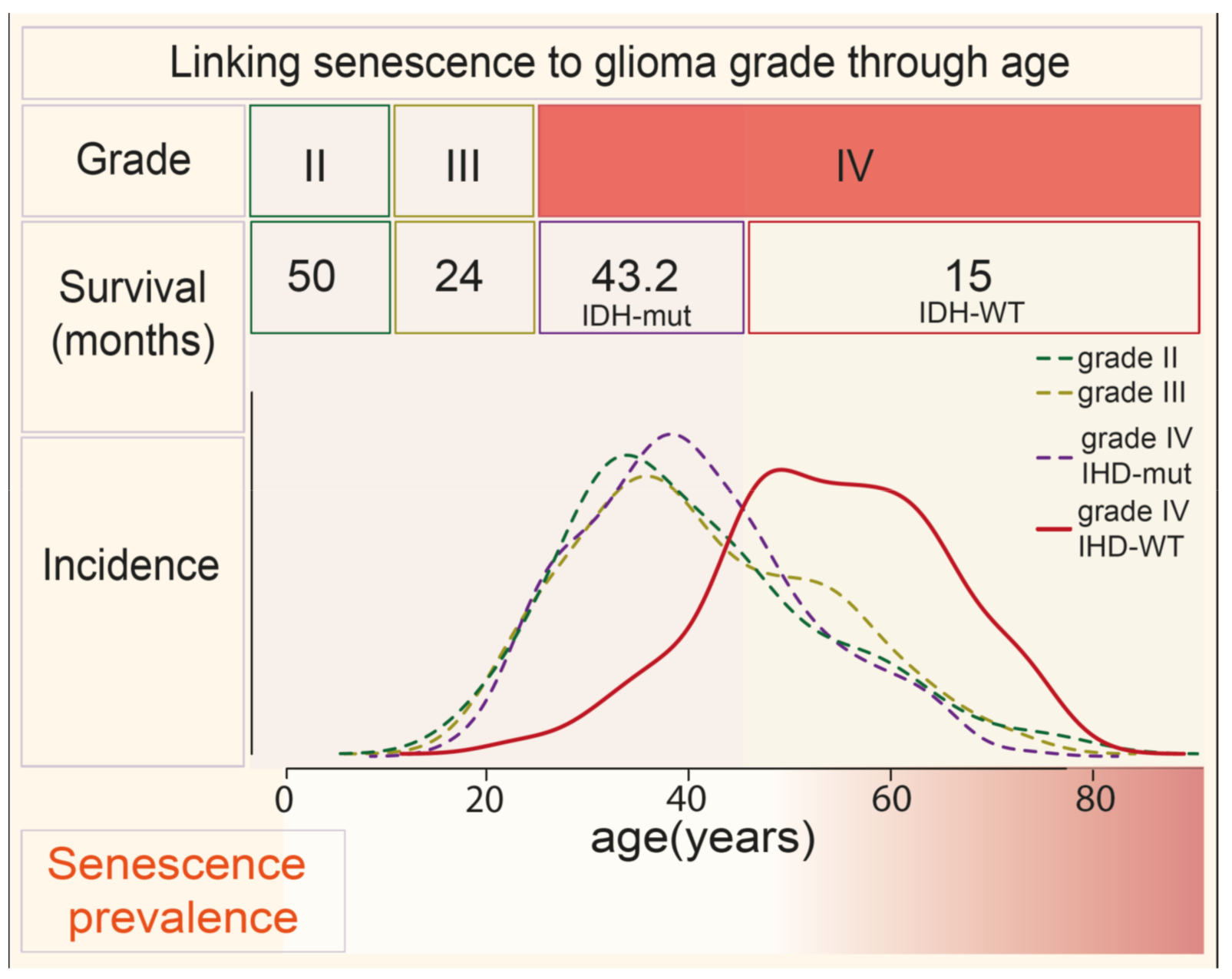
2. Aging Is a Risk Factor for Advanced GBM and Treatment Resistance—A Connection with Senescence?
3. Senescence Is a Causal Driver of Aging and Cancer
4. The Identity of the Tumor Cells in the Peritumoral Zone
4.1. Heterogeneity and Plasticity at the Tumor’s Edge by Pro-Inflammatory Signals
4.2. The GBM Tumor Micro-Environment (TME) Determines GBM Cancer Stemness
5. The Right Niche to Subsist in the Peritumoral Zone
5.1. Enhanced GBM Invasion Features
5.2. The Vascular Niche Supports Residual Invasion
5.3. The Neuron Niche Supports Growth
5.4. The Astrocyte Niche Promotes Invasion and Mesenchymal Transformation
6. Factors Secreted by Senescent Cells Create a Supportive Niche for GBM Recurrence
7. Conclusions: Senescence in the Tumor Niche Is Likely to Favor Advanced GBM and Post-Treatment Recurrence
8. Future Perspectives: Strategies to Target Senescence in Glioma
Author Contributions
Funding
Conflicts of Interest
References
- DeAngelis, L.M.; Wen, P.Y. Primary and Metastatic Tumors of the Nervous System. In Harrison’s Principles of Internal Medicine, 20th ed.; Shanahan, J.F., Davis, K.J., Eds.; McGraw-Hill Professional Publishing: New York, NY, USA, 2018; Chapter 86; p. 31. [Google Scholar]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Grech, N.; Dalli, T.; Mizzi, S.; Meilak, L.; Calleja, N.; Zrinzo, A. Rising Incidence of Glioblastoma Multiforme in a Well-Defined Population. Cureus 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Philips, A.; Henshaw, D.L.; Lamburn, G.; O’Carroll, M.J. Brain Tumours: Rise in Glioblastoma Multiforme Incidence in England 1995–2015 Suggests an Adverse Environmental or Lifestyle Factor. J. Environ. Public Health 2018, 2018, 1–10. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Tykocki, T.; Eltayeb, M. Ten-year survival in glioblastoma. A systematic review. J. Clin. Neurosci. 2018, 54, 7–13. [Google Scholar] [CrossRef]
- De Gooijer, M.C.; Navarro, M.G.; Bernards, R.; Wurdinger, T.; van Tellingen, O. An Experimenter’s Guide to Glioblastoma Invasion Pathways. Trends Mol. Med. 2018, 24, 763–780. [Google Scholar] [CrossRef]
- Chaichana, K.L.; Jusue-Torres, I.; Navarro-Ramirez, R.; Raza, S.M.; Pascual-Gallego, M.; Ibrahim, A.; Hernandez-Hermann, M.; Gomez, L.; Ye, X.; Weingart, J.D.; et al. Establishing percent resection and residual volume thresholds affecting survival and recurrence for patients with newly diagnosed intracranial glioblastoma. Neuro-Oncology 2014, 16, 113–122. [Google Scholar] [CrossRef]
- Lacroix, M.; Abi-Said, D.; Fourney, D.R.; Gokaslan, Z.L.; Shi, W.; Demonte, F.; Lang, F.F.; McCutcheon, I.E.; Hassenbusch, S.J.; Holland, E.; et al. A multivariate analysis of 416 patients with glioblastoma multiforme: Prognosis, extent of resection, and survival. J. Neurosurg. 2001, 95, 190–198. [Google Scholar] [CrossRef]
- Li, Y.M.; Suki, D.; Hess, K.R.; Sawaya, R. The influence of maximum safe resection of glioblastoma on survival in 1229 patients: Can we do better than gross-total resection? J. Neurosurg. 2016, 124, 977–988. [Google Scholar] [CrossRef]
- Sanai, N.; Polley, M.Y.; McDermott, M.W.; Parsa, A.T.; Berger, M.S. An extent of resection threshold for newly diagnosed glioblastomas. J. Neurosurg. 2011, 115, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.; Horton, J.; Scott, C.; Curran, W.; Rubin, P.; Fischbach, J.; Isaacson, S.; Rotman, M.; Asbell, S.; Nelson, J.; et al. Influence of location and extent of surgical resection on survival of patients with glioblastoma multiforme: Results of three consecutive radiation therapy oncology group (RTOG) clinical trials. Int. J. Radiat. Oncol. 1993, 26, 239–244. [Google Scholar] [CrossRef]
- Burger, P.C.; Heinz, E.R.; Shibata, T.; Kleihues, P. Topographic anatomy and CT correlations in the untreated glioblastoma multiforme. J. Neurosurg. 1988, 68, 698–704. [Google Scholar] [CrossRef]
- Kelly, P.J.; Daumas-Duport, C.; Kispert, D.B.; Kall, B.A.; Scheithauer, B.W.; Illig, J.J. Imaging-based stereotaxic serial biopsies in untreated intracranial glial neoplasms. J. Neurosurg. 1987, 66, 865–874. [Google Scholar] [CrossRef]
- Noone, A.M.; Howlader, N.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER Cancer Statistics Review, 1975–2015; National Cancer Institute: Bethesda, MD, USA, 2017. Available online: https://seer.cancer.gov/csr/1975_2015/ (accessed on 1 April 2018).
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef]
- Sherriff, J.; Tamangani, J.; Senthil, L.; Cruickshank, G.; Spooner, D.; Jones, B.; Brookes, C.; Sanghera, P. Patterns of relapse in glioblastoma multiforme following concomitant chemoradiotherapy with temozolomide. Br. J. Radiol. 2013, 86, 20120414. [Google Scholar] [CrossRef]
- Zhou, X.; Liao, X.; Zhang, B.; He, H.; Shui, Y.; Xu, W.; Jiang, C.; Shen, L.; Wei, Q. Recurrence patterns in patients with high-grade glioma following temozolomide-based chemoradiotherapy. Mol. Clin. Oncol. 2016, 5, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.-H.; Wang, Z.-F.; Pan, Z.-Y.; Péus, D.; Delgado-Fernandez, J.; Pallud, J.; Li, Z.-Q. A Meta-Analysis of Survival Outcomes Following Reoperation in Recurrent Glioblastoma: Time to Consider the Timing of Reoperation. Front. Neurol. 2019, 10, 286. [Google Scholar] [CrossRef]
- Nava, F.; Tramacere, I.; Fittipaldo, A.; Bruzzone, M.G.; DiMeco, F.; Fariselli, L.; Finocchiaro, G.; Pollo, B.; Salmaggi, A.; Silvani, A.; et al. Survival effect of first- and second-line treatments for patients with primary glioblastoma: A cohort study from a prospective registry, 1997-2010. Neuro-Oncology 2014, 16, 719–727. [Google Scholar] [CrossRef]
- Minniti, G.; Lombardi, G.; Paolini, S. Glioblastoma in Elderly Patients: Current Management and Future Perspectives. Cancers 2019, 11, 336. [Google Scholar] [CrossRef]
- Perry, J.R.; Laperriere, N.; O’Callaghan, C.J.; Brandes, A.A.; Menten, J.; Phillips, C.; Fay, M.; Nishikawa, R.; Cairncross, J.G.; Roa, W.; et al. Short-Course Radiation plus Temozolomide in Elderly Patients with Glioblastoma. N. Engl. J. Med. 2017, 376, 1027–1037. [Google Scholar] [CrossRef]
- Leonardi, G.C.; Accardi, G.; Monastero, R.; Nicoletti, F.; Libra, M. Ageing: From inflammation to cancer. Immun. Ageing 2018, 15, 1–7. [Google Scholar] [CrossRef]
- Dibattista, A.M.; Sierra, F.; Masliah, E. NIA workshop on senescence in brain aging and Alzheimer’s disease and its related dementias. GeroScience 2020, 42, 389–396. [Google Scholar] [CrossRef]
- Pekmezci, M.; Rice, T.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Hansen, H.; Sicotte, H.; Kollmeyer, T.M.; McCoy, L.S.; Sarkar, G.; et al. Adult infiltrating gliomas with WHO 2016 integrated diagnosis: Additional prognostic roles of ATRX and TERT. Acta Neuropathol. 2017, 133, 1001–1016. [Google Scholar] [CrossRef]
- Bhat, R.; Crowe, E.P.; Bitto, A.; Moh, M.; Katsetos, C.D.; Garcia, F.U.; Johnson, F.B.; Trojanowski, J.Q.; Sell, C.; Torres, C. Astrocyte Senescence as a Component of Alzheimer’s Disease. PLoS ONE 2012, 7, e45069. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; Van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147.e16. [Google Scholar] [CrossRef]
- Chinta, S.J.; Woods, G.; DeMaria, M.; Rane, A.; Zou, Y.; McQuade, A.; Rajagopalan, S.; Limbad, C.; Madden, D.T.; Campisi, J.; et al. Cellular Senescence Is Induced by the Environmental Neurotoxin Paraquat and Contributes to Neuropathology Linked to Parkinson’s Disease. Cell Rep. 2018, 22, 930–940. [Google Scholar] [CrossRef]
- Swenson, B.L.; Meyer, C.F.; Bussian, T.J.; Baker, D.J. Senescence in aging and disorders of the central nervous system. Transl. Med. Aging 2019, 3, 17–25. [Google Scholar] [CrossRef]
- Herbig, U.; Wei, W.; Dutriaux, A.; Jobling, W.A.; Sedivy, J.M. Real-time imaging of transcriptional activation in live cells reveals rapid up-regulation of the cyclin-dependent kinase inhibitor gene CDKN1A in replicative cellular senescence. Aging Cell 2008, 2, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Biran, A.; Zada, L.; Karam, P.A.; Vadai, E.; Roitman, L.; Ovadya, Y.; Porat, Z.; Krizhanovsky, V. Quantitative identification of senescent cells in aging and disease. Aging Cell 2017, 16, 661–671. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Schmitt, C.A. The dynamic nature of senescence in cancer. Nat. Cell Biol. 2019, 21, 94–101. [Google Scholar] [CrossRef]
- DeMaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2016, 7, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Choi, Y.W.; Lee, J.; Soh, E.Y.; Kim, J.-H.; Park, T.J. Senescent tumor cells lead the collective invasion in thyroid cancer. Nat. Commun. 2017, 8, 15208. [Google Scholar] [CrossRef]
- Asif, S.; Fatima, R.; Krc, R.; Bennett, J.; Raza, S. Comparative proteogenomic characterization of glioblastoma. CNS Oncol. 2019, 8, CNS37. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, Z.; Li, J.; Huang, T.; Wang, Y.; Chang, L.; Zheng, W.; Ma, Y.; Chen, F.; Gong, X.; et al. Genomic analysis of primary and recurrent gliomas reveals clinical outcome related molecular features. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Shin, Y.S.; Xue, M.; Matsutani, T.; Masui, K.; Yang, H.; Ikegami, S.; Gu, Y.; Herrmann, K.; Johnson, D.; et al. Single-Cell Phosphoproteomics Resolves Adaptive Signaling Dynamics and Informs Targeted Combination Therapy in Glioblastoma. Cancer Cell 2016, 29, 563–573. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; Decarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef]
- Halliday, J.; Helmy, K.; Pattwell, S.S.; Pitter, K.L.; LaPlant, Q.; Ozawa, T.; Holland, E.C. In vivo radiation response of proneural glioma characterized by protective p53 transcriptional program and proneural-mesenchymal shift. Proc. Natl. Acad. Sci. USA 2014, 111, 5248–5253. [Google Scholar] [CrossRef]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.C.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nat. Cell Biol. 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating Breast Tumor Cells Exhibit Dynamic Changes in Epithelial and Mesenchymal Composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef]
- Thomas, J.G.; Kerrigan, B.C.P.; Hossain, A.; Gumin, J.; Shinojima, N.; Nwajei, F.; Ezhilarasan, R.; Love, P.; Sulman, E.P.; Lang, F.F. Ionizing radiation augments glioma tropism of mesenchymal stem cells. J. Neurosurg. 2018, 128, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Kesanakurti, D.; Chetty, C.; Maddirela, D.R.; Gujrati, M.; Rao, J.S. Essential role of cooperative NF-κB and Stat3 recruitment to ICAM-1 intronic consensus elements in the regulation of radiation-induced invasion and migration in glioma. Oncogene 2012, 32, 5144–5155. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.J.; Nam, J.-K.; Kim, J.-H.; Choi, S.-H.; Lee, Y.-J. Endothelial-to-mesenchymal transition in anticancer therapy and normal tissue damage. Exp. Mol. Med. 2020, 52, 781–792. [Google Scholar] [CrossRef]
- Ahn, S.-H.; Park, H.; Ahn, Y.-H.; Kim, S.; Cho, M.-S.; Kang, J.L.; Choi, Y.-H. Necrotic cells influence migration and invasion of glioblastoma via NF-κB/AP-1-mediated IL-8 regulation. Sci. Rep. 2016, 6, 24552. [Google Scholar] [CrossRef]
- Priester, M.; Copanaki, E.; Vafaizadeh, V.; Hensel, S.; Bernreuther, C.; Glatzel, M.; Seifert, V.; Groner, B.; Kögel, D.; Weissenberger, J. STAT3 silencing inhibits glioma single cell infiltration and tumor growth. Neuro-Oncology 2013, 15, 840–852. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.-P.; Lee, Y.-W.; Chen, J.-T.; Lin, Y.-W.; Chen, R.-M. The Bradykinin-BDKRB1 Axis Regulates Aquaporin 4 Gene Expression and Consequential Migration and Invasion of Malignant Glioblastoma Cells via a Ca2+-MEK1-ERK1/2-NF-κB Mechanism. Cancers 2020, 12, 667. [Google Scholar] [CrossRef] [PubMed]
- Senft, C.; Priester, M.; Polacin, M.; Schröder, K.; Seifert, V.; Kögel, N.; Weissenberger, J. Inhibition of the JAK-2/STAT3 signaling pathway impedes the migratory and invasive potential of human glioblastoma cells. J. Neuro-Oncol. 2010, 101, 393–403. [Google Scholar] [CrossRef]
- Spiteri, I.; Caravagna, G.; Cresswell, G.; Vatsiou, A.; Nichol, D.; Acar, A.; Ermini, L.; Chkhaidze, K.; Werner, B.; Mair, R.; et al. Evolutionary dynamics of residual disease in human glioblastoma. Ann. Oncol. 2019, 30, 456–463. [Google Scholar] [CrossRef]
- Gill, B.J.; Pisapia, D.J.; Malone, H.R.; Goldstein, H.; Lei, L.; Sonabend, A.; Yun, J.; Samanamud, J.; Sims, J.S.; Banu, M.; et al. MRI-localized biopsies reveal subtype-specific differences in molecular and cellular composition at the margins of glioblastoma. Proc. Natl. Acad. Sci. USA 2014, 111, 12550–12555. [Google Scholar] [CrossRef]
- Dhruv, H.D.; Winslow, W.S.M.; Armstrong, B.; Tuncali, S.; Eschbacher, J.; Kislin, K.; Loftus, J.C.; Tran, N.L.; Berens, M.E. Reciprocal Activation of Transcription Factors Underlies the Dichotomy between Proliferation and Invasion of Glioma Cells. PLoS ONE 2013, 8, e72134. [Google Scholar] [CrossRef]
- Minata, M.; Audia, A.; Shi, J.; Lu, S.; Bernstock, J.; Pavlyukov, M.S.; Das, A.; Kim, S.-H.; Shin, Y.J.; Lee, Y.; et al. Phenotypic Plasticity of Invasive Edge Glioma Stem-like Cells in Response to Ionizing Radiation. Cell Rep. 2019, 26, 1893–1905.e7. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, M.; Inoue, A.; Ohnishi, T.; Kohno, S.; Ohue, S.; Matsumoto, S.; Suehiro, S.; Yamashita, D.; Ozaki, S.; Watanabe, H.; et al. Significance of Glioma Stem-Like Cells in the Tumor Periphery That Express High Levels of CD44 in Tumor Invasion, Early Progression, and Poor Prognosis in Glioblastoma. Stem Cells Int. 2018, 2018, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ferrandez, E.; Gutierrez, O.; Segundo, D.S.; Fernandez-Luna, J.L. NFκB activation in differentiating glioblastoma stem-like cells is promoted by hyaluronic acid signaling through TLR4. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, L.; Ruiz-Ontañon, P.; Vázquez-Barquero, A.; Lafarga, M.; Berciano, M.T.; Aldaz, B.; Grande, L.; Casafont, I.; Segura, V.; Robles, E.F.; et al. Blockade of the NFκB pathway drives differentiating glioblastoma-initiating cells into senescence both in vitro and in vivo. Oncogene 2011, 30, 3537–3548. [Google Scholar] [CrossRef]
- Medema, J.P. Cancer stem cells: The challenges ahead. Nat. Cell Biol. 2013, 15, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.V.; Filiz, G.; Daniel, P.M.; Hollande, F.; Dworkin, S.; Amiridis, S.; Kountouri, N.; Ng, W.; Morokoff, A.P.; Mantamadiotis, T. Expression of CD133 and CD44 in glioblastoma stem cells correlates with cell proliferation, phenotype stability and intra-tumor heterogeneity. PLoS ONE 2017, 12, e0172791. [Google Scholar] [CrossRef]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Beier, D.; Hau, P.; Proescholdt, M.A.; Lohmeier, A.; Wischhusen, J.; Oefner, P.J.; Aigner, L.; Brawanski, A.; Bogdahn, U.; Beier, C.P. CD133+ and CD133− Glioblastoma-Derived Cancer Stem Cells Show Differential Growth Characteristics and Molecular Profiles. Cancer Res. 2007, 67, 4010–4015. [Google Scholar] [CrossRef]
- Da Hora, C.C.; Pinkham, K.; Carvalho, L.; Zinter, M.; Tabet, E.; Nakano, I.; Tannous, B.A.; Badr, C.E. Sustained NF-κB-STAT3 signaling promotes resistance to Smac mimetics in Glioma stem-like cells but creates a vulnerability to EZH2 inhibition. Cell Death Discov. 2019, 5, 1–11. [Google Scholar] [CrossRef]
- Gimple, R.C.; Bhargava, S.; Dixit, D.; Rich, J.N. Glioblastoma stem cells: Lessons from the tumor hierarchy in a lethal cancer. Genes Dev. 2019, 33, 591–609. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nat. Cell Biol. 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Okolie, O.; Bago, J.R.; Schmid, R.S.; Irvin, D.M.; Bash, R.E.; Miller, C.R.; Hingtgen, S.D. Reactive astrocytes potentiate tumor aggressiveness in a murine glioma resection and recurrence model. Neuro-Oncology 2016, 18, 1622–1633. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Bartkova, J.; Hamerlik, P.; Stockhausen, M.-T.; Ehrmann, J.; Hlobilkova, A.; Laursen, H.; Kalita, O.; Kolar, Z.; Poulsen, H.S.; Broholm, H.; et al. Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene 2010, 29, 5095–5102. [Google Scholar] [CrossRef]
- Liu, W.-H.; Lin, J.-C.; Chou, Y.-C.; Li, M.-H.; Tsai, J.-T. CD44-associated radioresistance of glioblastoma in irradiated brain areas with optimal tumor coverage. Cancer Med. 2020, 9, 350–360. [Google Scholar] [CrossRef]
- Wang, F.; Zheng, Z.; Guan, J.; Qi, D.; Zhou, S.; Shen, X.; Wang, F.; Wenkert, D.; Kirmani, B.; Solouki, T.; et al. Identification of a panel of genes as a prognostic biomarker for glioblastoma. EBioMedicine 2018, 37, 68–77. [Google Scholar] [CrossRef]
- Velásquez, C.; Mansouri, S.; Mora, C.; Nassiri, F.; Suppiah, S.; Martino, J.; Zadeh, G.; Fernández-Luna, J.L. Molecular and Clinical Insights into the Invasive Capacity of Glioblastoma Cells. J. Oncol. 2019, 2019, 1–16. [Google Scholar] [CrossRef]
- Smith, C.L.; Kilic, O.; Schiapparelli, P.; Guerrero-Cazares, H.; Kim, D.-H.; Sedora-Roman, N.I.; Gupta, S.; O’Donnell, T.; Chaichana, K.L.; Rodriguez, F.J.; et al. Migration Phenotype of Brain-Cancer Cells Predicts Patient Outcomes. Cell Rep. 2016, 15, 2616–2624. [Google Scholar] [CrossRef] [PubMed]
- Alieva, M.; Leidgens, V.; Riemenschneider, M.J.; Klein, C.A.; Hau, P.; Van Rheenen, J. Intravital imaging of glioma border morphology reveals distinctive cellular dynamics and contribution to tumor cell invasion. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Gritsenko, P.G.; Atlasy, N.; Dieteren, C.E.J.; Navis, A.C.; Venhuizen, J.-H.; Veelken, C.; Schubert, D.; Acker-Palmer, A.; Westerman, B.A.; Wurdinger, T.; et al. p120-catenin-dependent collective brain infiltration by glioma cell networks. Nat. Cell Biol. 2020, 22, 97–107. [Google Scholar] [CrossRef]
- Griveau, A.; Seano, G.; Shelton, S.J.; Kupp, R.; Jahangiri, A.; Obernier, K.; Krishnan, S.; Lindberg, O.R.; Yuen, T.J.; Tien, A.-C.; et al. A Glial Signature and Wnt7 Signaling Regulate Glioma-Vascular Interactions and Tumor Microenvironment. Cancer Cell 2018, 33, 874–889.e7. [Google Scholar] [CrossRef]
- Montgomery, M.K.; Kim, S.H.; Dovas, A.; Zhao, H.T.; Goldberg, A.R.; Xu, W.; Yagielski, A.J.; Cambareri, M.K.; Patel, K.B.; Mela, A.; et al. Glioma-Induced Alterations in Neuronal Activity and Neurovascular Coupling during Disease Progression. Cell Rep. 2020, 31, 107500. [Google Scholar] [CrossRef]
- Zimmermann, D.R.; Dours-Zimmermann, M.T. Extracellular matrix of the central nervous system: From neglect to challenge. Histochem. Cell Biol. 2008, 130, 635–653. [Google Scholar] [CrossRef]
- Koh, I.; Cha, J.; Park, J.; Choi, J.; Kang, S.-G.; Kim, P. The mode and dynamics of glioblastoma cell invasion into a decellularized tissue-derived extracellular matrix-based three-dimensional tumor model. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Luo, N.; Knudson, W.; Askew, E.B.; Veluci, R.; Knudson, C.B. CD44 and Hyaluronan Promote the Bone Morphogenetic Protein 7 Signaling Response in Murine Chondrocytes. Arthritis Rheumatol. 2014, 66, 1547–1558. [Google Scholar] [CrossRef]
- Wu, R.-L.; Sedlmeier, G.; Kyjacova, L.; Schmaus, A.; Philipp, J.; Thiele, W.; Garvalov, B.K.; Sleeman, J.P. Hyaluronic acid-CD44 interactions promote BMP4/7-dependent Id1/3 expression in melanoma cells. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Kim, Y.; Kumar, S. CD44-Mediated Adhesion to Hyaluronic Acid Contributes to Mechanosensing and Invasive Motility. Mol. Cancer Res. 2014, 12, 1416–1429. [Google Scholar] [CrossRef] [PubMed]
- Bignami, A.; Hosley, M.; Dahl, D. Hyaluronic acid and hyaluronic acid-binding proteins in brain extracellular matrix. Brain Struct. Funct. 1993, 188, 419–433. [Google Scholar] [CrossRef]
- Serres, E.; Debarbieux, F.; Stanchi, F.; Maggiorella, L.; Grall, D.; Turchi, L.; Burelvandenbos, F.; Figarellabranger, D.; Virolle, T.; Rougon, G.; et al. Fibronectin expression in glioblastomas promotes cell cohesion, collective invasion of basement membrane in vitro and orthotopic tumor growth in mice. Oncogene 2014, 33, 3451–3462. [Google Scholar] [CrossRef]
- Yu, Q.; Xue, Y.; Liu, J.; Xi, Z.; Li, Z.; Liu, Y. Fibronectin Promotes the Malignancy of Glioma Stem-Like Cells Via Modulation of Cell Adhesion, Differentiation, Proliferation and Chemoresistance. Front. Mol. Neurosci. 2018, 11, 130. [Google Scholar] [CrossRef]
- Ellert-Miklaszewska, A.; Poleszak, K.; Pasierbinska, M.; Kaminska, B. Integrin Signaling in Glioma Pathogenesis: From Biology to Therapy. Int. J. Mol. Sci. 2020, 21, 888. [Google Scholar] [CrossRef] [PubMed]
- Kassis, J.; Lauffenburger, U.A.; Turner, T.; Wells, A. Tumor invasion as dysregulated cell motility. Semin. Cancer Biol. 2001, 11, 105–117. [Google Scholar] [CrossRef]
- Farin, A.; Suzuki, S.O.; Weiker, M.; Goldman, J.E.; Bruce, J.N.; Canoll, P. Transplanted glioma cells migrate and proliferate on host brain vasculature: A dynamic analysis. Glia 2006, 53, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, T.; Liu, S.; Yoshida, D.; Teramoto, A. The expression of matrix metalloproteinase-2 and-9 in human gliomas of different pathological grades. Brain Tumor Pathol. 2003, 20, 65–72. [Google Scholar] [CrossRef]
- Zhang, H.; Ma, Y.; Wang, H.; Xu, L.; Yu, Y. MMP-2 expression and correlation with pathology and MRI of glioma. Oncol. Lett. 2018, 17, 1826–1832. [Google Scholar] [CrossRef]
- Steen, P.E.V.D.; Dubois, B.; Nelissen, I.; Rudd, P.M.; Dwek, R.A.; Opdenakker, G. Biochemistry and Molecular Biology of Gelatinase B or Matrix Metalloproteinase-9 (MMP-9). Crit. Rev. Biochem. Mol. Biol. 2002, 37, 375–536. [Google Scholar] [CrossRef] [PubMed]
- Buszczak, M.; Inaba, M.; Yamashita, Y.M. Signaling by Cellular Protrusions: Keeping the Conversation Private. Trends Cell Biol. 2016, 26, 526–534. [Google Scholar] [CrossRef]
- Djuzenova, C.S.; Fiedler, V.; Memmel, S.; Katzer, A.; Hartmann, S.; Krohne, G.; Zimmermann, H.; Scholz, C.-J.; Polat, B.; Flentje, M.; et al. Actin cytoskeleton organization, cell surface modification and invasion rate of 5 glioblastoma cell lines differing in PTEN and p53 status. Exp. Cell Res. 2015, 330, 346–357. [Google Scholar] [CrossRef]
- Paw, I.; Carpenter, R.C.; Watabe, K.; Debinski, W.; Lo, H.-W. Mechanisms regulating glioma invasion. Cancer Lett. 2015, 362, 1–7. [Google Scholar] [CrossRef]
- Leclerc, C.; Haeich, J.; Aulestia, F.J.; Kilhoffer, M.-C.; Miller, A.L.; Néant, I.; Webb, S.E.; Schaeffer, E.; Junier, M.-P.; Chneiweiss, H.; et al. Calcium signaling orchestrates glioblastoma development: Facts and conjunctures. Biochim. Biophys. Acta (BBA) Bioenergy 2016, 1863, 1447–1459. [Google Scholar] [CrossRef]
- Yang, W.; Wu, P.-F.; Ma, J.-X.; Liao, M.-J.; Xu, L.-S.; Yi, L. TRPV4 activates the Cdc42/N-wasp pathway to promote glioblastoma invasion by altering cellular protrusions. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, M.D.M.; Dharmawardhane, S. Targeting Rac and Cdc42 GTPases in Cancer. Cancer Res. 2018, 78, 3101–3111. [Google Scholar] [CrossRef]
- Okura, H.; Golbourn, B.J.; Shahzad, U.; Agnihotri, S.; Sabha, N.; Krieger, J.R.; Figueiredo, C.A.; Chalil, A.; Landon-Brace, N.; Riemenschneider, A.; et al. A role for activated Cdc42 in glioblastoma multiforme invasion. Oncotarget 2016, 7, 56958–56975. [Google Scholar] [CrossRef]
- Caspani, E.M.; Crossley, P.H.; Redondo-Garcia, C.; Martinez, S. Glioblastoma: A Pathogenic Crosstalk between Tumor Cells and Pericytes. PLoS ONE 2014, 9, e101402. [Google Scholar] [CrossRef]
- Krusche, B.; Ottone, C.; Clements, M.P.; Johnstone, E.R.; Goetsch, K.; Lieven, H.; Mota, S.G.; Singh, P.; Khadayate, S.; Ashraf, A.; et al. EphrinB2 drives perivascular invasion and proliferation of glioblastoma stem-like cells. eLife 2016, 5, 4565. [Google Scholar] [CrossRef] [PubMed]
- Montana, V.; Sontheimer, H. Bradykinin Promotes the Chemotactic Invasion of Primary Brain Tumors. J. Neurosci. 2011, 31, 4858–4867. [Google Scholar] [CrossRef] [PubMed]
- Seifert, S.; Sontheimer, H. Bradykinin enhances invasion of malignant glioma into the brain parenchyma by inducing cells to undergo amoeboid migration. J. Physiol. 2014, 592, 5109–5127. [Google Scholar] [CrossRef]
- Umans, R.A.; Kate, M.T.; Pollock, C.; Sontheimer, H. Fishing for Contact: Modeling Perivascular Glioma Invasion in the Zebrafish Brain. ACS Pharmacol. Transl. Sci. 2020. [Google Scholar] [CrossRef]
- McCoy, M.G.; Nyanyo, D.; Hung, C.K.; Goerger, J.P.; Zipfel, W.R.; Williams, R.M.; Nishimura, N.; Fischbach, C. Endothelial cells promote 3D invasion of GBM by IL-8-dependent induction of cancer stem cell properties. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Sharma, I.; Singh, A.; Siraj, F.; Saxena, S. IL-8/CXCR1/2 signalling promotes tumor cell proliferation, invasion and vascular mimicry in glioblastoma. J. Biomed. Sci. 2018, 25, 1–13. [Google Scholar] [CrossRef]
- Raychaudhuri, B.; Vogelbaum, M.A. IL-8 is a mediator of NF-κB induced invasion by gliomas. J. Neuro-Oncol. 2010, 101, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Infanger, D.W.; Cho, Y.; Lopez, B.S.; Mohanan, S.; Liu, S.C.; Gursel, D.; Boockvar, J.A.; Fischbach, C. Glioblastoma Stem Cells Are Regulated by Interleukin-8 Signaling in a Tumoral Perivascular Niche. Cancer Res. 2013, 73, 7079–7089. [Google Scholar] [CrossRef] [PubMed]
- Piao, Y.; Lu, L.; De Groot, J. AMPA receptors promote perivascular glioma invasion via β1 integrin–dependent adhesion to the extracellular matrix. Neuro-Oncology 2009, 11, 260–273. [Google Scholar] [CrossRef]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A.; Tang, Y.; Nagaraja, S.; Gibson, E.M.; Mount, C.W.; Polepalli, J.; Mitra, S.S.; et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell 2015, 161, 803–816. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Tam, L.T.; Woo, P.J.; Lennon, J.; Nagaraja, S.; Gillespie, S.M.; Ni, J.; Duveau, D.Y.; Morris, P.J.; Zhao, J.J.; et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nat. Cell Biol. 2017, 549, 533–537. [Google Scholar] [CrossRef]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Körber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef]
- Zeng, Q.; Michael, I.P.; Zhang, P.; Saghafinia, S.; Knott, G.; Jiao, W.; McCabe, B.D.; Galván, J.A.; Robinson, H.P.C.; Zlobec, I.; et al. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature 2019, 573, 526–531. [Google Scholar] [CrossRef]
- Buckingham, S.C.; Campbell, S.L.; Haas, B.R.; Montana, V.; Robel, S.; Ogunrinu, T.; Sontheimer, H. Glutamate release by primary brain tumors induces epileptic activity. Nat. Med. 2011, 17, 1269–1274. [Google Scholar] [CrossRef]
- Komuro, H.; Rakic, P. Modulation of neuronal migration by NMDA receptors. Science 1993, 260, 95–97. [Google Scholar] [CrossRef]
- Dalley, C.B.; Wroblewska, B.; Wolfe, B.B.; Wroblewski, J.T. The Role of Metabotropic Glutamate Receptor 1 Dependent Signaling in Glioma Viability. J. Pharmacol. Exp. Ther. 2018, 367, 59–70. [Google Scholar] [CrossRef]
- Lange, F.; Weßlau, K.; Porath, K.; Hörnschemeyer, J.; Bergner, C.; Krause, B.J.; Mullins, C.S.; Linnebacher, M.; Köhling, R.; Kirschstein, T. AMPA receptor antagonist perampanel affects glioblastoma cell growth and glutamate release in vitro. PLoS ONE 2019, 14, e0211644. [Google Scholar] [CrossRef]
- Takano, T.; Lin, J.H.-C.; Arcuino, G.; Gao, Q.; Yang, J.; Nedergaard, M. Glutamate release promotes growth of malignant gliomas. Nat. Med. 2001, 7, 1010–1015. [Google Scholar] [CrossRef]
- Oh, M.C.; Kim, J.M.; Safaee, M.; Kaur, G.; Sun, M.Z.; Kaur, R.; Celli, A.; Mauro, T.M.; Parsa, A.T. Overexpression of Calcium-Permeable Glutamate Receptors in Glioblastoma Derived Brain Tumor Initiating Cells. PLoS ONE 2012, 7, e47846. [Google Scholar] [CrossRef]
- Tan, H.W.S.; Sim, A.Y.L.; Long, Y.C. Glutamine metabolism regulates autophagy-dependent mTORC1 reactivation during amino acid starvation. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef]
- Buccarelli, M.; Marconi, M.; Pacioni, S.; De Pasqualis, I.; D’Alessandris, Q.G.; Martini, M.; Ascione, B.; Malorni, W.; LaRocca, L.M.; Pallini, R.; et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Nazio, F.; Bordi, M.; Cianfanelli, V.; Locatelli, F.; Cecconi, F. Autophagy and cancer stem cells: Molecular mechanisms and therapeutic applications. Cell Death Differ. 2019, 26, 690–702. [Google Scholar] [CrossRef]
- Wang, J.; Xu, S.-L.; Duan, J.-J.; Yi, L.; Guo, Y.-F.; Shi, Y.; Li, L.; Yang, Z.-Y.; Liao, X.-M.; Cai, J.; et al. Invasion of white matter tracts by glioma stem cells is regulated by a NOTCH1–SOX2 positive-feedback loop. Nat. Neurosci. 2019, 22, 91–105. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Astrocyte Reactivity: Subtypes, States, and Functions in CNS Innate Immunity. Trends Immunol. 2020, 41, 758–770. [Google Scholar] [CrossRef]
- Duncombe, J.; Lennen, R.J.; Jansen, M.A.; Marshall, I.; Wardlaw, J.M.; Horsburgh, K. Ageing causes prominent neurovascular dysfunction associated with loss of astrocytic contacts and gliosis. Neuropathol. Appl. Neurobiol. 2017, 43, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Ba, B.T.K.; Iliff, J.J.; Xia, M.; Wang, M.; Wei, H.S.; Bs, D.Z.; Xie, L.; Kang, H.; Xu, Q.; Liew, J.A.; et al. Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol. 2014, 76, 845–861. [Google Scholar] [CrossRef]
- Noell, S.; Wolburg-Buchholz, K.; Mack, A.F.; Ritz, R.; Tatagiba, M.; Beschorner, R.; Wolburg, H.; Fallier-Becker, P. Dynamics of expression patterns of AQP4, dystroglycan, agrin and matrix metalloproteinases in human glioblastoma. Cell Tissue Res. 2012, 347, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S.; Smith, A.J.; Phuan, P.-W.; Tradtrantip, L.; Anderson, M.O. The aquaporin-4 water channel as a potential drug target in neurological disorders. Expert Opin. Ther. Targets 2017, 21, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Watkins, S.; Robel, S.; Kimbrough, I.F.; Robert, S.M.; Ellisdavies, G.C.R.; Sontheimer, H. Disruption of astrocyte–vascular coupling and the blood–brain barrier by invading glioma cells. Nat. Commun. 2014, 5, 1–15. [Google Scholar] [CrossRef]
- Le, D.M.; Besson, A.; Fogg, D.K.; Choi, K.-S.; Waisman, D.M.; Goodyer, C.G.; Rewcastle, B.; Yong, V.W. Exploitation of Astrocytes by Glioma Cells to Facilitate Invasiveness: A Mechanism Involving Matrix Metalloproteinase-2 and the Urokinase-Type Plasminogen Activator–Plasmin Cascade. J. Neurosci. 2003, 23, 4034–4043. [Google Scholar] [CrossRef] [PubMed]
- Rath, B.H.; Fair, J.M.; Jamal, M.; Camphausen, K.; Tofilon, P.J. Astrocytes Enhance the Invasion Potential of Glioblastoma Stem-Like Cells. PLoS ONE 2013, 8, e54752. [Google Scholar] [CrossRef]
- Shabtay-Orbach, A.; Amit, M.; Binenbaum, Y.; Na’Ara, S.; Gil, Z. Paracrine regulation of glioma cells invasion by astrocytes is mediated by glial-derived neurotrophic factor. Int. J. Cancer 2015, 137, 1012–1020. [Google Scholar] [CrossRef]
- Heiland, D.H.; Ravi, V.M.; Behringer, S.P.; Frenking, J.H.; Wurm, J.; Joseph, K.; Garrelfs, N.W.C.; Strähle, J.; Heynckes, S.; Grauvogel, J.; et al. Tumor-associated reactive astrocytes aid the evolution of immunosuppressive environment in glioblastoma. Nat. Commun. 2019, 10, 2541. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-K.; Jin, X.; Sohn, Y.-W.; Jin, X.; Jeon, H.-Y.; Kim, E.-J.; Ham, S.W.; Jeon, H.-M.; Chang, S.-Y.; Oh, S.-Y.; et al. Tumoral RANKL activates astrocytes that promote glioma cell invasion through cytokine signaling. Cancer Lett. 2014, 353, 194–200. [Google Scholar] [CrossRef]
- Fournié, J.-J.; Poupot, M. The Pro-tumorigenic IL-33 Involved in Antitumor Immunity: A Yin and Yang Cytokine. Front. Immunol. 2018, 9, 2506. [Google Scholar] [CrossRef]
- Zhang, J.-F.; Wang, P.; Yan, Y.-J.; Li, Y.; Guan, M.-W.; Yu, J.-J.; Wang, X.-D. IL-33 enhances glioma cell migration and invasion by upregulation of MMP2 and MMP9 via the ST2-NF-κB pathway. Oncol. Rep. 2017, 38, 2033–2042. [Google Scholar] [CrossRef]
- Zhang, J.-F.; Tao, T.; Wang, K.; Zhang, G.-X.; Yan, Y.; Lin, H.-R.; Li, Y.; Guan, M.-W.; Yu, J.-J.; Wang, X.-D. IL-33/ST2 axis promotes glioblastoma cell invasion by accumulating tenascin-C. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.-P.; Wang, J.; Xi, S.-Y.; Ni, X.-R.; Chen, Y.-S.; Yu, Y.-J.; Cen, Z.-W.; Yu, Z.-H.; Chen, F.-R.; Guo, C.-C.; et al. Tenascin-c mediated vasculogenic mimicry formation via regulation of MMP2/MMP9 in glioma. Cell Death Dis. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Haage, V.; Elmadany, N.; Roll, L.; Faissner, A.; Gutmann, D.H.; Semtner, M.; Kettenmann, H. Tenascin C regulates multiple microglial functions involving TLR4 signaling and HDAC1. Brain Behav. Immun. 2019, 81, 470–483. [Google Scholar] [CrossRef]
- Theodosis, D.; Pierre, K.; Cadoret, M.; Allard, M.; Faissner, A.; Poulain, D. Expression of high levels of the extracellular matrix glycoprotein, tenascin-C, in the normal adult hypothalamoneurohypophysial system. J. Comp. Neurol. 1997, 379, 386–398. [Google Scholar] [CrossRef]
- Leins, A.; Riva, P.; Lindstedt, R.; Davidoff, M.S.; Mehraein, P.; Weis, S. Expression of tenascin-C in various human brain tumors and its relevance for survival in patients with astrocytoma. Cancer 2003, 98, 2430–2439. [Google Scholar] [CrossRef] [PubMed]
- De Boeck, A.; Ahn, B.Y.; D’Mello, C.; Lun, X.; Menon, S.V.; AlShehri, M.M.; Szulzewsky, F.; Shen, Y.; Khan, L.; Dang, N.H.; et al. Glioma-derived IL-33 orchestrates an inflammatory brain tumor microenvironment that accelerates glioma progression. Nat. Commun. 2020, 11, 1–24. [Google Scholar] [CrossRef]
- Angel, I.; Kerman, O.P.; Rousso-Noori, L.; Friedmann-Morvinski, D. Tenascin C promotes cancer cell plasticity in mesenchymal glioblastoma. Oncogene 2020, 39, 6990–7004. [Google Scholar] [CrossRef] [PubMed]
- Saggu, R.; Schumacher, T.; Gerich, F.; Rakers, C.; Tai, K.; Delekate, A.; Petzold, G.C. Astroglial NF-kB contributes to white matter damage and cognitive impairment in a mouse model of vascular dementia. Acta Neuropathol. Commun. 2016, 4, 1–10. [Google Scholar] [CrossRef]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the senescence-associated secretory phenotype by NF- B promotes senescence and enhances chemosensitivity. Genes Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef]
- Le, O.N.L.; Rodier, F.; Fontaine, F.; Coppé, J.-P.; Campisi, J.; DeGregori, J.; Laverdière, C.; Kokta, V.; Haddad, E.; Beauséjour, C.M. Ionizing radiation-induced long-term expression of senescence markers in mice is independent of p53 and immune status. Aging Cell 2010, 9, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Aasland, D.; Götzinger, L.; Hauck, L.; Berte, N.; Meyer, J.; Effenberger, M.; Schneider, S.; Reuber, E.E.; Roos, W.P.; Tomicic, M.T.; et al. Temozolomide Induces Senescence and Repression of DNA Repair Pathways in Glioblastoma Cells via Activation of ATR–CHK1, p21, and NF-κB. Cancer Res. 2019, 79, 99–113. [Google Scholar] [CrossRef]
- Cargill, R.; Kohama, S.G.; Struve, J.; Su, W.; Banine, F.; Witkowski, E.; Back, S.A.; Sherman, L.S. Astrocytes in aged nonhuman primate brain gray matter synthesize excess hyaluronan. Neurobiol. Aging 2012, 33, 830.e13–830.e24. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; De Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; DeMaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660.e4. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, M.M.; Erikson, G.A.; Shokhirev, M.N.; Allen, N.J. The Aging Astrocyte Transcriptome from Multiple Regions of the Mouse Brain. Cell Rep. 2018, 22, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Zhang, J.; Huang, Y.; Fang, Y.; Lyu, J.; Shen, Y. The age-related changes and differences in energy metabolism and glutamate-glutamine recycling in the d-gal-induced and naturally occurring senescent astrocytes in vitro. Exp. Gerontol. 2019, 118, 9–18. [Google Scholar] [CrossRef]
- Limbad, C.; Oron, T.R.; Alimirah, F.; Davalos, A.R.; Tracy, T.E.; Gan, L.; Desprez, P.-Y.; Campisi, J. Astrocyte senescence promotes glutamate toxicity in cortical neurons. PLoS ONE 2020, 15, e0227887. [Google Scholar] [CrossRef]
- Hou, J.; Yun, Y.; Xue, J.; Sun, M.; Kim, S. D-galactose induces astrocytic aging and contributes to astrocytoma progression and chemoresistance via cellular senescence. Mol. Med. Rep. 2019, 20, 4111–4118. [Google Scholar] [CrossRef]
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 2018, 14, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Wyld, L.; Bellantuono, I.; Tchkonia, T.; Morgan, J.; Turner, O.; Foss, F.; George, J.; Danson, S.; Kirkland, J.L. Senescence and Cancer: A Review of Clinical Implications of Senescence and Senotherapies. Cancers 2020, 12, 2134. [Google Scholar] [CrossRef]
- Rincheval, V.; Renaud, F.; Lemaire, C.; Godefroy, N.; Krejbich-Trotot, P.; Boulo, V.; Mignotte, B.; Vayssière, J.-L. Bcl-2 can promote p53-dependent senescence versus apoptosis without affecting the G1/S transition. Biochem. Biophys. Res. Commun. 2002, 298, 282–288. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Barbosa, I.A.M.; et al. Senescence-associated reprogramming promotes cancer stemness. Nat. Cell Biol. 2018, 553, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Olson, I.; Mansour, M.; Carlstrom, L.P.; Sutiwisesak, R.; Saber, R.; Burns, T.C. Selective Vulnerability of Senescent Glioblastoma Cells to Bcl-XL Inhibition (preprint). Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Harmouch, E.; Seitlinger, J.; Chaddad, H.; Ubeaud-Sequier, G.; Barths, J.; Saidu, S.; Désaubry, L.; Grandemange, S.; Massfelder, T.; Fuhrmann, G.; et al. Flavagline synthetic derivative induces senescence in glioblastoma cancer cells without being toxic to healthy astrocytes. Sci. Rep. 2020, 10, 1–16. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Role in Glioma Progression | Type of Component | Name of the SASP Component | Cell Type |
|---|---|---|---|
| Invasion | ECM | Fibronectin | Astrocyte |
| Invasion | ECM | Hyaluronan | Astrocyte |
| Invasion | ECM | MMP2 | Astrocyte |
| Invasion | ECM | MMP9 | Astrocyte |
| Invasion | Ion balance | Calcium | |
| Invasion | Cytokine | IL6 | Astrocyte, endothelial cell |
| Invasion | Cytokine | IL8 | Astrocyte, endothelial cell |
| Glioma cell proliferation | Nutrients | Glutamate | Astrocyte |
| GSC maintenance | Cytokine | IL33 | Astrocyte |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Putavet, D.A.; de Keizer, P.L.J. Residual Disease in Glioma Recurrence: A Dangerous Liaison with Senescence. Cancers 2021, 13, 1560. https://doi.org/10.3390/cancers13071560
Putavet DA, de Keizer PLJ. Residual Disease in Glioma Recurrence: A Dangerous Liaison with Senescence. Cancers. 2021; 13(7):1560. https://doi.org/10.3390/cancers13071560
Chicago/Turabian StylePutavet, Diana A., and Peter L. J. de Keizer. 2021. "Residual Disease in Glioma Recurrence: A Dangerous Liaison with Senescence" Cancers 13, no. 7: 1560. https://doi.org/10.3390/cancers13071560
APA StylePutavet, D. A., & de Keizer, P. L. J. (2021). Residual Disease in Glioma Recurrence: A Dangerous Liaison with Senescence. Cancers, 13(7), 1560. https://doi.org/10.3390/cancers13071560