
Epigenetic Regulation of MicroRNA Clusters and Families during Tumor Development


Simple Summary
Abstract
1. History of microRNA Discovery
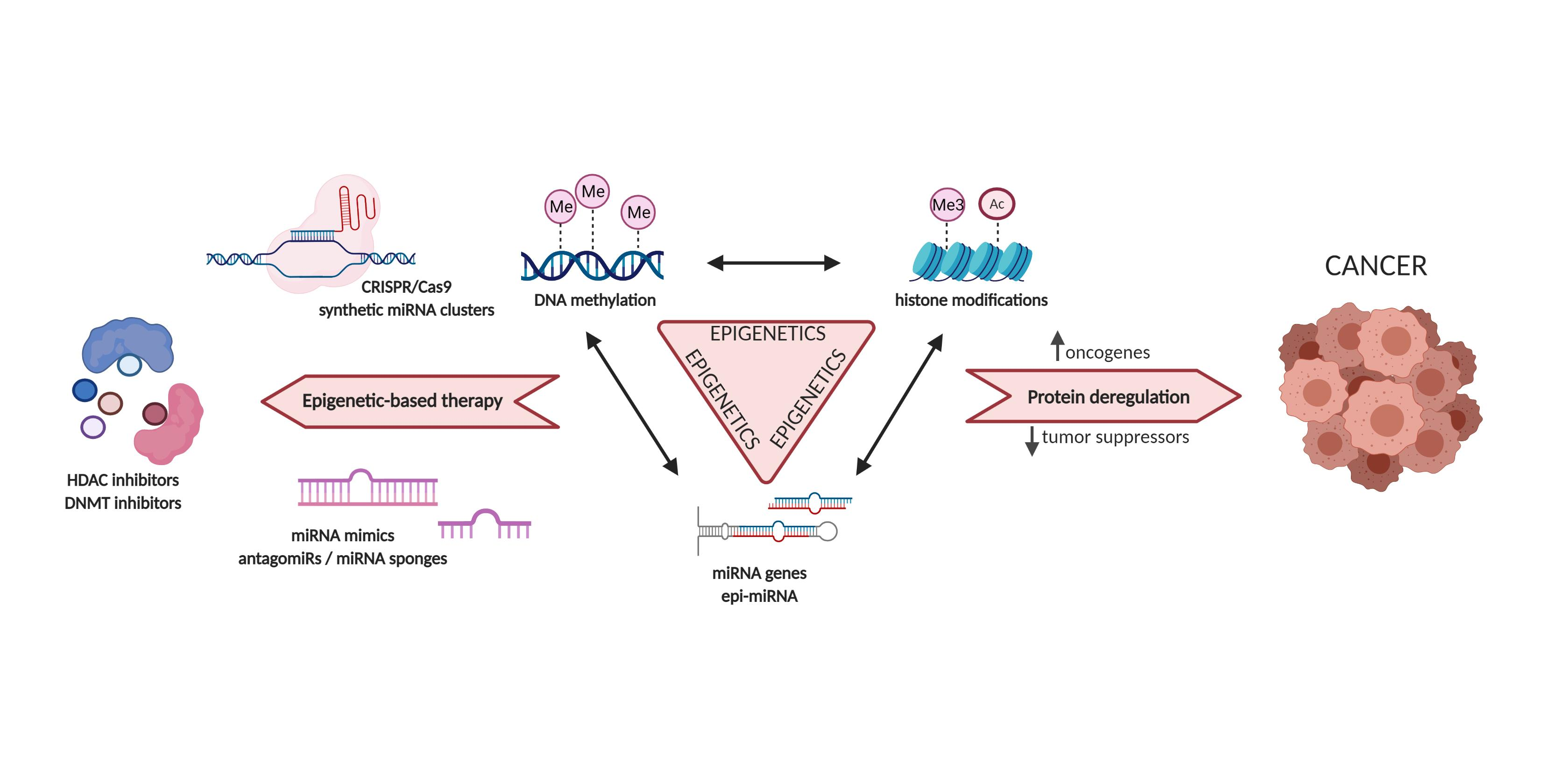
1.1. Biogenesis of microRNAs
1.2. Regulation of Gene Expression by microRNAs
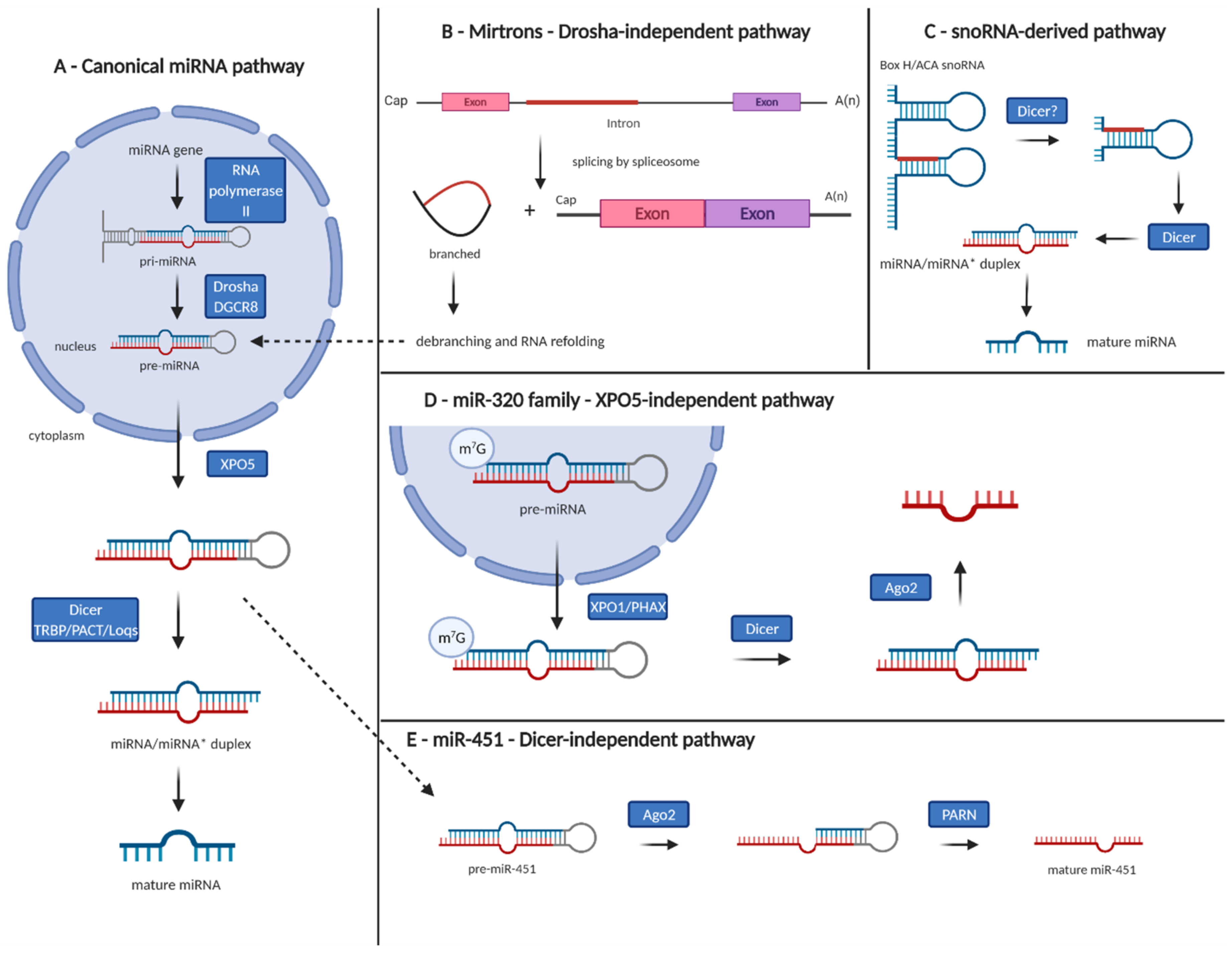
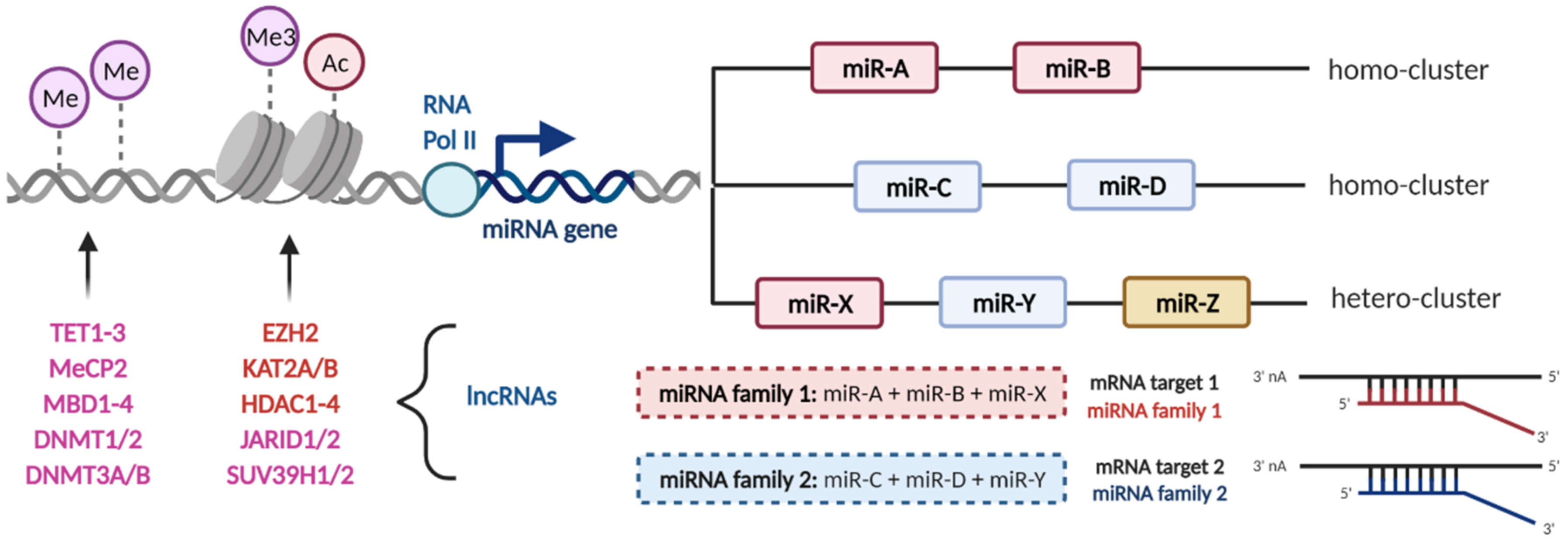
2. MicroRNA Clusters and Families
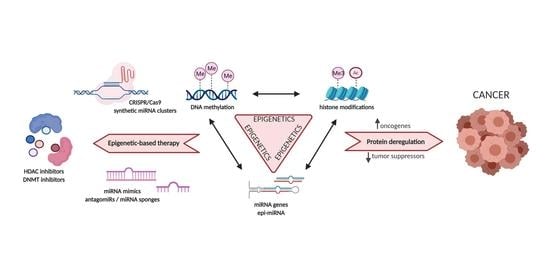
3. The Interconnection between microRNAs and Epigenetics
4. Epigenetic Regulation of microRNA Clusters and Families during Tumor Development
4.1. Let-7-5p/98-5p Family, miR-125-5p Family, miR-99-5p/100-5p Family
4.2. miR-34-5p/449-5p Family, miR-34b-5p/449c-5p Family
4.3. The miR-141-3p/200a-3p Family, miR-200ab-5p Family, miR-200bc-3p/429 Family, miR-200c-5p/550a-3p Family
4.4. mir-17~92a-1 Cluster, mir-106a~363 Cluster
4.5. miR-15-5p/16-5p/195-5p/424-5p/497-5p Family
4.6. miR-23-3p Family, mir-23b~24-1 Cluster, mir-23a~24-2 Cluster
4.7. miR-130-3p/301-3p/454-3p Family
4.8. miR-29-3p Family
5. Clinical Utility of microRNA Clusters and Families and Epigenetic-Based Therapeutics
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dunham, I.; Kundaje, A.; Aldred, S.F.; Collins, P.J.; Davis, C.A.; Doyle, F.; Epstein, C.B.; Frietze, S.; Harrow, J.; Kaul, R.; et al. An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 489, 57–74. [Google Scholar]
- Boland, C.R. Non-Coding RNA: It’s Not Junk. Dig. Dis. Sci. 2017, 62, 1107–1109. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. Elegans Heterochronic Gene Lin-4 Encodes Small RNAs with Antisense Complementarity to Lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional Regulation of the Heterochronic Gene Lin-14 by Lin-4 Mediates Temporal Pattern Formation in C. Elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef]
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquinelli, A.E.; Bettinger, J.C.; Rougvie, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-Nucleotide Let-7 RNA Regulates Developmental Timing in Caenorhabditis Elegans. Nature 2000, 403, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Pasquinelli, A.E.; Reinhart, B.J.; Slack, F.; Martindale, M.Q.; Kuroda, M.I.; Maller, B.; Hayward, D.C.; Ball, E.E.; Degnan, B.; Müller, P.; et al. Conservation of the Sequence and Temporal Expression of Let-7 Heterochronic Regulatory RNA. Nature 2000, 408, 86–89. [Google Scholar] [CrossRef]
- Su, J.-L.; Chen, P.-S.; Johansson, G.; Kuo, M.-L. Function and Regulation of Let-7 Family MicroRNAs. Microrna 2012, 1, 34–39. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. MiRBase: From MicroRNA Sequences to Function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-H.; Shao, P.; Zhou, H.; Chen, Y.-Q.; Qu, L.-H. DeepBase: A Database for Deeply Annotating and Mining Deep Sequencing Data. Nucleic Acids Res. 2010, 38, D123–D130. [Google Scholar] [CrossRef] [PubMed]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The MicroRNA.Org Resource: Targets and Expression. Nucleic Acids Res. 2008, 36, D149–D153. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Moxon, S.; Marshall, M.; Khanna, A.; Eddy, S.R.; Bateman, A. Rfam: Annotating Non-Coding RNAs in Complete Genomes. Nucleic Acids Res. 2005, 33, D121–D124. [Google Scholar] [CrossRef] [PubMed]
- Alexiou, P.; Vergoulis, T.; Gleditzsch, M.; Prekas, G.; Dalamagas, T.; Megraw, M.; Grosse, I.; Sellis, T.; Hatzigeorgiou, A.G. MiRGen 2.0: A Database of MicroRNA Genomic Information and Regulation. Nucleic Acids Res. 2010, 38, D137–D141. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting Effective MicroRNA Target Sites in Mammalian MRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Krek, A.; Grün, D.; Poy, M.N.; Wolf, R.; Rosenberg, L.; Epstein, E.J.; MacMenamin, P.; da Piedade, I.; Gunsalus, K.C.; Stoffel, M.; et al. Combinatorial MicroRNA Target Predictions. Nat. Genet. 2005, 37, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Sethupathy, P.; Corda, B.; Hatzigeorgiou, A.G. TarBase: A Comprehensive Database of Experimentally Supported Animal MicroRNA Targets. RNA 2006, 12, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Dweep, H.; Sticht, C.; Pandey, P.; Gretz, N. MiRWalk—Database: Prediction of Possible MiRNA Binding Sites by “Walking” the Genes of Three Genomes. J. Biomed. Inform. 2011, 44, 839–847. [Google Scholar] [CrossRef]
- Maragkakis, M.; Alexiou, P.; Papadopoulos, G.L.; Reczko, M.; Dalamagas, T.; Giannopoulos, G.; Goumas, G.; Koukis, E.; Kourtis, K.; Simossis, V.A.; et al. Accurate MicroRNA Target Prediction Correlates with Protein Repression Levels. BMC Bioinform. 2009, 10, 295. [Google Scholar] [CrossRef]
- Miranda, K.C.; Huynh, T.; Tay, Y.; Ang, Y.-S.; Tam, W.-L.; Thomson, A.M.; Lim, B.; Rigoutsos, I. A Pattern-Based Method for the Identification of MicroRNA Binding Sites and Their Corresponding Heteroduplexes. Cell 2006, 126, 1203–1217. [Google Scholar] [CrossRef]
- Ghorai, A.; Ghosh, U. MiRNA Gene Counts in Chromosomes Vary Widely in a Species and Biogenesis of MiRNA Largely Depends on Transcription or Post-Transcriptional Processing of Coding Genes. Front. Genet. 2014, 5, 100. [Google Scholar] [CrossRef]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human MicroRNA Genes Are Frequently Located at Fragile Sites and Genomic Regions Involved in Cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar] [CrossRef] [PubMed]
- Olena, A.F.; Patton, J.G. Genomic Organization of MicroRNAs. J. Cell Physiol. 2010, 222, 540–545. [Google Scholar] [CrossRef]
- Wang, G.; Wang, Y.; Shen, C.; Huang, Y.; Huang, K.; Huang, T.H.M.; Nephew, K.P.; Li, L.; Liu, Y. RNA Polymerase II Binding Patterns Reveal Genomic Regions Involved in MicroRNA Gene Regulation. PLoS ONE 2010, 5, e13798. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Gregory, R.I.; Yan, K.-P.; Amuthan, G.; Chendrimada, T.; Doratotaj, B.; Cooch, N.; Shiekhattar, R. The Microprocessor Complex Mediates the Genesis of MicroRNAs. Nature 2004, 432, 235–240. [Google Scholar] [CrossRef]
- Park, J.-E.; Heo, I.; Tian, Y.; Simanshu, D.K.; Chang, H.; Jee, D.; Patel, D.J.; Kim, V.N. Dicer Recognizes the 5′ End of RNA for Efficient and Accurate Processing. Nature 2011, 475, 201–205. [Google Scholar] [CrossRef]
- Liu, J.; Carmell, M.A.; Rivas, F.V.; Marsden, C.G.; Thomson, J.M.; Song, J.-J.; Hammond, S.M.; Joshua-Tor, L.; Hannon, G.J. Argonaute2 Is the Catalytic Engine of Mammalian RNAi. Science 2004, 305, 1437–1441. [Google Scholar] [CrossRef] [PubMed]
- Yeo, J.H.C.; Chong, M.M.W. Many Routes to a Micro RNA. IUBMB Life 2011, 63, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Okamura, K.; Hagen, J.W.; Duan, H.; Tyler, D.M.; Lai, E.C. The Mirtron Pathway Generates MicroRNA-Class Regulatory RNAs in Drosophila. Cell 2007, 130, 89–100. [Google Scholar] [CrossRef]
- Matera, A.G.; Terns, R.M.; Terns, M.P. Non-Coding RNAs: Lessons from the Small Nuclear and Small Nucleolar RNAs. Nat. Rev. Mol. Cell Biol. 2007, 8, 209–220. [Google Scholar] [CrossRef]
- Cheloufi, S.; Dos Santos, C.O.; Chong, M.M.W.; Hannon, G.J. A Dicer-Independent MiRNA Biogenesis Pathway That Requires Ago Catalysis. Nature 2010, 465, 584–589. [Google Scholar] [CrossRef]
- Kretov, D.A.; Walawalkar, I.A.; Mora-Martin, A.; Shafik, A.M.; Moxon, S.; Cifuentes, D. Ago2-Dependent Processing Allows MiR-451 to Evade the Global MicroRNA Turnover Elicited during Erythropoiesis. Mol. Cell 2020, 78, 317–328.e6. [Google Scholar] [CrossRef]
- Xie, M.; Li, M.; Vilborg, A.; Lee, N.; Shu, M.-D.; Yartseva, V.; Šestan, N.; Steitz, J.A. Mammalian 5′-Capped MicroRNA Precursors That Generate a Single MicroRNA. Cell 2013, 155, 1568–1580. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and Specific Genetic Interference by Double-Stranded RNA in Caenorhabditis Elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Zamore, P.D. RNA Interference: Big Applause for Silencing in Stockholm. Cell 2006, 127, 1083–1086. [Google Scholar] [CrossRef] [PubMed]
- Boivin, V.; Deschamps-Francoeur, G.; Scott, M.S. Protein Coding Genes as Hosts for Noncoding RNA Expression. Semin. Cell Dev. Biol. 2018, 75, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Shih, I.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of Mammalian MicroRNA Targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef]
- Brennecke, J.; Stark, A.; Russell, R.B.; Cohen, S.M. Principles of MicroRNA-Target Recognition. PLoS Biol. 2005, 3, e85. [Google Scholar] [CrossRef]
- Lytle, J.R.; Yario, T.A.; Steitz, J.A. Target MRNAs Are Repressed as Efficiently by MicroRNA-Binding Sites in the 5′ UTR as in the 3′ UTR. Proc. Natl. Acad. Sci. USA 2007, 104, 9667–9672. [Google Scholar] [CrossRef]
- Tay, Y.; Zhang, J.; Thomson, A.M.; Lim, B.; Rigoutsos, I. MicroRNAs to Nanog, Oct4 and Sox2 Coding Regions Modulate Embryonic Stem Cell Differentiation. Nature 2008, 455, 1124–1128. [Google Scholar] [CrossRef]
- Kong, Y.; Han, J.-H. MicroRNA: Biological and Computational Perspective. Genom. Proteom. Bioinform. 2005, 3, 62–72. [Google Scholar] [CrossRef]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian MicroRNAs Predominantly Act to Decrease Target MRNA Levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, S.W.; Guo, H.; McGeary, S.E.; Rodriguez-Mias, R.A.; Shin, C.; Baek, D.; Hsu, S.; Ghoshal, K.; Villén, J.; Bartel, D.P. MRNA Destabilization Is the Dominant Effect of Mammalian MicroRNAs by the Time Substantial Repression Ensues. Mol. Cell 2014, 56, 104–115. [Google Scholar] [CrossRef]
- Djuranovic, S.; Nahvi, A.; Green, R. MiRNA-Mediated Gene Silencing by Translational Repression Followed by MRNA Deadenylation and Decay. Science 2012, 336, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, S.; Tong, Y.; Steitz, J.A. Switching from Repression to Activation: MicroRNAs Can up-Regulate Translation. Science 2007, 318, 1931–1934. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-C.; Liu, L.-Z.; Addison, J.B.; Wonderlin, W.F.; Ivanov, A.V.; Ruppert, J.M. A KLF4–MiRNA-206 Autoregulatory Feedback Loop Can Promote or Inhibit Protein Translation Depending upon Cell Context. Mol. Cell Biol. 2011, 31, 2513–2527. [Google Scholar] [CrossRef] [PubMed]
- Place, R.F.; Li, L.-C.; Pookot, D.; Noonan, E.J.; Dahiya, R. MicroRNA-373 Induces Expression of Genes with Complementary Promoter Sequences. Proc. Natl. Acad. Sci. USA 2008, 105, 1608–1613. [Google Scholar] [CrossRef] [PubMed]
- Eiring, A.M.; Harb, J.G.; Neviani, P.; Garton, C.; Oaks, J.J.; Spizzo, R.; Liu, S.; Schwind, S.; Santhanam, R.; Hickey, C.J.; et al. MiR-328 Functions as an RNA Decoy to Modulate HnRNP E2 Regulation of MRNA Translation in Leukemic Blasts. Cell 2010, 140, 652–665. [Google Scholar] [CrossRef]
- Valinezhad Orang, A.; Safaralizadeh, R.; Kazemzadeh-Bavili, M. Mechanisms of MiRNA-Mediated Gene Regulation from Common Downregulation to MRNA-Specific Upregulation. Int. J. Genom. 2014, 2014, 970607. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Zhao, Y.; Zhang, H.; Yang, S.; Chen, F. Integrated Evolutionary Analysis of Human MiRNA Gene Clusters and Families Implicates Evolutionary Relationships. Gene 2014, 534, 24–32. [Google Scholar] [CrossRef]
- Yuan, X.; Liu, C.; Yang, P.; He, S.; Liao, Q.; Kang, S.; Zhao, Y. Clustered MicroRNAs’ Coordination in Regulating Protein-Protein Interaction Network. BMC Syst. Biol. 2009, 3, 65. [Google Scholar] [CrossRef]
- Kabekkodu, S.P.; Shukla, V.; Varghese, V.K.; Adiga, D.; Vethil Jishnu, P.; Chakrabarty, S.; Satyamoorthy, K. Cluster MiRNAs and Cancer: Diagnostic, Prognostic and Therapeutic Opportunities. Wiley Interdiscip. Rev. RNA 2020, 11, e1563. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Mao, Y.; Hu, L.; Wu, Y.; Ji, Z. MiRClassify: An Advanced Web Server for MiRNA Family Classification and Annotation. Comput. Biol. Med. 2014, 45, 157–160. [Google Scholar] [CrossRef]
- Roush, S.; Slack, F.J. The Let-7 Family of MicroRNAs. Trends Cell Biol. 2008, 18, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Desvignes, T.; Batzel, P.; Berezikov, E.; Eilbeck, K.; Eppig, J.T.; McAndrews, M.S.; Singer, A.; Postlethwait, J.H. MicroRNA Nomenclature: A View Incorporating Genetic Origins, Biosynthetic Pathways, and Sequence Variants. Trends Genet. 2015, 31, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Ding, J.; Zhou, S.; Guan, J. MiRFam: An Effective Automatic MiRNA Classification Method Based on n-Grams and a Multiclass SVM. BMC Bioinform. 2011, 12, 216. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, D.; Kriventseva, E.V.; Rahman, N.; Vejnar, C.E.; Zdobnov, E.M. MiROrtho: Computational Survey of MicroRNA Genes. Nucleic Acids Res. 2009, 37, D111–D117. [Google Scholar] [CrossRef]
- Wuchty, S.; Arjona, D.; Bozdag, S.; Bauer, P.O. Involvement of MicroRNA Families in Cancer. Nucleic Acids Res. 2012, 40, 8219–8226. [Google Scholar] [CrossRef]
- Gruber Pfeifer, G.P. Defining Driver DNA Methylation Changes in Human Cancer. Int. J. Mol. Sci. 2018, 19, 1166. [Google Scholar] [CrossRef]
- Pastor, W.A.; Aravind, L.; Rao, A. TETonic Shift: Biological Roles of TET Proteins in DNA Demethylation and Transcription. Nat. Rev. Mol. Cell Biol. 2013, 14, 341–356. [Google Scholar] [CrossRef]
- Yang, X.-J.; Seto, E. HATs and HDACs: From Structure, Function and Regulation to Novel Strategies for Therapy and Prevention. Oncogene 2007, 26, 5310–5318. [Google Scholar] [CrossRef]
- Simon, J.A.; Kingston, R.E. Occupying Chromatin: Polycomb Mechanisms for Getting to Genomic Targets, Stopping Transcriptional Traffic, and Staying Put. Mol. Cell 2013, 49, 808–824. [Google Scholar] [CrossRef]
- Plath, K.; Fang, J.; Mlynarczyk-Evans, S.K.; Cao, R.; Worringer, K.A.; Wang, H.; de la Cruz, C.C.; Otte, A.P.; Panning, B.; Zhang, Y. Role of Histone H3 Lysine 27 Methylation in X Inactivation. Science 2003, 300, 131–135. [Google Scholar] [CrossRef]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.-C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long Non-Coding RNA HOTAIR Reprograms Chromatin State to Promote Cancer Metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef]
- Yap, K.L.; Li, S.; Muñoz-Cabello, A.M.; Raguz, S.; Zeng, L.; Mujtaba, S.; Gil, J.; Walsh, M.J.; Zhou, M.-M. Molecular Interplay of the Noncoding RNA ANRIL and Methylated Histone H3 Lysine 27 by Polycomb CBX7 in Transcriptional Silencing of INK4a. Mol. Cell 2010, 38, 662–674. [Google Scholar] [CrossRef]
- Zhang, Z.-K.; Davies, K.P.; Allen, J.; Zhu, L.; Pestell, R.G.; Zagzag, D.; Kalpana, G.V. Cell Cycle Arrest and Repression of Cyclin D1 Transcription by INI1/HSNF5. Mol. Cell Biol. 2002, 22, 5975–5988. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Miller, E.L.; Ronan, J.L.; Ho, W.Q.; Jothi, R.; Crabtree, G.R. EsBAF Facilitates Pluripotency by Conditioning the Genome for LIF/STAT3 Signalling and by Regulating Polycomb Function. Nat. Cell Biol. 2011, 13, 903–913. [Google Scholar] [CrossRef]
- Orlando, K.A.; Nguyen, V.; Raab, J.R.; Walhart, T.; Weissman, B.E. Remodeling the Cancer Epigenome: Mutations in the SWI/SNF Complex Offer New Therapeutic Opportunities. Expert Rev. Anticancer Ther. 2019, 19, 375–391. [Google Scholar] [CrossRef]
- Kunej, T.; Godnic, I.; Ferdin, J.; Horvat, S.; Dovc, P.; Calin, G.A. Epigenetic Regulation of MicroRNAs in Cancer: An Integrated Review of Literature. Mutat Res. 2011, 717, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Glaich, O.; Parikh, S.; Bell, R.E.; Mekahel, K.; Donyo, M.; Leader, Y.; Shayevitch, R.; Sheinboim, D.; Yannai, S.; Hollander, D.; et al. DNA Methylation Directs MicroRNA Biogenesis in Mammalian Cells. Nat. Commun. 2019, 10, 5657. [Google Scholar] [CrossRef] [PubMed]
- Scott, G.K.; Mattie, M.D.; Berger, C.E.; Benz, S.C.; Benz, C.C. Rapid Alteration of MicroRNA Levels by Histone Deacetylase Inhibition. Cancer Res. 2006, 66, 1277–1281. [Google Scholar] [CrossRef] [PubMed]
- Gruber, A.J.; Zavolan, M. Modulation of Epigenetic Regulators and Cell Fate Decisions by MiRNAs. Epigenomics 2013, 5, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Saetrom, P.; Snøve, O.; Rossi, J.J. MicroRNA-Directed Transcriptional Gene Silencing in Mammalian Cells. Proc. Natl. Acad. Sci. USA 2008, 105, 16230–16235. [Google Scholar] [CrossRef] [PubMed]
- Baer, C.; Claus, R.; Frenzel, L.P.; Zucknick, M.; Park, Y.J.; Gu, L.; Weichenhan, D.; Fischer, M.; Pallasch, C.P.; Herpel, E.; et al. Extensive Promoter DNA Hypermethylation and Hypomethylation Is Associated with Aberrant MicroRNA Expression in Chronic Lymphocytic Leukemia. Cancer Res. 2012, 72, 3775–3785. [Google Scholar] [CrossRef]
- Vaira, V.; Elli, F.; Forno, I.; Guarnieri, V.; Verdelli, C.; Ferrero, S.; Scillitani, A.; Vicentini, L.; Cetani, F.; Mantovani, G.; et al. The MicroRNA Cluster C19MC Is Deregulated in Parathyroid Tumours. J. Mol. Endocrinol. 2012, 49, 115–124. [Google Scholar] [CrossRef]
- Nojima, M.; Matsui, T.; Tamori, A.; Kubo, S.; Shirabe, K.; Kimura, K.; Shimada, M.; Utsunomiya, T.; Kondo, Y.; Iio, E.; et al. Global, Cancer-Specific MicroRNA Cluster Hypomethylation Was Functionally Associated with the Development of Non-B Non-C Hepatocellular Carcinoma. Mol. Cancer 2016, 15, 31. [Google Scholar] [CrossRef]
- Lujambio, A.; Calin, G.A.; Villanueva, A.; Ropero, S.; Sánchez-Céspedes, M.; Blanco, D.; Montuenga, L.M.; Rossi, S.; Nicoloso, M.S.; Faller, W.J.; et al. A MicroRNA DNA Methylation Signature for Human Cancer Metastasis. Proc. Natl. Acad. Sci. USA 2008, 105, 13556–13561. [Google Scholar] [CrossRef]
- Chen, X.; He, D.; Dong, X.D.; Dong, F.; Wang, J.; Wang, L.; Tang, J.; Hu, D.-N.; Yan, D.; Tu, L. MicroRNA-124a Is Epigenetically Regulated and Acts as a Tumor Suppressor by Controlling Multiple Targets in Uveal Melanoma. Invest. Ophthalmol Vis. Sci. 2013, 54, 2248–2256. [Google Scholar] [CrossRef]
- Sampath, D.; Liu, C.; Vasan, K.; Sulda, M.; Puduvalli, V.K.; Wierda, W.G.; Keating, M.J. Histone Deacetylases Mediate the Silencing of MiR-15a, MiR-16, and MiR-29b in Chronic Lymphocytic Leukemia. Blood 2012, 119, 1162–1172. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Yang, F.; Chen, B.; Lu, Z.; Huo, X.; Zhou, W.; Wang, F.; Sun, S. The Histone Deacetylase 4/SP1/Microrna-200a Regulatory Network Contributes to Aberrant Histone Acetylation in Hepatocellular Carcinoma. Hepatology 2011, 54, 2025–2035. [Google Scholar] [CrossRef]
- Lu, L.; Katsaros, D.; de la Longrais, I.A.R.; Sochirca, O.; Yu, H. Hypermethylation of Let-7a-3 in Epithelial Ovarian Cancer Is Associated with Low Insulin-like Growth Factor-II Expression and Favorable Prognosis. Cancer Res. 2007, 67, 10117–10122. [Google Scholar] [CrossRef]
- Nishi, M.; Eguchi-Ishimae, M.; Wu, Z.; Gao, W.; Iwabuki, H.; Kawakami, S.; Tauchi, H.; Inukai, T.; Sugita, K.; Hamasaki, Y.; et al. Suppression of the Let-7b MicroRNA Pathway by DNA Hypermethylation in Infant Acute Lymphoblastic Leukemia with MLL Gene Rearrangements. Leukemia 2013, 27, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.-Q.; Huang, Y.-M.; Xiao, H.-F. Expression Analysis and Epigenetics of MicroRNA let-7b in Acute Lymphoblastic Leukemia. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2015, 23, 1535–1541. [Google Scholar] [PubMed]
- Brueckner, B.; Stresemann, C.; Kuner, R.; Mund, C.; Musch, T.; Meister, M.; Sültmann, H.; Lyko, F. The Human Let-7a-3 Locus Contains an Epigenetically Regulated MicroRNA Gene with Oncogenic Function. Cancer Res. 2007, 67, 1419–1423. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.-H.; Yao, D.-M.; Yang, L.; Ma, J.-C.; Wen, X.-M.; Yang, J.; Guo, H.; Li, X.-X.; Qian, W.; Lin, J.; et al. Hypomethylation of Let-7a-3 Is Associated with Poor Prognosis in Myelodysplastic Syndrome. Leuk. Lymphoma 2017, 58, 96–103. [Google Scholar] [CrossRef]
- Aure, M.R.; Leivonen, S.-K.; Fleischer, T.; Zhu, Q.; Overgaard, J.; Alsner, J.; Tramm, T.; Louhimo, R.; Alnæs, G.I.G.; Perälä, M.; et al. Individual and Combined Effects of DNA Methylation and Copy Number Alterations on MiRNA Expression in Breast Tumors. Genome Biol. 2013, 14, R126. [Google Scholar] [CrossRef]
- Bi, C.; Chung, T.-H.; Huang, G.; Zhou, J.; Yan, J.; Ahmann, G.J.; Fonseca, R.; Chng, W.J. Genome-Wide Pharmacologic Unmasking Identifies Tumor Suppressive MicroRNAs in Multiple Myeloma. Oncotarget 2015, 6, 26508–26518. [Google Scholar] [CrossRef]
- Chen, H.; Xu, Z. Hypermethylation-Associated Silencing of MiR-125a and MiR-125b: A Potential Marker in Colorectal Cancer. Dis. Markers 2015, 2015, 345080. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Chen, Q.; Shen, J.; Lv, C.; Cai, L. Epigenetic Silenced MiR-125a-5p Could Be Self-Activated through Targeting Suv39H1 in Gastric Cancer. J. Cell Mol. Med. 2018, 22, 4721–4731. [Google Scholar] [CrossRef]
- Xia, Z.; Qiu, D.; Deng, J.; Jiao, X.; Yang, R.; Sun, Z.; Wan, X.; Li, J. Methylation-Induced Downregulation and Tumor-Suppressive Role of MicroRNA-98 in Glioma through Targeting Sal-like Protein 4. Int. J. Mol. Med. 2018, 41, 2651–2659. [Google Scholar] [CrossRef]
- Mitra, D.; Das, P.M.; Huynh, F.C.; Jones, F.E. Jumonji/ARID1 B (JARID1B) Protein Promotes Breast Tumor Cell Cycle Progression through Epigenetic Repression of MicroRNA Let-7e. J. Biol. Chem. 2011, 286, 40531–40535. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Tsujii, M.; Wang, J.; Kondo, J.; Akasaka, T.; Jin, Y.; Li, W.; Nakamura, T.; Nishida, T.; Iijima, H.; et al. CagA Mediates Epigenetic Regulation to Attenuate Let-7 Expression in Helicobacter Pylori-Related Carcinogenesis. Gut 2013, 62, 1536–1546. [Google Scholar] [CrossRef] [PubMed]
- Jing, P.; Zhao, N.; Ye, M.; Zhang, Y.; Zhang, Z.; Sun, J.; Wang, Z.; Zhang, J.; Gu, Z. Protein Arginine Methyltransferase 5 Promotes Lung Cancer Metastasis via the Epigenetic Regulation of MiR-99 Family/FGFR3 Signaling. Cancer Lett. 2018, 427, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Vrba, L.; Muñoz-Rodríguez, J.L.; Stampfer, M.R.; Futscher, B.W. MiRNA Gene Promoters Are Frequent Targets of Aberrant DNA Methylation in Human Breast Cancer. PLoS ONE 2013, 8, e54398. [Google Scholar] [CrossRef] [PubMed]
- Au, S.L.-K.; Wong, C.C.-L.; Lee, J.M.-F.; Fan, D.N.-Y.; Tsang, F.H.; Ng, I.O.-L.; Wong, C.-M. Enhancer of Zeste Homolog 2 Epigenetically Silences Multiple Tumor Suppressor MicroRNAs to Promote Liver Cancer Metastasis. Hepatology 2012, 56, 622–631. [Google Scholar] [CrossRef]
- Strmsek, Z.; Kunej, T. Data Integration of 104 Studies Related with MicroRNA Epigenetics Revealed That MiR-34 Gene Family Is Silenced by DNA Methylation in the Highest Number of Cancer Types. Discoveries 2014, 2, e18. [Google Scholar] [CrossRef]
- Lodygin, D.; Tarasov, V.; Epanchintsev, A.; Berking, C.; Knyazeva, T.; Körner, H.; Knyazev, P.; Diebold, J.; Hermeking, H. Inactivation of MiR-34a by Aberrant CpG Methylation in Multiple Types of Cancer. Cell Cycle 2008, 7, 2591–2600. [Google Scholar] [CrossRef]
- Chim, C.S.; Wong, K.Y.; Qi, Y.; Loong, F.; Lam, W.L.; Wong, L.G.; Jin, D.Y.; Costello, J.F.; Liang, R. Epigenetic Inactivation of the MiR-34a in Hematological Malignancies. Carcinogenesis 2010, 31, 745–750. [Google Scholar] [CrossRef]
- Vogt, M.; Munding, J.; Grüner, M.; Liffers, S.-T.; Verdoodt, B.; Hauk, J.; Steinstraesser, L.; Tannapfel, A.; Hermeking, H. Frequent Concomitant Inactivation of MiR-34a and MiR-34b/c by CpG Methylation in Colorectal, Pancreatic, Mammary, Ovarian, Urothelial, and Renal Cell Carcinomas and Soft Tissue Sarcomas. Virchows Arch. 2011, 458, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, Z.; Gao, Y.; Li, N.; Li, B.; Tan, F.; Tan, X.; Lu, N.; Sun, Y.; Sun, J.; et al. DNA Hypermethylation of MicroRNA-34b/c Has Prognostic Value for Stage I Non-Small Cell Lung Cancer. Cancer Biol. Ther. 2011, 11, 490–496. [Google Scholar] [CrossRef]
- Chen, X.; Hu, H.; Guan, X.; Xiong, G.; Wang, Y.; Wang, K.; Li, J.; Xu, X.; Yang, K.; Bai, Y. CpG Island Methylation Status of MiRNAs in Esophageal Squamous Cell Carcinoma. Int. J. Cancer 2012, 130, 1607–1613. [Google Scholar] [CrossRef]
- Kubo, T.; Toyooka, S.; Tsukuda, K.; Sakaguchi, M.; Fukazawa, T.; Soh, J.; Asano, H.; Ueno, T.; Muraoka, T.; Yamamoto, H.; et al. Epigenetic Silencing of MicroRNA-34b/c Plays an Important Role in the Pathogenesis of Malignant Pleural Mesothelioma. Clin. Cancer Res. 2011, 17, 4965–4974. [Google Scholar] [CrossRef]
- Xie, K.; Liu, J.; Chen, J.; Dong, J.; Ma, H.; Liu, Y.; Hu, Z. Methylation-Associated Silencing of MicroRNA-34b in Hepatocellular Carcinoma Cancer. Gene 2014, 543, 101–107. [Google Scholar] [CrossRef]
- Shen, Z.; Zhou, C.; Li, J.; Ye, D.; Li, Q.; Wang, J.; Cui, X.; Chen, X.; Bao, T.; Duan, S. Promoter Hypermethylation of MiR-34a Contributes to the Risk, Progression, Metastasis and Poor Survival of Laryngeal Squamous Cell Carcinoma. Gene 2016, 593, 272–276. [Google Scholar] [CrossRef]
- Xu, K.; Chen, B.; Li, B.; Li, C.; Zhang, Y.; Jiang, N.; Lang, B. DNMT3B Silencing Suppresses Migration and Invasion by Epigenetically Promoting MiR-34a in Bladder Cancer. Aging 2020, 12, 23668–23683. [Google Scholar] [PubMed]
- Suzuki, H.; Yamamoto, E.; Nojima, M.; Kai, M.; Yamano, H.-O.; Yoshikawa, K.; Kimura, T.; Kudo, T.; Harada, E.; Sugai, T.; et al. Methylation-Associated Silencing of MicroRNA-34b/c in Gastric Cancer and Its Involvement in an Epigenetic Field Defect. Carcinogenesis 2010, 31, 2066–2073. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Chang, H.; Chen, J.; Zhang, Q.; Yu, X.; Mi, M. 3,6-Dihydroxyflavone Regulates MicroRNA-34a through DNA Methylation. BMC Cancer 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jiang, T.; Shao, L.; Liu, Y.; Zheng, C.; Zhong, Y.; Zhang, J.; Chang, Q. Mir-449a, a Potential Diagnostic Biomarker for WNT Group of Medulloblastoma. J. Neurooncol. 2016, 129, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Bach, D.-H.; Kim, D.; Bae, S.Y.; Kim, W.K.; Hong, J.-Y.; Lee, H.-J.; Rajasekaran, N.; Kwon, S.; Fan, Y.; Luu, T.-T.-T.; et al. Targeting Nicotinamide N-Methyltransferase and MiR-449a in EGFR-TKI-Resistant Non-Small-Cell Lung Cancer Cells. Mol. Ther. Nucleic Acids 2018, 11, 455–467. [Google Scholar] [CrossRef]
- Lin, L.; Jiang, H.; Huang, M.; Hou, X.; Sun, X.; Jiang, X.; Dong, X.; Sun, X.; Zhou, B.; Qiao, H. Depletion of Histone Deacetylase 1 Inhibits Metastatic Abilities of Gastric Cancer Cells by Regulating the MiR-34a/CD44 Pathway. Oncol. Rep. 2015, 34, 663–672. [Google Scholar] [CrossRef]
- Liu, X.; Yu, Y.; Zhang, J.; Lu, C.; Wang, L.; Liu, P.; Song, H. HDAC1 Silencing in Ovarian Cancer Enhances the Chemotherapy Response. Cell Physiol. Biochem. 2018, 48, 1505–1518. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-M.; Liu, Y.; Wei, H.-Y.; Lv, K.-Z.; Fu, P.-F. Large Intergenic Non-Coding RNA-ROR Reverses Gemcitabine-Induced Autophagy and Apoptosis in Breast Cancer Cells. Oncotarget 2016, 7, 59604–59617. [Google Scholar] [CrossRef]
- Yang, X.; Feng, M.; Jiang, X.; Wu, Z.; Li, Z.; Aau, M.; Yu, Q. MiR-449a and MiR-449b Are Direct Transcriptional Targets of E2F1 and Negatively Regulate PRb-E2F1 Activity through a Feedback Loop by Targeting CDK6 and CDC25A. Genes Dev. 2009, 23, 2388–2393. [Google Scholar] [CrossRef]
- You, J.; Zhang, Y.; Li, Y.; Fang, N.; Liu, B.; Zu, L.; Zhou, Q. MiR-449a Suppresses Cell Invasion by Inhibiting MAP2K1 in Non-Small Cell Lung Cancer. Am. J. Cancer Res. 2015, 5, 2730–2744. [Google Scholar] [PubMed]
- Buurman, R.; Gürlevik, E.; Schäffer, V.; Eilers, M.; Sandbothe, M.; Kreipe, H.; Wilkens, L.; Schlegelberger, B.; Kühnel, F.; Skawran, B. Histone Deacetylases Activate Hepatocyte Growth Factor Signaling by Repressing MicroRNA-449 in Hepatocellular Carcinoma Cells. Gastroenterology 2012, 143, 811–820. [Google Scholar] [CrossRef]
- Yun, M.R.; Lim, S.M.; Kim, S.-K.; Choi, H.M.; Pyo, K.-H.; Kim, S.K.; Lee, J.M.; Lee, Y.W.; Choi, J.W.; Kim, H.R.; et al. Enhancer Remodeling and MicroRNA Alterations Are Associated with Acquired Resistance to ALK Inhibitors. Cancer Res. 2018, 78, 3350–3362. [Google Scholar] [CrossRef]
- Gurbuz, V.; Kiliccioglu, I.; Dikmen, A.U.; Bilen, C.Y.; Sozen, S.; Konac, E. Comparative Analysis of Epi-MiRNA Expression Levels in Local/Locally Advanced and Metastatic Prostate Cancer Patients. Gene 2020, 758, 144963. [Google Scholar] [CrossRef]
- Varghese, V.K.; Shukla, V.; Kabekkodu, S.P.; Pandey, D.; Satyamoorthy, K. DNA Methylation Regulated MicroRNAs in Human Cervical Cancer. Mol. Carcinog. 2018, 57, 370–382. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Song, K.; Han, C.; Zhang, J.; Lu, L.; Chen, W.; Wu, T. Epigenetic Silencing of MiRNA-34a in Human Cholangiocarcinoma via EZH2 and DNA Methylation: Impact on Regulation of Notch Pathway. Am. J. Pathol. 2017, 187, 2288–2299. [Google Scholar] [CrossRef] [PubMed]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A Reciprocal Repression between ZEB1 and Members of the MiR-200 Family Promotes EMT and Invasion in Cancer Cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef]
- Wee, E.J.H.; Peters, K.; Nair, S.S.; Hulf, T.; Stein, S.; Wagner, S.; Bailey, P.; Lee, S.Y.; Qu, W.J.; Brewster, B.; et al. Mapping the Regulatory Sequences Controlling 93 Breast Cancer-Associated MiRNA Genes Leads to the Identification of Two Functional Promoters of the Hsa-Mir-200b Cluster, Methylation of Which Is Associated with Metastasis or Hormone Receptor Status in Advanced Breast Cancer. Oncogene 2012, 31, 4182–4195. [Google Scholar] [PubMed]
- Wiklund, E.D.; Bramsen, J.B.; Hulf, T.; Dyrskjøt, L.; Ramanathan, R.; Hansen, T.B.; Villadsen, S.B.; Gao, S.; Ostenfeld, M.S.; Borre, M.; et al. Coordinated Epigenetic Repression of the MiR-200 Family and MiR-205 in Invasive Bladder Cancer. Int. J. Cancer 2011, 128, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Shindo, T.; Niinuma, T.; Nishiyama, N.; Shinkai, N.; Kitajima, H.; Kai, M.; Maruyama, R.; Tokino, T.; Masumori, N.; Suzuki, H. Epigenetic Silencing of MiR-200b Is Associated with Cisplatin Resistance in Bladder Cancer. Oncotarget 2018, 9, 24457–24469. [Google Scholar] [CrossRef]
- Li, A.; Omura, N.; Hong, S.-M.; Vincent, A.; Walter, K.; Griffith, M.; Borges, M.; Goggins, M. Pancreatic Cancers Epigenetically Silence SIP1 and Hypomethylate and Overexpress MiR-200a/200b in Association with Elevated Circulating MiR-200a and MiR-200b Levels. Cancer Res. 2010, 70, 5226–5237. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.-C.; Lin, C.-C.; Shih, T.-C.; Tseng, R.-J.; Yu, M.-C.; Lin, Y.-J.; Hsieh, S.-Y. The MiR-200b-ZEB1 Circuit Regulates Diverse Stemness of Human Hepatocellular Carcinoma. Mol. Carcinog. 2017, 56, 2035–2047. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Lu, F.; Xiong, P.; Pan, M.; Zhang, Z.; Lin, X.; Pan, M.; Huang, H. WIPF1 Antagonizes the Tumor Suppressive Effect of MiR-141/200c and Is Associated with Poor Survival in Patients with PDAC. J. Exp. Clin. Cancer Res. 2018, 37, 167. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Chatterjee, A.; Das, D.; Ray, A.; Singh, R.; Chattopadhyay, E.; Sarkar, N.D.; Eccles, M.; Pal, M.; Maitra, A.; et al. Genome-Wide MiRNA Methylome Analysis in Oral Cancer: Possible Biomarkers Associated with Patient Survival. Epigenomics 2019, 11, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Qu, X.; Zhao, C.; Xu, L.; Hou, K.; Liu, Y.; Zhang, N.; Feng, J.; Shi, S.; Zhang, L.; et al. FEN1 Mediates MiR-200a Methylation and Promotes Breast Cancer Cell Growth via MET and EGFR Signaling. Faseb J. 2019, 33, 10717–10730. [Google Scholar] [CrossRef]
- Yu, Y.; Wu, J.; Guan, L.; Qi, L.; Tang, Y.; Ma, B.; Zhan, J.; Wang, Y.; Fang, W.; Zhang, H. Kindlin 2 Promotes Breast Cancer Invasion via Epigenetic Silencing of the MicroRNA200 Gene Family. Int. J. Cancer 2013, 133, 1368–1379. [Google Scholar] [CrossRef]
- Pang, Y.; Liu, J.; Li, X.; Xiao, G.; Wang, H.; Yang, G.; Li, Y.; Tang, S.-C.; Qin, S.; Du, N.; et al. MYC and DNMT3A-Mediated DNA Methylation Represses MicroRNA-200b in Triple Negative Breast Cancer. J. Cell Mol. Med. 2018, 22, 6262–6274. [Google Scholar] [CrossRef]
- Song, S.J.; Poliseno, L.; Song, M.S.; Ala, U.; Webster, K.; Ng, C.; Beringer, G.; Brikbak, N.J.; Yuan, X.; Cantley, L.C.; et al. MicroRNA-Antagonism Regulates Breast Cancer Stemness and Metastasis via TET Family Dependent Chromatin Remodeling. Cell 2013, 154, 311–324. [Google Scholar] [CrossRef]
- Hu, X.; Zhang, L.; Mao, S.-Q.; Li, Z.; Chen, J.; Zhang, R.-R.; Wu, H.-P.; Gao, J.; Guo, F.; Liu, W.; et al. Tet and TDG Mediate DNA Demethylation Essential for Mesenchymal-to-Epithelial Transition in Somatic Cell Reprogramming. Cell Stem Cell 2014, 14, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.M.; Kim, S.G.; Yang, H.-J.; Lim, J.H.; Cho, N.-Y.; Kim, W.H.; Kim, J.S.; Jung, H.C. Helicobacter Pylori Eradication Can Reverse the Methylation-Associated Regulation of MiR-200a/b in Gastric Carcinogenesis. Gut Liver 2020, 14, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.M.; O’Neill, K.M.; McKenna, M.M.; Walsh, C.P.; McKenna, D.J. Regulation of MiR-200c and MiR-141 by Methylation in Prostate Cancer. Prostate 2016, 76, 1146–1159. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.-Y.; Wright, J.A.; Attema, J.L.; Gregory, P.A.; Bert, A.G.; Smith, E.; Thomas, D.; Lopez, A.F.; Drew, P.A.; Khew-Goodall, Y.; et al. Epigenetic Modulation of the MiR-200 Family Is Associated with Transition to a Breast Cancer Stem-Cell-like State. J. Cell Sci. 2013, 126, 2256–2266. [Google Scholar] [CrossRef] [PubMed]
- Ning, X.; Shi, Z.; Liu, X.; Zhang, A.; Han, L.; Jiang, K.; Kang, C.; Zhang, Q. DNMT1 and EZH2 Mediated Methylation Silences the MicroRNA-200b/a/429 Gene and Promotes Tumor Progression. Cancer Lett. 2015, 359, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, G.; Liu, J. Long Noncoding RNA PVT1 Promotes Cervical Cancer Progression through Epigenetically Silencing MiR-200b. APMIS 2016, 124, 649–658. [Google Scholar] [CrossRef]
- Sui, C.-J.; Zhou, Y.-M.; Shen, W.-F.; Dai, B.-H.; Lu, J.-J.; Zhang, M.-F.; Yang, J.-M. Long Noncoding RNA GIHCG Promotes Hepatocellular Carcinoma Progression through Epigenetically Regulating MiR-200b/a/429. J. Mol. Med. 2016, 94, 1281–1296. [Google Scholar] [CrossRef] [PubMed]
- Enkhbaatar, Z.; Terashima, M.; Oktyabri, D.; Tange, S.; Ishimura, A.; Yano, S.; Suzuki, T. KDM5B Histone Demethylase Controls Epithelial-Mesenchymal Transition of Cancer Cells by Regulating the Expression of the MicroRNA-200 Family. Cell Cycle 2013, 12, 2100–2112. [Google Scholar] [CrossRef]
- Roy, S.S.; Gonugunta, V.K.; Bandyopadhyay, A.; Rao, M.K.; Goodall, G.J.; Sun, L.-Z.; Tekmal, R.R.; Vadlamudi, R.K. Significance of PELP1/HDAC2/MiR-200 Regulatory Network in EMT and Metastasis of Breast Cancer. Oncogene 2014, 33, 3707–3716. [Google Scholar] [CrossRef]
- Chen, D.-Q.; Pan, B.-Z.; Huang, J.-Y.; Zhang, K.; Cui, S.-Y.; De, W.; Wang, R.; Chen, L.-B. HDAC 1/4-Mediated Silencing of MicroRNA-200b Promotes Chemoresistance in Human Lung Adenocarcinoma Cells. Oncotarget 2014, 5, 3333–3349. [Google Scholar] [CrossRef]
- Mizuguchi, Y.; Specht, S.; Lunz, J.G.; Isse, K.; Corbitt, N.; Takizawa, T.; Demetris, A.J. Cooperation of P300 and PCAF in the Control of MicroRNA 200c/141 Transcription and Epithelial Characteristics. PLoS ONE 2012, 7, e32449. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, F.; Yuan, J.; Yuan, S.; Zhou, W.; Huo, X.; Xu, D.; Bi, H.; Wang, F.; Sun, S. Epigenetic Activation of the MiR-200 Family Contributes to H19-Mediated Metastasis Suppression in Hepatocellular Carcinoma. Carcinogenesis 2013, 34, 577–586. [Google Scholar] [CrossRef]
- Khuu, C.; Utheim, T.P.; Sehic, A. The Three Paralogous MicroRNA Clusters in Development and Disease, MiR-17-92, MiR-106a-363, and MiR-106b-25. Science 2016, 2016, 1379643. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Wu, Q.; Ni, Z.; Lei, C.; Li, T.; Shi, Y. MiR-19a/b and MeCP2 Repress Reciprocally to Regulate Multidrug Resistance in Gastric Cancer Cells. Int. J. Mol. Med. 2018, 42, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Wan, Y.; Xie, D.; Wang, Y.; Wei, J.; Yan, Q.; Lu, P.; Mo, L.; Xie, J.; Yang, S.; et al. DNMT1 Mediates Chemosensitivity by Reducing Methylation of MiRNA-20a Promoter in Glioma Cells. Exp. Mol. Med. 2015, 47, e182. [Google Scholar] [CrossRef] [PubMed]
- Yuan, R.; Zhi, Q.; Zhao, H.; Han, Y.; Gao, L.; Wang, B.; Kou, Z.; Guo, Z.; He, S.; Xue, X.; et al. Upregulated Expression of MiR-106a by DNA Hypomethylation Plays an Oncogenic Role in Hepatocellular Carcinoma. Tumour Biol. 2015, 36, 3093–3100. [Google Scholar] [CrossRef]
- Yuan, R.; Wang, G.; Xu, Z.; Zhao, H.; Chen, H.; Han, Y.; Wang, B.; Zhou, J.; Hu, H.; Guo, Z.; et al. Up-Regulated Circulating MiR-106a by DNA Methylation Promised a Potential Diagnostic and Prognostic Marker for Gastric Cancer. Anticancer Agents Med. Chem. 2016, 16, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Chen, S.; Niu, Y.; Liu, M.; Zhang, J.; Yang, Z.; Gao, P.; Wang, W.; Han, X.; Sun, G. Functional Significance and Therapeutic Potential of MiRNA-20b-5p in Esophageal Squamous Cell Carcinoma. Mol. Ther. Nucleic Acids 2020, 21, 315–331. [Google Scholar] [CrossRef]
- Go, H.; Jang, J.-Y.; Kim, C.-W.; Huh, J.; Kim, P.-J.; Jeon, Y.K. Identification of MicroRNAs Modulated by DNA Hypomethylating Drugs in Extranodal NK/T-Cell Lymphoma. Leuk. Lymphoma 2020, 61, 66–74. [Google Scholar] [CrossRef]
- Wong, K.K. DNMT1 as a Therapeutic Target in Pancreatic Cancer: Mechanisms and Clinical Implications. Cell Oncol. 2020, 43, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Zagorac, S.; Alcala, S.; Fernandez Bayon, G.; Bou Kheir, T.; Schoenhals, M.; González-Neira, A.; Fernandez Fraga, M.; Aicher, A.; Heeschen, C.; Sainz, B. DNMT1 Inhibition Reprograms Pancreatic Cancer Stem Cells via Upregulation of the MiR-17-92 Cluster. Cancer Res. 2016, 76, 4546–4558. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.A.; Majid, S.; Rittsteuer, C.; de Semir, D.; Bezrookove, V.; Tong, S.; Nosrati, M.; Sagebiel, R.; Miller, J.R., 3rd; Kashani-Sabet, M. The Role of MiR-18b in MDM2-P53 Pathway Signaling and Melanoma Progression. J. Natl. Cancer Inst. 2013, 105, 433–442. [Google Scholar] [CrossRef]
- Jin, L.; Cai, Q.; Wang, S.; Wang, S.; Wang, J.; Quan, Z. Long Noncoding RNA PVT1 Promoted Gallbladder Cancer Proliferation by Epigenetically Suppressing MiR-18b-5p via DNA Methylation. Cell Death Dis. 2020, 11, 871. [Google Scholar] [CrossRef]
- Mi, S.; Li, Z.; Chen, P.; He, C.; Cao, D.; Elkahloun, A.; Lu, J.; Pelloso, L.A.; Wunderlich, M.; Huang, H.; et al. Aberrant Overexpression and Function of the MiR-17-92 Cluster in MLL-Rearranged Acute Leukemia. Proc. Natl. Acad. Sci. USA 2010, 107, 3710–3715. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, Y.; Dou, J.; Guo, Y.; He, J.; Li, L.; Liu, X.; Chen, R.; Deng, R.; Huang, J.; et al. Acetylation of AGO2 Promotes Cancer Progression by Increasing Oncogenic MiR-19b Biogenesis. Oncogene 2019, 38, 1410–1431. [Google Scholar] [CrossRef] [PubMed]
- Lepore, I.; Dell’Aversana, C.; Pilyugin, M.; Conte, M.; Nebbioso, A.; De Bellis, F.; Tambaro, F.P.; Izzo, T.; Garcia-Manero, G.; Ferrara, F.; et al. HDAC Inhibitors Repress BARD1 Isoform Expression in Acute Myeloid Leukemia Cells via Activation of MiR-19a and/or b. PLoS ONE 2013, 8, e83018. [Google Scholar]
- Lovat, F.; Nigita, G.; Distefano, R.; Nakamura, T.; Gasparini, P.; Tomasello, L.; Fadda, P.; Ibrahimova, N.; Catricalà, S.; Palamarchuk, A.; et al. Combined Loss of Function of Two Different Loci of MiR-15/16 Drives the Pathogenesis of Acute Myeloid Leukemia. Proc. Natl. Acad. Sci. USA 2020, 117, 12332–12340. [Google Scholar] [CrossRef]
- Li, D.; Zhao, Y.; Liu, C.; Chen, X.; Qi, Y.; Jiang, Y.; Zou, C.; Zhang, X.; Liu, S.; Wang, X.; et al. Analysis of MiR-195 and MiR-497 Expression, Regulation and Role in Breast Cancer. Clin. Cancer Res. 2011, 17, 1722–1730. [Google Scholar] [CrossRef]
- Tao, S.; Li, H.; Ma, X.; Lian, B.; He, J.; Gao, Y.; Li, J. Methylation-Mediated Silencing of MicroRNA-497 Promotes Breast Cancer Progression Through Up-Regulation of Mucin1. Front. Oncol. 2020, 10, 552099. [Google Scholar] [CrossRef]
- Liu, J.; Li, Y.; Zou, Y.; Zhang, J.; An, J.; Guo, J.; Ma, M.; Dai, D. MicroRNA-497 Acts as a Tumor Suppressor in Gastric Cancer and Is Downregulated by DNA Methylation. Oncol. Rep. 2017, 38, 497–505. [Google Scholar] [CrossRef][Green Version]
- He, X.-X.; Kuang, S.-Z.; Liao, J.-Z.; Xu, C.-R.; Chang, Y.; Wu, Y.-L.; Gong, J.; Tian, D.-A.; Guo, A.-Y.; Lin, J.-S. The Regulation of MicroRNA Expression by DNA Methylation in Hepatocellular Carcinoma. Mol. Biosyst. 2015, 11, 532–539. [Google Scholar] [CrossRef]
- Ma, X.; Zou, L.; Chen, Z.; Li, X.; Wei, L.; Wu, X. Demethylation of MiR-195 Suppresses Prostate Cancer Cell Proliferation, Migration and Invasion. FEBS Open Biol. 2020, 10, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Li, M.; Ouyang, Y.; Tan, Z.; Jiang, Y. MiR-424 Functions as a Tumor Suppressor in Glioma Cells and Is down-Regulated by DNA Methylation. J. Neurooncol. 2017, 133, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Devor, E.J.; Cha, E.; Warrier, A.; Miller, M.D.; Gonzalez-Bosquet, J.; Leslie, K.K. The MiR-503 Cluster Is Coordinately under-Expressed in Endometrial Endometrioid Adenocarcinoma and Targets Many Oncogenes, Cell Cycle Genes, DNA Repair Genes and Chemotherapy Response Genes. Oncol. Targets Ther. 2018, 11, 7205–7211. [Google Scholar] [CrossRef]
- Li, T.; Li, Y.; Gan, Y.; Tian, R.; Wu, Q.; Shu, G.; Yin, G. Methylation-Mediated Repression of MiR-424/503 Cluster Promotes Proliferation and Migration of Ovarian Cancer Cells through Targeting the Hub Gene KIF23. Cell Cycle 2019, 18, 1601–1618. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, X.; Lin, J.; Lwin, T.; Wright, G.; Moscinski, L.C.; Dalton, W.S.; Seto, E.; Wright, K.; Sotomayor, E.; et al. Myc Represses MiR-15a/MiR-16-1 Expression through Recruitment of HDAC3 in Mantle Cell and Other Non-Hodgkin B-Cell Lymphomas. Oncogene 2012, 31, 3002–3008. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-Q.; Chen, C.-S.; Chen, J.-J.; Zhou, L.-P.; Xu, H.-L.; Jin, W.-W.; Wu, J.-B.; Gao, S.-M. Histone Deacetylases Inhibitor Trichostatin A Increases the Expression of Dleu2/MiR-15a/16-1 via HDAC3 in Non-Small Cell Lung Cancer. Mol. Cell Biochem. 2013, 383, 137–148. [Google Scholar] [CrossRef]
- Zhao, N.; Li, S.; Wang, R.; Xiao, M.; Meng, Y.; Zeng, C.; Fang, J.-H.; Yang, J.; Zhuang, S.-M. Expression of MicroRNA-195 Is Transactivated by Sp1 but Inhibited by Histone Deacetylase 3 in Hepatocellular Carcinoma Cells. Biochim. Biophys. Acta 2016, 1859, 933–942. [Google Scholar] [CrossRef]
- Shen, C.-J.; Cheng, Y.-M.; Wang, C.-L. LncRNA PVT1 Epigenetically Silences MiR-195 and Modulates EMT and Chemoresistance in Cervical Cancer Cells. J. Drug Target. 2017, 25, 637–644. [Google Scholar] [CrossRef]
- He, Y.; Meng, C.; Shao, Z.; Wang, H.; Yang, S. MiR-23a Functions as a Tumor Suppressor in Osteosarcoma. Cell Physiol. Biochem. 2014, 34, 1485–1496. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Z.-X.; Chen, S.; Qiu, G.-B.; Xu, Z.-M.; Fu, W.-N. Methylation Status of SP1 Sites within MiR-23a-27a-24-2 Promoter Region Influences Laryngeal Cancer Cell Proliferation and Apoptosis. Biomed. Res. Int. 2016, 2016, 2061248. [Google Scholar] [CrossRef]
- Majid, S.; Dar, A.A.; Saini, S.; Arora, S.; Shahryari, V.; Zaman, M.S.; Chang, I.; Yamamura, S.; Tanaka, Y.; Deng, G.; et al. MiR-23b Represses Proto-Oncogene Src Kinase and Functions as Methylation-Silenced Tumor Suppressor with Diagnostic and Prognostic Significance in Prostate Cancer. Cancer Res. 2012, 72, 6435–6446. [Google Scholar] [CrossRef] [PubMed]
- Campos-Viguri, G.E.; Jiménez-Wences, H.; Peralta-Zaragoza, O.; Torres-Altamirano, G.; Soto-Flores, D.G.; Hernández-Sotelo, D.; Alarcón-Romero, L.D.C.; Jiménez-López, M.A.; Illades-Aguiar, B.; Fernández-Tilapa, G. MiR-23b as a Potential Tumor Suppressor and Its Regulation by DNA Methylation in Cervical Cancer. Infect. Agent Cancer 2015, 10, 42. [Google Scholar] [CrossRef] [PubMed]
- Fulciniti, M.; Amodio, N.; Bandi, R.L.; Cagnetta, A.; Samur, M.K.; Acharya, C.; Prabhala, R.; D’Aquila, P.; Bellizzi, D.; Passarino, G.; et al. MiR-23b/SP1/c-Myc Forms a Feed-Forward Loop Supporting Multiple Myeloma Cell Growth. Blood Cancer J. 2016, 6, e380. [Google Scholar] [CrossRef] [PubMed]
- Grossi, I.; Arici, B.; Portolani, N.; De Petro, G.; Salvi, A. Clinical and Biological Significance of MiR-23b and MiR-193a in Human Hepatocellular Carcinoma. Oncotarget 2017, 8, 6955–6969. [Google Scholar] [CrossRef] [PubMed]
- Yeung, C.L.A.; Tsang, T.Y.; Yau, P.L.; Kwok, T.T. Human Papillomavirus Type 16 E6 Suppresses MicroRNA-23b Expression in Human Cervical Cancer Cells through DNA Methylation of the Host Gene C9orf3. Oncotarget 2017, 8, 12158–12173. [Google Scholar] [CrossRef]
- Barros-Silva, D.; Costa-Pinheiro, P.; Duarte, H.; Sousa, E.J.; Evangelista, A.F.; Graça, I.; Carneiro, I.; Martins, A.T.; Oliveira, J.; Carvalho, A.L.; et al. MicroRNA-27a-5p Regulation by Promoter Methylation and MYC Signaling in Prostate Carcinogenesis. Cell Death Dis. 2018, 9, 167. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, Y.; Liu, A.; Tang, X. MiR-27b Is Epigenetically Downregulated in Tamoxifen Resistant Breast Cancer Cells Due to Promoter Methylation and Regulates Tamoxifen Sensitivity by Targeting HMGB3. Biochem. Biophys. Res. Commun. 2016, 477, 768–773. [Google Scholar] [CrossRef]
- Zhang, C.; Zou, Y.; Dai, D.-Q. Downregulation of MicroRNA-27b-3p via Aberrant DNA Methylation Contributes to Malignant Behavior of Gastric Cancer Cells by Targeting GSPT1. Biomed. Pharm. 2019, 119, 109417. [Google Scholar] [CrossRef]
- Yao, J.; Li, Z.; Yang, Z.; Xue, H.; Chang, H.; Zhang, X.; Li, T.; Guo, K. Long Noncoding RNA TOB1-AS1, an Epigenetically Silenced Gene, Functioned as a Novel Tumor Suppressor by Sponging MiR-27b in Cervical Cancer. Am. J. Cancer Res. 2018, 8, 1483–1498. [Google Scholar] [PubMed]
- Hashimoto, Y.; Shiina, M.; Kato, T.; Yamamura, S.; Tanaka, Y.; Majid, S.; Saini, S.; Shahryari, V.; Kulkarni, P.; Dasgupta, P.; et al. The Role of MiR-24 as a Race Related Genetic Factor in Prostate Cancer. Oncotarget 2017, 8, 16581–16593. [Google Scholar] [CrossRef]
- Bharathy, N.; Berlow, N.E.; Wang, E.; Abraham, J.; Settelmeyer, T.P.; Hooper, J.E.; Svalina, M.N.; Ishikawa, Y.; Zientek, K.; Bajwa, Z.; et al. The HDAC3-SMARCA4-MiR-27a Axis Promotes Expression of the PAX3:FOXO1 Fusion Oncogene in Rhabdomyosarcoma. Sci. Signal. 2018, 11, eaau7632. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.J.; Liu, Z.B.; Wang, W.G.; Sun, C.B.; Wei, P.; Yang, Y.L.; You, M.J.; Yu, B.H.; Li, X.Q.; Zhou, X.Y. HDAC6 Regulates MicroRNA-27b That Suppresses Proliferation, Promotes Apoptosis and Target MET in Diffuse Large B-Cell Lymphoma. Leukemia 2018, 32, 703–711. [Google Scholar] [CrossRef]
- Yang, C.; Cai, J.; Wang, Q.; Tang, H.; Cao, J.; Wu, L.; Wang, Z. Epigenetic Silencing of MiR-130b in Ovarian Cancer Promotes the Development of Multidrug Resistance by Targeting Colony-Stimulating Factor 1. Gynecol. Oncol. 2012, 124, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Ramalho-Carvalho, J.; Graça, I.; Gomez, A.; Oliveira, J.; Henrique, R.; Esteller, M.; Jerónimo, C. Downregulation of MiR-130b~301b Cluster Is Mediated by Aberrant Promoter Methylation and Impairs Cellular Senescence in Prostate Cancer. J. Hematol. Oncol. 2017, 10, 43. [Google Scholar] [CrossRef]
- Fort, R.S.; Mathó, C.; Oliveira-Rizzo, C.; Garat, B.; Sotelo-Silveira, J.R.; Duhagon, M.A. An Integrated View of the Role of MiR-130b/301b MiRNA Cluster in Prostate Cancer. Exp. Hematol. Oncol. 2018, 7, 10. [Google Scholar] [CrossRef]
- Bao, X.; Ren, T.; Huang, Y.; Sun, K.; Wang, S.; Liu, K.; Zheng, B.; Guo, W. Knockdown of Long Non-Coding RNA HOTAIR Increases MiR-454-3p by Targeting Stat3 and Atg12 to Inhibit Chondrosarcoma Growth. Cell Death Dis. 2017, 8, e2605. [Google Scholar] [CrossRef]
- Li, B.-L.; Lu, W.; Lu, C.; Qu, J.; Yang, T.; Yan, Q.; Wan, X. CpG Island Hypermethylation-Associated Silencing of MicroRNAs Promotes Human Endometrial Cancer. Cancer Cell Int. 2013, 13, 44. [Google Scholar] [CrossRef]
- Mazzoccoli, L.; Robaina, M.C.; Apa, A.G.; Bonamino, M.; Pinto, L.W.; Queiroga, E.; Bacchi, C.E.; Klumb, C.E. MiR-29 Silencing Modulates the Expression of Target Genes Related to Proliferation, Apoptosis and Methylation in Burkitt Lymphoma Cells. J. Cancer Res. Clin. Oncol. 2018, 144, 483–497. [Google Scholar] [CrossRef]
- Cui, H.; Wang, L.; Gong, P.; Zhao, C.; Zhang, S.; Zhang, K.; Zhou, R.; Zhao, Z.; Fan, H. Deregulation between MiR-29b/c and DNMT3A Is Associated with Epigenetic Silencing of the CDH1 Gene, Affecting Cell Migration and Invasion in Gastric Cancer. PLoS ONE 2015, 10, e0123926. [Google Scholar] [CrossRef]
- Li, H.; Liu, G.; Pan, K.; Miao, X.; Xie, Y. Methylation-Induced Downregulation and Tumor Suppressive Role of MicroRNA-29b in Gastric Cancer through Targeting LASP1. Oncotarget 2017, 8, 95880–95895. [Google Scholar] [CrossRef]
- Teng, Y.; Zuo, X.; Hou, M.; Zhang, Y.; Li, C.; Luo, W.; Li, X. A Double-Negative Feedback Interaction between MicroRNA-29b and DNMT3A/3B Contributes to Ovarian Cancer Progression. Cell Physiol. Biochem. 2016, 39, 2341–2352. [Google Scholar] [CrossRef]
- Wang, L.; Mu, N.; Qu, N. Methylation of the MiR-29b-3p Promoter Contributes to Angiogenesis, Invasion, and Migration in Pancreatic Cancer. Oncol. Rep. 2021, 45, 65–72. [Google Scholar] [CrossRef]
- Hu, S.; Yao, Y.; Hu, X.; Zhu, Y. LncRNA DCST1-AS1 Downregulates MiR-29b through Methylation in Glioblastoma (GBM) to Promote Cancer Cell Proliferation. Clin. Transl. Oncol. 2020, 22, 2230–2235. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wu, L.-C.; Pang, J.; Santhanam, R.; Schwind, S.; Wu, Y.-Z.; Hickey, C.J.; Yu, J.; Becker, H.; Maharry, K.; et al. Sp1/NFkappaB/HDAC/MiR-29b Regulatory Network in KIT-Driven Myeloid Leukemia. Cancer Cell 2010, 17, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Tarighat, S.S.; Santhanam, R.; Frankhouser, D.; Radomska, H.S.; Lai, H.; Anghelina, M.; Wang, H.; Huang, X.; Alinari, L.; Walker, A.; et al. The Dual Epigenetic Role of PRMT5 in Acute Myeloid Leukemia: Gene Activation and Repression via Histone Arginine Methylation. Leukemia 2016, 30, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Amodio, N.; Stamato, M.A.; Gullà, A.M.; Morelli, E.; Romeo, E.; Raimondi, L.; Pitari, M.R.; Ferrandino, I.; Misso, G.; Caraglia, M.; et al. Therapeutic Targeting of MiR-29b/HDAC4 Epigenetic Loop in Multiple Myeloma. Mol. Cancer Ther. 2016, 15, 1364–1375. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, X.; Fiskus, W.; Lin, J.; Lwin, T.; Rao, R.; Zhang, Y.; Chan, J.C.; Fu, K.; Marquez, V.E.; et al. Coordinated Silencing of MYC-Mediated MiR-29 by HDAC3 and EZH2 as a Therapeutic Target of Histone Modification in Aggressive B-Cell Lymphomas. Cancer Cell 2012, 22, 506–523. [Google Scholar] [CrossRef]
- Muraoka, T.; Soh, J.; Toyooka, S.; Aoe, K.; Fujimoto, N.; Hashida, S.; Maki, Y.; Tanaka, N.; Shien, K.; Furukawa, M.; et al. The Degree of MicroRNA-34b/c Methylation in Serum-Circulating DNA Is Associated with Malignant Pleural Mesothelioma. Lung Cancer 2013, 82, 485–490. [Google Scholar] [CrossRef]
- Sato, H.; Soh, J.; Aoe, K.; Fujimoto, N.; Tanaka, S.; Namba, K.; Torigoe, H.; Shien, K.; Yamamoto, H.; Tomida, S.; et al. Droplet Digital PCR as a Novel System for the Detection of MicroRNA-34b/c Methylation in Circulating DNA in Malignant Pleural Mesothelioma. Int. J. Oncol. 2019, 54, 2139–2148. [Google Scholar] [CrossRef] [PubMed]
- Harada, T.; Yamamoto, E.; Yamano, H.; Nojima, M.; Maruyama, R.; Kumegawa, K.; Ashida, M.; Yoshikawa, K.; Kimura, T.; Harada, E.; et al. Analysis of DNA Methylation in Bowel Lavage Fluid for Detection of Colorectal Cancer. Cancer Prev. Res. 2014, 7, 1002–1010. [Google Scholar] [CrossRef]
- Ohtsubo, K.; Miyake, K.; Arai, S.; Fukuda, K.; Yanagimura, N.; Suzuki, C.; Otani, S.; Adachi, Y.; Tanimoto, A.; Nishiyama, A.; et al. Aberrant Methylation of Tumor Suppressive MiRNAs in Bile from Patients With Pancreaticobiliary Diseases. Anticancer Res. 2019, 39, 5449–5459. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting Epigenetic Regulators for Cancer Therapy: Mechanisms and Advances in Clinical Trials. Signal. Transduct. Target. Ther. 2019, 4, 1–39. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.L.; Perfler, B.; Hatzl, S.; Mayer, M.-C.; Wurm, S.; Uhl, B.; Reinisch, A.; Klymiuk, I.; Tierling, S.; Pregartner, G.; et al. Micro-RNA-125a Mediates the Effects of Hypomethylating Agents in Chronic Myelomonocytic Leukemia. Clin. Epigenetics 2021, 13, 1–10. [Google Scholar] [CrossRef]
- Soung, Y.H.; Chung, H.; Yan, C.; Fesler, A.; Kim, H.; Oh, E.-S.; Ju, J.; Chung, J. Therapeutic Potential of Chemically Modified MiR-489 in Triple-Negative Breast Cancers. Cancers 2020, 12, 2209. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.S.; Sharp, P.A. MicroRNA Sponges: Progress and Possibilities. RNA 2010, 16, 2043–2050. [Google Scholar] [CrossRef]
- Wang, Z. The Principles of MiRNA-Masking Antisense Oligonucleotides Technology. Methods Mol. Biol. 2011, 676, 43–49. [Google Scholar]
- Hong, D.S.; Kang, Y.-K.; Borad, M.; Sachdev, J.; Ejadi, S.; Lim, H.Y.; Brenner, A.J.; Park, K.; Lee, J.-L.; Kim, T.-Y.; et al. Phase 1 Study of MRX34, a Liposomal MiR-34a Mimic, in Patients with Advanced Solid Tumours. Br. J. Cancer 2020, 122, 1630–1637. [Google Scholar] [CrossRef]
- Van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.J.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.J.; Bailey, D.L.; et al. Safety and Activity of MicroRNA-Loaded Minicells in Patients with Recurrent Malignant Pleural Mesothelioma: A First-in-Man, Phase 1, Open-Label, Dose-Escalation Study. Lancet Oncol. 2017, 18, 1386–1396. [Google Scholar] [CrossRef]
- Wang, T.; Xie, Y.; Tan, A.; Li, S.; Xie, Z. Construction and Characterization of a Synthetic MicroRNA Cluster for Multiplex RNA Interference in Mammalian Cells. ACS Synth Biol. 2016, 5, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- González-Vallinas, M.; Rodríguez-Paredes, M.; Albrecht, M.; Sticht, C.; Stichel, D.; Gutekunst, J.; Pitea, A.; Sass, S.; Sánchez-Rivera, F.J.; Lorenzo-Bermejo, J.; et al. Epigenetically Regulated Chromosome 14q32 MiRNA Cluster Induces Metastasis and Predicts Poor Prognosis in Lung Adenocarcinoma Patients. Mol. Cancer Res. 2018, 16, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Yang, S.; Zhao, Y.; Zhang, H.; Wu, Q.; Chen, F. Global Analysis of MiRNA Gene Clusters and Gene Families Reveals Dynamic and Coordinated Expression. Biomed. Res. Int. 2014, 2014, 782490. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lin, Y.; Gu, C.; Yang, J. FCMDAP: Using MiRNA Family and Cluster Information to Improve the Prediction Accuracy of Disease Related MiRNAs. BMC Syst. Biol. 2019, 13, 26. [Google Scholar] [CrossRef] [PubMed]
- Dai, E.; Yu, X.; Zhang, Y.; Meng, F.; Wang, S.; Liu, X.; Liu, D.; Wang, J.; Li, X.; Jiang, W. EpimiR: A Database of Curated Mutual Regulation between MiRNAs and Epigenetic Modifications. Database 2014, 2014, bau023. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| DNA Methylation | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cancer Type | miRNA | miRNA Cluster (miRBase 22.1) | Methylation Status | Epigenetic Regulator | miRNA Family (miRBase 22.1) | Seed Region # | Associated Processes/Targets Clinical Outcomes | Reference |
| GC | miR-125a | mir-99b~125a (chr19) | HyperMet | - | miR-125a-3p miR-125-5p | CAGGUGA CCCUGAG | Migration, invasion, proliferation, progression | [89] |
| CRC | miR-125a | mir-99b~125a (chr19) | HyperMet | - | miR-125a-3p miR-125-5p | CAGGUGA CCCUGAG | OS | [88] |
| LC | let-7a-3 | let-7a-3~let-7b (chr22) | HyperMet | DNMT1/3B | let-7a-3p/98-3p let-7-5p/98-5p | UAUACAA GAGGUAG | Proliferation, cell adhesion | [84] |
| OC | let-7a-3 | let-7a-3~let-7b (chr22) | HypoMet | - | let-7a-3p/98-3p let-7-5p/98-5p | UAUACAA GAGGUAG | IGF2, OS | [81] |
| BC | let-7e | mir-99b~125a (chr19) | HyperMet | - | let-7e-3p let-7-5p/98-5p | UAUACGG GAGGUAG | Cell viability, apoptosis, MAPK1, EZH2, OS, subtype | [86] |
| Gliomas | miR-98 | let-7f-2~98 (chrX) | HyperMet | - | let-7a-3p/98-3p let-7-5p/98-5p | UAUACAA GAGGUAG | Migration, invasion, SALL4, aggressiveness, OS | [90] |
| MM | miR-125a | mir-99b~125a (chr19) | HyperMet | - | miR-125a-3p miR-125-5p | CAGGUGA CCCUGAG | - | [87] |
| ALL | let-7b | let-7a-3~let-7b (chr22) | HyperMet | - | let-7a-3p/98-3p let-7-5p/98-5p | UAUACAA GAGGUAG | Apoptosis, cell cycle | [83] |
| MDS | let-7a-3 | let-7a-3~let-7b (chr22) | HypoMet | - | let-7a-3p/98-3p let-7-5p/98-5p | UAUACAA GAGGUAG | Age, survival | [85] |
| Histone Modifications | ||||||||
| Cancer Type | miRNA | miRNA Cluster (miRBase 22.1) | Histone Modification | Epigenetic Regulator | miRNA Family (miRBase 22.1) | Seed Region # | Associated Processes/Targets Clinical Outcomes | Reference |
| BC | let-7a-3 | let-7a-3~let-7b (chr22) | H3K27me3 | - | let-7a-3p/98-3p let-7-5p/98-5p | UAUACAA GAGGUAG | - | [94] |
| let-7b | let-7a-3~let-7b (chr22) | H3K27me3 | - | let-7a-3p/98-3p let-7-5p/98-5p | UAUACAA GAGGUAG | - | [94] | |
| let-7e | mir-99b~125 (chr19) | H3K4me3 | JARID1B | let-7e-3p let-7-5p/98-5p | UAUACGG GAGGUAG | Proliferation, CCND1 | [91] | |
| LC | miR-99a | mir-99a~let-7c (chr21) | H4R3me2 | PRMT5 | miR-99-3p miR-99-5p/100-5p | AAGCUCG ACCCGUA | cell growth, FGFR3, metastasis, stage, LNP, OS | [93] |
| miR-99b | mir-99b~125a (chr19) | H4R3me2 | PRMT5 | miR-99-3p miR-99-5p/100-5p | AAGCUCG ACCCGUA | cell growth, FGFR3, metastasis, stage, LNP, OS | [93] | |
| miR-100 | mir-100~let-7a-2 (chr11) | H4R3me2 | PRMT5 | miR-100-3p miR-99-5p/100-5p | AAGCUUG ACCCGUA | cell growth, FGFR3, metastasis, stage, LNP, OS | [93] | |
| HCC | let-7c | mir-99a~let-7c (chr21) | H3K27me3 | EZH2 | let-7c-3p let-7-5p/98-5p | UGUACAA GAGGUAG | Liver metastasis | [95] |
| DNA Methylation | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cancer Type | miRNA | miRNA Cluster (miRBase 22.1) | Methylation Status | Epigenetic Regulator | miRNA Family (miRBase 22.1) | Seed Region # | Associated Processes/Targets Clinical Outcomes | Reference |
| PAC | miR-34a | - | HyperMet | - | miR-34a-3p miR-34-5p/449-5p | AAUCAGC GGCAGUG | Senescence, cell cycle, CDK6 | [97,99] |
| miR-34b | mir-34b~34c (chr11) | HyperMet | - | miR-34b-3p miR-34b-5p/449c-5p | AAUCACU AGGCAGU | - | [99] | |
| miR-34c | mir-34b~34c (chr11) | HyperMet | - | miR-34c-3p miR-34-5p/449-5p | AUCACUA GGCAGUG | - | [99] | |
| PC | miR-34a | - | Hyper/ HypoMet | - | miR-34a-3p miR-34-5p/449-5p | AAUCAGC GGCAGUG | Senescence, cell cycle, CDK6, stage, Gleason score | [97,117] |
| miR-34b | mir-34b~34c (chr11) | HypoMet | - | miR-34b-3p miR-34b-5p/449c-5p | AAUCACU AGGCAGU | PSA level | [117] | |
| miR-34c | mir-34b~34c (chr11) | HypoMet | - | miR-34c-3p miR-34-5p/449-5p | AUCACUA GGCAGUG | PSA level | [117] | |
| GC | miR-34b | mir-34b~34c (chr11) | HyperMet | - | miR-34b-3p miR-34b-5p/449c-5p | AAUCACU AGGCAGU | Cell growth | [106] |
| miR-34c | mir-34b~34c (chr11) | HyperMet | - | miR-34c-3p miR-34-5p/449-5p | AUCACUA GGCAGUG | Cell growth | [106] | |
| CRC | miR-34a | - | HyperMet | - | miR-34a-3p miR-34-5p/449-5p | AAUCAGC GGCAGUG | p53 mutation status | [97,99] |
| miR-34b | mir-34b~34c (chr11) | HyperMet | - | miR-34b-3p miR-34b-5p/449c-5p | AAUCACU AGGCAGU | - | [99] | |
| miR-34c | mir-34b~34c (chr11) | HyperMet | - | miR-34c-3p miR-34-5p/449-5p | AUCACUA GGCAGUG | - | [99] | |
| OC | miR-34a | - | HyperMet | - | miR-34a-3p miR-34-5p/449-5p | AAUCAGC GGCAGUG | - | [99] |
| miR-34b | mir-34b~34c (chr11) | HyperMet | - | miR-34b-3p miR-34b-5p/449c-5p | AAUCACU AGGCAGU | - | [99] | |
| miR-34c | mir-34b~34c (chr11) | HyperMet | - | miR-34c-3p miR-34-5p/449-5p | AUCACUA GGCAGUG | - | [99] | |
| BC | miR-34a | - | HyperMet HypoMet | DNMT1 TET1 | miR-34a-3p miR-34-5p/449-5p | AAUCAGC GGCAGUG | - | [97,99,107] |
| miR-34b | mir-34b~34c (chr11) | HyperMet | - | miR-34b-3p miR-34b-5p/449c-5p | AAUCACU AGGCAGU | - | [99] | |
| miR-34c | mir-34b~34c (chr11) | HyperMet | - | miR-34c-3p miR-34-5p/449-5p | AUCACUA GGCAGUG | - | [99] | |
| CC | miR-34c | mir-34b~34c (chr11) | HypoMet | - | miR-34c-3p miR-34-5p/449-5p | AUCACUA GGCAGUG | - | [118] |
| UCA | miR-34a | - | HyperMet | - | miR-34a-3p miR-34-5p/449-5p | AAUCAGC GGCAGUG | - | [99] |
| miR-34b | mir-34b~34c (chr11) | HyperMet | - | miR-34b-3p miR-34b-5p/449c-5p | AAUCACU AGGCAGU | - | [99] | |
| miR-34c | mir-34b~34c (chr11) | HyperMet | - | miR-34c-3p miR-34-5p/449-5p | AUCACUA GGCAGUG | - | [99] | |
| RCC | miR-34a | - | HyperMet | - | miR-34a-3p miR-34-5p/449-5p | AAUCAGC GGCAGUG | - | [97,99] |
| miR-34b | mir-34b~34c (chr11) | HyperMet | - | miR-34b-3p miR-34b-5p/449c-5p | AAUCACU AGGCAGU | - | [99] | |
| miR-34c | mir-34b~34c (chr11) | HyperMet | - | miR-34c-3p miR-34-5p/449-5p | AUCACUA GGCAGUG | - | [99] | |
| STS | miR-34a | - | HyperMet | - | miR-34a-3p miR-34-5p/449-5p | AAUCAGC GGCAGUG | - | [99] |
| miR-34b | mir-34b~34c (chr11) | HyperMet | - | miR-34b-3p miR-34b-5p/449c-5p | AAUCACU AGGCAGU | - | [99] | |
| miR-34c | mir-34b~34c (chr11) | HyperMet | - | miR-34c-3p miR-34-5p/449-5p | AUCACUA GGCAGUG | - | [99] | |
| ESCC | miR-34a | - | HyperMet | - | miR-34a-3p miR-34-5p/449-5p | AAUCAGC GGCAGUG | - | [101] |
| miR-34b | mir-34b~34c (chr11) | HyperMet | - | miR-34b-3p miR-34b-5p/449c-5p | AAUCACU AGGCAGU | Early stages, ESCC differentiation | [100,101] | |
| miR-34c | mir-34b~34c (chr11) | HyperMet | - | miR-34c-3p miR-34-5p/449-5p | AUCACUA GGCAGUG | Early stages, ESCC differentiation | [100,101] | |
| MPM | miR-34a | - | HyperMet | - | miR-34a-3p miR-34-5p/449-5p | AAUCAGC GGCAGUG | - | [102] |
| miR-34b | mir-34b~34c (chr11) | HyperMet | - | miR-34b-3p miR-34b-5p/449c-5p | AAUCACU AGGCAGU | Cell cycle, migration, invasion, motility, apoptosis | [102] | |
| miR-34c | mir-34b~34c (chr11) | HyperMet | - | miR-34c-3p miR-34-5p/449-5p | AUCACUA GGCAGUG | Cell cycle, migration, invasion, motility, apoptosis | [102] | |
| MB | miR-449a | mir-449c~449a (chr5) | HyperMet | - | miR-34-5p/449-5p | GGCAGUG | WNT group | [108] |
| LSCC | miR-34a | - | HyperMet | - | miR-34a-3p miR-34-5p/449-5p | AAUCAGC GGCAGUG | Smoking, stage, LNP, OS | [104] |
| Melanoma | miR-34a | - | HyperMet | - | miR-34a-3p miR-34-5p/449-5p | AAUCAGC GGCAGUG | - | [97] |
| BLC | miR-34a | - | HyperMet | DNMT3B | miR-34a-3p miR-34-5p/449-5p | AAUCAGC GGCAGUG | Migration, invasion, HNF4γ, NOTCH1 | [97,105] |
| HCC | miR-34b | mir-34b~34c (chr11) | HyperMet | - | miR-34b-3p miR-34b-5p/449c-5p | AAUCACU AGGCAGU | - | [103] |
| LC | miR-34a | - | HyperMet | - | miR-34a-3p miR-34-5p/449-5p | AAUCAGC GGCAGUG | - | [97] |
| NKTCL | miR-34a | - | HyperMet | - | miR-34a-3p miR-34-5p/449-5p | AAUCAGC GGCAGUG | - | [98] |
| NHL | miR-34a | - | HyperMet | - | miR-34a-3p miR-34-5p/449-5p | AAUCAGC GGCAGUG | - | [98] |
| Histone Modifications | ||||||||
| Cancer Type | miRNA | miRNA Cluster (miRBase 22.1) | Histone Modification | Epigenetic Regulator | miRNA Family (miRBase 22.1) | Seed Region # | Associated Processes/Targets Clinical Outcomes | Reference |
| BC | miR-34a | - | H3Ac | Linc-ROR | miR-34a-3p miR-34-5p/449-5p | AAUCAGC GGCAGUG | Gemcitabine-induced autophagy, apoptosis | [112] |
| miR-34b | mir-34b~34c (chr11) | H3K27me3 | - | miR-34b-3p miR-34b-5p/449c-5p | AAUCACU AGGCAGU | - | [94] | |
| miR-34c | mir-34b~34c (chr11) | H3K27me3 | - | miR-34c-3p miR-34-5p/449-5p | AUCACUA GGCAGUG | - | [94] | |
| LC | miR-449a | mir-449c~449a (chr5) | H3K27me3 | SUZ12 | miR-34-5p/449-5p | GGCAGUG | Metastasis, LNP, MAP2K1 | [114] |
| CHC | miR-34a | - | H3K27me3 | EZH2 | miR-34a-3p miR-34-5p/449-5p | AAUCAGC GGCAGUG | Proliferation, clonogenicity, tumor growth, NOTCH1/2, JAGGED1 | [119] |
| GC | miR-34a | - | HAc | HDAC1 | miR-34a-3p miR-34-5p/449-5p | AAUCAGC GGCAGUG | Metastasis, CD44, prognosis | [110] |
| OC | miR-34a | - | HAc | HDAC1 | miR-34a-3p miR-34-5p/449-5p | AAUCAGC GGCAGUG | Proliferation, apoptosis, chemosensitivity | [111] |
| OSS | miR-449a | mir-449c~449a (chr5) | H3K27me3 | - | miR-34-5p/449-5p | GGCAGUG | Cell cycle, CDK6, CDC25A | [113] |
| miR-449b | mir-449c~449a (chr5) | H3K27me3 | - | miR-449b-3p miR-34-5p/449-5p | AGCCACA GGCAGUG | Cell cycle, CDK6, CDC25A | [113] | |
| HCC | miR-449a | mir-449c~449a (chr5) | HAc | HDAC1-3 | miR-34-5p/449-5p | GGCAGUG | Proliferation, apoptosis, growth, c-MET | [115] |
| DNA Methylation | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cancer Type | miRNA | miRNA Cluster (miRBase 22.1) | Methylation Status | Epigenetic Regulator | miRNA Family (miRBase 22.1) | Seed Region # | Associated Processes/Targets Clinical Outcomes | Reference |
| PAC | miR-200a | mir-200b~429 (chr1) | HypoMet | - | miR-141-3p/200a-3p miR-200ab-5p | AACACUG AUCUUAC | Serum, SIP1 methylation | [124,126] |
| miR-200b | mir-200b~429 (chr1) | HypoMet | - | miR-200bc-3p/429 miR-200ab-5p | AAUACUG AUCUUAC | Serum, SIP1 methylation | [124,126] | |
| miR-200c | mir-200c~141 (chr12) | HyperMet | - | miR-200bc-3p/429 miR-200c-5p/550a-3p | AAUACUG GUCUUAC | Invasion, WIPF1, OS | [126] | |
| miR-141 | mir-200c~141 (chr12) | HyperMet | - | miR-141-3p/200a-3p miR-141-5p | AACACUG AUCUUCC | Invasion, WIPF1, OS | [126] | |
| miR-429 | mir-200b~429 (chr1) | HypoMet | - | miR-200bc-3p/429 | AAUACUG | - | [126] | |
| PC | miR-200c | mir-200c~141 (chr12) | HyperMet | DNMT1 | miR-200bc-3p/429 miR-200c-5p/550a-3p | AAUACUG GUCUUAC | Apoptosis, clonogenicity, cell growth, DNMT3A, TET1/3 | [134] |
| miR-141 | mir-200c~141 (chr12) | HyperMet | DNMT1 | miR-141-3p/200a-3p miR-141-5p | AACACUG AUCUUCC | DNMT3A, TET1/3 | [134] | |
| GC | miR-200a | mir-200b~429 (chr1) | HyperMet | - | miR-141-3p/200a-3p miR-200ab-5p | AACACUG AUCUUAC | H. pylori infection | [133] |
| miR-200b | mir-200b~429 (chr1) | HyperMet | - | miR-200bc-3p/429 miR-200ab-5p | AAUACUG AUCUUAC | H. pylori infection | [133] | |
| BLC | miR-200a | mir-200b~429 (chr1) | HyperMet | - | miR-141-3p/200a-3p miR-200ab-5p | AACACUG AUCUUAC | Invasiveness, differentiation | [122] |
| miR-200b | mir-200b~429 (chr1) | HyperMet | - | miR-200bc-3p/429 miR-200ab-5p | AAUACUG AUCUUAC | Invasiveness, differentiation, CDDP sensitivity, OS | [122,123] | |
| miR-200c | mir-200c~141 (chr12) | HyperMet | - | miR-200bc-3p/429 miR-200c-5p/550a-3p | AAUACUG GUCUUAC | Invasiveness, differentiation, prognosis of T1 tumors | [122] | |
| HCC | miR-200b | mir-200b~429 (chr1) | HyperMet | - | miR-200bc-3p/429 miR-200ab-5p | AAUACUG AUCUUAC | Prognosis, BMI1, ZEB1 | [125] |
| BC | miR-200b | mir-200b~429 (chr1) | HyperMet HypoMet | DNMT3A/ Kindlin 2/MYC TET1-3 | miR-200bc-3p/429 miR-200ab-5p | AAUACUG AUCUUAC | Invasion, metastasis, SOX2, CD133, EMT, BC subtype | [121,129,130,131,132] |
| miR-200a | mir-200b~429 (chr1) | HyperMet HypoMet | DNMT3A/ FEN1/PCNA TET1-3 | miR-141-3p/200a-3p miR-200ab-5p | AACACUG AUCUUAC | Proliferation, MET, EGFR | [128,131,132] | |
| CC | miR-200b | mir-200b~429 (chr1) | HypoMet | - | miR-200bc-3p/429 miR-200ab-5p | AAUACUG AUCUUAC | HIPK3, RBBP6 | [118] |
| oral | miR-200a | mir-200b~429 (chr1) | HyperMet | - | miR-141-3p/200a-3p miR-200ab-5p | AACACUG AUCUUAC | OS, smokers/chewers | [127] |
| miR-200b | mir-200b~429 (chr1) | HyperMet | - | miR-200bc-3p/429 miR-200ab-5p | AAUACUG AUCUUAC | OS, smokers/chewers | [127] | |
| MM | miR-200c | mir-200c~141 (chr12) | HyperMet | - | miR-200bc-3p/429 miR-200c-5p/550a-3p | AAUACUG GUCUUAC | Proliferation, migration, colony formation | [87] |
| Histone Modifications | ||||||||
| Cancer Type | miRNA | miRNA Cluster (miRBase 22.1) | Histone Modification | Epigenetic Regulator | miRNA Family (miRBase 22.1) | Seed Region # | Associated Processes/Targets Clinical Outcomes | Reference |
| BC | miR-200a | mir-200b~429 (chr1) | H3K27me3 HAc | EZH2/SUZ12 HDAC2/PELP1 | miR-141-3p/200a-3p miR-200ab-5p | AACACUG AUCUUAC | CSCs, metastasis, ZEB1/2, OS | [135,140] |
| miR-200b | mir-200b~429 (chr1) | H3K27me3 | EZH2/SUZ12 | miR-200bc-3p/429 miR-200ab-5p | AAUACUG AUCUUAC | CSCs | [135] | |
| miR-429 | mir-200b~429 (chr1) | H3K27me3 | EZH2/SUZ12 | miR-200bc-3p/429 | AAUACUG | CSCs | [135] | |
| miR-141 | mir-200c~141 (chr12) | HAc | HDAC2/PELP1 | miR-141-3p/200a-3p miR-141-5p | AACACUG AUCUUCC | metastasis, ZEB1/2, OS | [140] | |
| LC | miR-200a | mir-200b~429 (chr1) | H3K4me3 | JARID1B | miR-200bc-3p/429 miR-200ab-5p | AAUACUG AUCUUAC | EMT, ZEB1/2 | [139] |
| miR-200c | mir-200c~141 (chr12) | H3K4me3 | JARID1B | miR-200bc-3p/429 miR-200c-5p/550a-3p | AAUACUG GUCUUAC | EMT, ZEB1/2 | [139] | |
| miR-200b | mir-200b~429 (chr1) | H3Ac | HDAC1/4 | miR-200bc-3p/429 miR-200ab-5p | AAUACUG AUCUUAC | Proliferation, apoptosis, cell cycle, chemoresistance, E2F3 | [141] | |
| GC | miR-200a | mir-200b~429 (chr1) | H3K27me3 | EZH2 | miR-141-3p/200a-3p miR-200ab-5p | AACACUG AUCUUAC | Disease progression | [136] |
| miR-200b | mir-200b~429 (chr1) | H3K27me3 | EZH2 | miR-200bc-3p/429 miR-200ab-5p | AAUACUG AUCUUAC | Disease progression | [136] | |
| miR-429 | mir-200b~429 (chr1) | H3K27me3 | EZH2 | miR-200bc-3p/429 | AAUACUG | Disease progression | [136] | |
| CC | miR-200b | mir-200b~429 (chr1) | H3K27me3 | EZH2/PVT1 | miR-200bc-3p/429 miR-200ab-5p | AAUACUG AUCUUAC | Proliferation, cell cycle, migration | [137] |
| HCC | miR-200b | mir-200b~429 (chr1) | H3K27me3 H3Ac | EZH2/GIHCG hnRNP U/PCAF/ RNA Pol II/H19 | miR-200bc-3p/429 miR-200ab-5p | AAUACUG AUCUUAC | Proliferation, migration, liver metastasis, EMT, tumor size, stage, OS | [95,138,143] |
| miR-200a | mir-200b~429 (chr1) | H3K27me3 H3Ac | EZH2/GIHCG hnRNP U/PCAF/ RNA Pol II/H19 | miR-141-3p/200a-3p miR-200ab-5p | AACACUG AUCUUAC | Proliferation, migration, metastasis, EMT, tumor size, stage, OS | [138,143] | |
| miR-429 | mir-200b~429 (chr1) | H3K27me3 H3Ac | EZH2/GIHCG hnRNP U/PCAF/ RNA Pol II/H19 | miR-200bc-3p/429 | AAUACUG | Proliferation, migration, metastasis, EMT, tumor size, stage, OS | [138,143] | |
| miR-200c | mir-200c~141 (chr12) | H3Ac | hnRNP U/PCAF/ RNA Pol II/H19 | miR-200bc-3p/429 miR-200c-5p/550a-3p | AAUACUG GUCUUAC | EMT, metastasis | [143] | |
| miR-141 | mir-200c~141 (chr12) | H3Ac | hnRNP U/PCAF/ RNA Pol II/H19 | miR-141-3p/200a-3p miR-141-5p | AACACUG AUCUUCC | EMT, metastasis | [143] | |
| glioma | miR-200a | mir-200b~429 (chr1) | H3K27me3 | EZH2 | miR-141-3p/200a-3p miR-200ab-5p | AACACUG AUCUUAC | Disease progression | [136] |
| miR-200b | mir-200b~429 (chr1) | H3K27me3 | EZH2 | miR-200bc-3p/429 miR-200ab-5p | AAUACUG AUCUUAC | Disease progression | [136] | |
| miR-429 | mir-200b~429 (chr1) | H3K27me3 | EZH2 | miR-200bc-3p/429 | AAUACUG | Disease progression | [136] | |
| DNA Methylation | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cancer Type | miRNA | miRNA Cluster (miRBase 22.1) | Methylation Status | Epigenetic Regulator | miRNA Family (miRBase 22.1) | Seed Region # | Associated Processes/Targets Clinical Outcomes | Reference |
| PAC | miR-19a | mir-17~92a-1 (chr13) | HyperMet | DNMT1 | miR-19-3p miR-19-5p | GUGCAAA GUUUUGC | CSCs | [152] |
| miR-19b | mir-17~92a-1 (chr13) | HyperMet | DNMT1 | miR-19-3p miR-19-5p | GUGCAAA GUUUUGC | CSCs | [152] | |
| GC | miR-19a | mir-17~92a-1 (chr13) | HypoMet | MeCP2 | miR-19-3p miR-19-5p | GUGCAAA GUUUUGC | MeCP2, MDR | [145] |
| miR-19b | mir-17~92a-1 (chr13) mir-106a~363 (chrX) | HypoMet | MeCP2 | miR-19-3p miR-19-5p | GUGCAAA GUUUUGC | MeCP2, MDR | [145] | |
| miR-106a | mir-106a~363 (chrX) | HypoMet | - | miR-106a-3p miR-17-5p/519-3p | UGCAAUG AAAGUGC | LNP, stage, diagnosis | [148] | |
| HCC | miR-106a | mir-106a~363 (chrX) | HypoMet | - | miR-106a-3p miR-17-5p/519-3p | UGCAAUG AAAGUGC | Invasiveness, cell cycle, apoptosis, TP53INP1, CDKN1A | [147] |
| glioma | miR-20a | mir-17~92a-1 (chr13) | HypoMet | DNMT1 | miR-20a-3p miR-17-5p/519-3p | CUGCAUU AAAGUGC | TMZ-resistance, apoptosis, cell cycle, tumor growth, LRIG1 | [146] |
| ESCC | miR-20b | mir-106a~363 (chrX) | HypoMet | - | miR-20b-3p miR-17-5p/519-3p | CUGUAGU AAAGUGC | Proliferation, migration, RB1, TP53INP1, LNP, stage, OS | [149] |
| melanoma | miR-18b | mir-106a~363 (chrX) | HyperMet | - | miR-18b-3p miR-18-5p | GCCCUAA AAGGUGC | colony formation, apoptosis, tumor growth, EMT, MDM2 | [153] |
| GB | miR-18b | mir-106a~363 (chrX) | HyperMet | DNMT1/EZH2/PVT1 | miR-18b-3p miR-18-5p | GCCCUAA AAGGUGC | Proliferation, HIF1α, prognosis | [154] |
| NKTCL | miR-20b | mir-106a~363 (chrX) | HyperMet | - | miR-20b-3p miR-17-5p/519-3p | CUGUAGU AAAGUGC | STAT3 | [150] |
| Histone Modifications | ||||||||
| Cancer Type | miRNA | miRNA Cluster (miRBase 22.1) | Histone Modification | Epigenetic Regulator | miRNA Family (miRBase 22.1) | Seed Region # | Associated Processes/Targets Clinical Outcomes | Reference |
| ALL | miR-17 | mir-17~92a-1 (chr13) | H3ac H3K4me3 | - | miR-17-3p miR-17-5p/519-3p | CUGCAGU AAAGUGC | Proliferation, apoptosis, colony formation, MLL-rear. | [155] |
| miR-18a | mir-17~92a-1 (chr13) | H3ac H3K4me3 | - | miR-18a-3p miR-18-5p | CUGCCCU AAGGUGC | Proliferation, apoptosis, colony formation, MLL-rear. | [155] | |
| miR-19a | mir-17~92a-1 (chr13) | H3ac H3K4me3 | - | miR-19-3p miR-19-5p | GUGCAAA GUUUUGC | Proliferation, apoptosis, colony formation, MLL-rear. | [155] | |
| miR-20a | mir-17~92a-1 (chr13) | H3ac H3K4me3 | - | miR-20a-3p mir-17-5p/519-3p | CUGCAUU AAAGUGC | Proliferation, apoptosis, colony formation, MLL-rear. | [155] | |
| miR-19b-1 | mir-17~92a-1 (chr13) | H3ac H3K4me3 | - | miR-19-3p miR-19-5p | GUGCAAA GUUUUGC | Proliferation, apoptosis, colony formation, MLL-rear. | [155] | |
| miR-92a-1 | mir-17~92a-1 (chr13) | H3ac H3K4me3 | - | miR-25-3p/367-3p miR-92a-1-5p | AUUGCAC GGUUGGG | Proliferation, apoptosis, colony formation, MLL-rear. | [155] | |
| DLBCL | miR-19a | mir-17~92a-1 (chr13) | HAc | HDAC1/2 | miR-19-3p miR-19-5p | GUGCAAA GUUUUGC | BARD1 Vorinostat treatment | [157] |
| miR-19b | mir-17~92a-1 (chr13) mir-106a~363 (chrX) | HAc | HDAC1/2 | miR-19-3p miR-19-5p | GUGCAAA GUUUUGC | BARD1 Vorinostat treatment | [157] | |
| DNA Methylation | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cancer Type | miRNA | miRNA Cluster (miRBase 22.1) | Methylation Status | Epigenetic Regulator | miRNA Family (miRBase 22.1) | Seed Region # | Associated Processes/Targets Clinical Outcomes | Reference |
| PC | miR-195 | mir-497~195 (chr17) | HyperMet | - | miR-16-2-3p/195-3p miR-15-5p/497-5p | CAAUAUU AGCAGCA | Proliferation, migration, EMT | [163] |
| GC | miR-497 | mir-497~195 (chr17) | HyperMet | - | miR-497-3p miR-15-5p/497-5p | AAACCAC AGCAGCA | Proliferation, invasion, apoptosis, RAF1, stage | [161] |
| HCC | miR-195 | mir-497~195 (chr17) | HyperMet | - | miR-16-2-3p/195-3p miR-15-5p/497-5p | CAAUAUU AGCAGCA | - | [162] |
| miR-497 | mir-497~195 (chr17) | HyperMet | - | miR-497-3p miR-15-5p/497-5p | AAACCAC AGCAGCA | - | [162] | |
| OC | miR-424 | mir-424~450b (chrX) | HyperMet | - | miR-424-3p miR-15-5p/497-5p | AAAACGU AGCAGCA | Proliferation, migration, KIF23 | [166] |
| BC | miR-195 | mir-497~195 (chr17) | HyperMet | - | miR-16-2-3p/195-3p miR-15-5p/497-5p | CAAUAUU AGCAGCA | Proliferation, invasion, RAF1, CCND1 | [159] |
| miR-497 | mir-497~195 (chr17) | HyperMet | - | miR-497-3p miR-15-5p/497-5p | AAACCAC AGCAGCA | Proliferation, invasion, RAF1, CCND1, MUC1 | [159,160] | |
| glioma | miR-424 | mir-424~450b (chrX) | HyperMet | - | miR-424-3p miR-15-5p/497-5p | AAAACGU AGCAGCA | Invasion, apoptosis, grade, IDH mutation | [164] |
| CC | miR-424 | mir-424~450b (chrX) | HyperMet | - | miR-424-3p miR-15-5p/497-5p | AAAACGU AGCAGCA | HIPK3, RBBP6 | [118] |
| EEA | miR-424 | mir-424~450b (chrX) | HyperMet | - | miR-424-3p miR-15-5p/497-5p | AAAACGU AGCAGCA | CCND1, RICTOR | [165] |
| AML | miR-15a | mir-15a~16-1 (chr13) | HyperMet | - | miR-15a-3p miR-15-5p/497-5p | AGGCCAU AGCAGCA | Prognosis, diagnosis BCL2, ROR1 | [158] |
| miR-15b | mir-15b~16-2 (chr3) | HyperMet | - | miR-15b-3p miR-15-5p/497-5p | GAAUCAU AGCAGCA | Prognosis, diagnosis BCL2, ROR1 | [158] | |
| Histone Modifications | ||||||||
| Cancer Type | miRNA | miRNA Cluster (miRBase 22.1) | Histone Modification | Epigenetic Regulator | miRNA Family (miRBase 22.1) | Seed Region # | Associated Processes/Targets Clinical Outcomes | Reference |
| LC | miR-15a | mir-15a~16-1 (chr13) | HAc | HDAC3 | miR-15a-3p miR-15-5p/497-5p | AGGCCAU AGCAGCA | Cell growth, colony formation, BCL2 | [168] |
| miR-16 | mir-15a~16-1 (chr13) | HAc | HDAC3 | miR-16-1-3p miR-15-5p/497-5p | CAGUAUU AGCAGCA | Cell growth, colony formation, BCL2 | [168] | |
| CC | miR-195 | mir-497~195 (chr17) | H3K27me3 | EZH2/PVT1 | miR-16-2-3p/195-3p miR-15-5p/497-5p | CAAUAUU AGCAGCA | EMT, paclitaxel sensitivity | [170] |
| HCC | miR-195 | mir-497~195 (chr17) | HAc | HDAC3/SP1 | miR-16-2-3p/195-3p miR-15-5p/497-5p | CAAUAUU AGCAGCA | - | [169] |
| CLL | miR-15a | mir-15a~16-1 (chr13) | H3K4me2 | HDACs | miR-15a-3p miR-15-5p/497-5p | AGGCCAU AGCAGCA | Apoptosis, MCL-1 | [79] |
| miR-16 | mir-15a~16-1 (chr13) mir-15b~16-2 (chr3) | H3K4me2 | HDACs | miR-16-1-3p miR-16-2-3p/195-3p miR-15-5p/497-5p | CAGUAUU CAAUAUU AGCAGCA | Apoptosis, MCL-1 | [79] | |
| NHL | miR-15 | mir-15a~16-1 (chr13) | HAc | HDAC3 | miR-15a-3p miR-15-5p/497-5p | AGGCCAU AGCAGCA | c-MYC expression-associated | [167] |
| miR-16 | mir-15a~16-1 (chr13) | HAc | HDAC3 | miR-16-1-3p miR-15-5p/497-5p | CAGUAUU AGCAGCA | c-MYC expression-associated | [167] | |
| DNA Methylation | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cancer Type | miRNA | miRNA Cluster (miRBase 22.1) | Methylation Status | Epigenetic Regulator | miRNA Family (miRBase 22.1) | Seed Region # | Associated Processes/Targets Clinical Outcomes | Reference |
| PC | miR-23b | mir-23b~24-1 (chr9) | HyperMet | - | miR-23-3p miR-23-5p | UCACAUU GGGUUCC | Proliferation, migration, cell cycle, colony formation, EMT, SRC, AKT, OS, RFS | [173] |
| miR-27a | mir-23a~24-2 (chr19) | Hyper/ HypoMet | - | miR-27-3p miR-27a-5p | UCACAGU GGGCUUA | Cell growth, stage | [178] | |
| miR-24 | mir-23b~24-1 (chr9) mir-23a~24-2 (chr19) | HyperMet | - | miR-24-3p miR-24-5p | GGCUCAG GCCUACU | Cell growth, apoptosis, racial difference, AR, IGF1, IGFBP5, ETV1 | [182] | |
| GC | miR-27b | mir-23b~24-1 (chr9) | HyperMet | - | miR-27-3p miR-27b-5p | UCACAGU GAGCUUA | Proliferation, invasion, GSPT1, stage, tumor size | [180] |
| HCC | miR-23a | mir-23a~24-2 (chr19) | HypoMet | - | miR-23-3p miR-23-5p | UCACAUU GGGUUCC | - | [162] |
| miR-27a | mir-23a~24-2 (chr19) | HypoMet | - | miR-27-3p miR-27a-5p | UCACAGU GGGCUUA | - | [162] | |
| miR-23b | mir-23b~24-1 (chr9) | HyperMet | - | miR-23-3p miR-23-5p | UCACAUU GGGUUCC | Proliferation, migration | [176] | |
| BC | miR-27b | mir-23b~24-1 (chr9) | HyperMet | - | miR-27-3p miR-27b-5p | UCACAGU GAGCUUA | Invasion, EMT, tamoxifen resistance, HMGB3 | [179] |
| CC | miR-23b | mir-23b~24-1 (chr9) | HyperMet | DNMT1 | miR-23-3p miR-23-5p | UCACAUU GGGUUCC | Apoptosis, uPA, ZEB1, c-MET, HPV-16 E6, C9orf3 | [174,177] |
| OSS | miR-23a | mir-23a~24-2 (chr19) | HyperMet | - | miR-23-3p miR-23-5p | UCACAUU GGGUUCC | Proliferation, migration, invasion, RUNX2, CXCL12 | [171] |
| MM | miR-23b | mir-23b~24-1 (chr9) | HyperMet | - | miR-23-3p miR-23-5p | UCACAUU GGGUUCC | Proliferation, apoptosis, colony formation, SP1 | [175] |
| WM | miR-23b | mir-23b~24-1 (chr9) | HyperMet | - | miR-23-3p miR-23-5p | UCACAUU GGGUUCC | Proliferation, apoptosis, colony formation, SP1 | [175] |
| Histone Modifications | ||||||||
| Cancer Type | miRNA | miRNA Cluster (miRBase 22.1) | Histone Modification | Epigenetic Regulator | miRNA Family (miRBase 22.1) | Seed Region # | Associated Processes/Targets Clinical Outcomes | Reference |
| BC | miR-130a | - | H3K27me3 | - | miR-130-3p/454-3p miR-23-3p | AGUGCAA UCACAUU | - | [94] |
| RMS | miR-27a | mir-23a~24-2 (chr19) | HAc | HDAC3/ SMARCA4 | miR-27-3p miR-27a-5p | UCACAGU GGGCUUA | PAX3:FOXO1 fusion, chemosensitivity, entinostat | [183] |
| DLBCL | miR-27b | mir-23b~24-1 (chr9) | HAc | HDAC6 | miR-27-3p miR-27b-5p | UCACAGU GAGCUUA | Proliferation, viability, MET, OS | [184] |
| DNA Methylation | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cancer Type | miRNA | miRNA Cluster (miRBase 22.1) | Methylation Status | Epigenetic Regulator | miRNA Family (miRBase 22.1) | Seed Region # | Associated Processes/Targets Clinical Outcomes | Reference |
| PC | miR-130b | mir-301b~130b (chr22) | HyperMet | - | miR-130-3p/454-3p miR-130b-5p | AGUGCAA CUCUUUC | DNA-damage response, senescence | [186] |
| miR-301b | mir-301b~130b (chr22) | HyperMet | - | miR-130-3p/454-3p miR-301a-5p/301b-5p | AGUGCAA CUCUGAC | DNA-damage response, senescence | [186] | |
| OC | miR-130b | mir-301b~130b (chr22) | HyperMet | - | miR-130-3p/454-3p miR-130b-5p | AGUGCAA CUCUUUC | CSF-1, differentiation, stage, MDR | [185] |
| CHS | miR-454 | - | HyperMet | DNMT1/EZH2/HOTAIR | miR-130-3p/454-3p miR-454-5p | AGUGCAA CCCUAUC | Apoptosis, autophagy, STAT3, ATG12 | [188] |
| PAC | miR-29b | mir-29b-2~29c (chr1) | HyperMet | DNMT1 | miR-29-3p miR-29b-2-5p | AGCACCA UGGUUUC | Migration, angiogenesis | [194] |
| GC | miR-29b | mir-29b-2~29c (chr1) | HyperMet | DNMT3A | miR-29-3p miR-29b-2-5p | AGCACCA UGGUUUC | Proliferation, migration, LASP1, DNMT3A, CDH1, prognosis | [191,192] |
| miR-29c | mir-29b-2~29c (chr1) | HyperMet | DNMT3A | miR-29-3p miR-29c-5p | AGCACCA GACCGAU | Migration, DNMT3A, CDH1 | [191] | |
| OC | miR-29b | mir-29b-2~29c (chr1) | HyperMet | DNMT3A/B | miR-29-3p miR-29b-2-5p | AGCACCA UGGUUUC | DNMT3A/B | [193] |
| GBM | miR-29b | mir-29b-2~29c (chr1) | HyperMet | DCST1-AS1 | miR-29-3p miR-29b-2-5p | AGCACCA UGGUUUC | Proliferation, OS | [195] |
| BL | miR-29a | mir-29b-1~29a (chr7) | HyperMet | DNMT3B | miR-29-3p miR-29a-5p | AGCACCA CUGAUUU | Cell cycle, apoptosis CDK6, DNMT3B, TCL-1, MCL-1 | [190] |
| miR-29b | mir-29b-1~29a (chr7) mir-29b-2~29c (chr1) | HyperMet | DNMT3B | miR-29-3p miR-29b-1-5p miR-29b-2-5p | AGCACCA CUGGUUU UGGUUUC | Cell cycle, apoptosis CDK6, DNMT3B, TCL-1, MCL-1 | [190] | |
| miR-29c | mir-29b-2~29c (chr1) | HyperMet | - | miR-29-3p miR-29c-5p | AGCACCA GACCGAU | Cell cycle, apoptosis CDK6, DNMT3B, TCL-1, MCL-1 | [190] | |
| Histone Modifications | ||||||||
| Cancer Type | miRNA | miRNA Cluster (miRBase 22.1) | Histone Modification | Epigenetic Regulator | miRNA Family (miRBase 22.1) | Seed Region # | Associated Processes/Targets Clinical Outcomes | Reference |
| BC | miR-130a | - | H3K27me3 | - | miR-130-3p/454-3p miR-23-3p | AGUGCAA UCACAUU | - | [94] |
| CLL | miR-29b | mir-29b-1~29a (chr7) mir-29b-2~29c (chr1) | H3K4me2 | HDACs | miR-29-3p miR-29b-1-5p miR-29b-2-5p | AGCACCA CUGGUUU UGGUUUC | Apoptosis, MCL-1 | [79] |
| AML | miR-29b | mir-29b-1~29a (chr7) mir-29b-2~29c (chr1) | HAc H4R3me2 | HDAC1/3 PRMT5 | miR-29-3p miR-29b-1-5p miR-29b-2-5p | AGCACCA CUGGUUU UGGUUUC | SP1, KIT mutations, FLT3 | [196,197] |
| MCL | miR-29a | mir-29b-1~29a (chr7) | H4Ac H3K27me3 | HDAC3 PRC2/EZH2 | miR-29-3p miR-29a-5p | AGCACCA CUGAUUU | Viability, colony formation, IGFR1R, CDK6 | [199] |
| miR-29b | mir-29b-1~29a (chr7) mir-29b-2~29c (chr1) | H4Ac H3K27me3 | HDAC3 PRC2/EZH2 | miR-29-3p miR-29b-1-5p miR-29b-2-5p | AGCACCA CUGGUUU UGGUUUC | Viability, colony formation, IGFR1R, CDK6 | [199] | |
| miR-29c | mir-29b-2~29c (chr1) | H4Ac H3K27me3 | HDAC3 PRC2/EZH2 | miR-29-3p miR-29c-5p | AGCACCA GACCGAU | Viability, colony formation, IGFR1R, CDK6 | [199] | |
| MM | miR-29b | mir-29b-1~29a (chr7) mir-29b-2~29c (chr1) | HAc | HDAC4 | miR-29-3p miR-29b-1-5p miR-29b-2-5p | AGCACCA CUGGUUU UGGUUUC | Cell survival, migration SP1, MCL-1 | [198] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gregorova, J.; Vychytilova-Faltejskova, P.; Sevcikova, S. Epigenetic Regulation of MicroRNA Clusters and Families during Tumor Development. Cancers 2021, 13, 1333. https://doi.org/10.3390/cancers13061333
Gregorova J, Vychytilova-Faltejskova P, Sevcikova S. Epigenetic Regulation of MicroRNA Clusters and Families during Tumor Development. Cancers. 2021; 13(6):1333. https://doi.org/10.3390/cancers13061333
Chicago/Turabian StyleGregorova, Jana, Petra Vychytilova-Faltejskova, and Sabina Sevcikova. 2021. "Epigenetic Regulation of MicroRNA Clusters and Families during Tumor Development" Cancers 13, no. 6: 1333. https://doi.org/10.3390/cancers13061333
APA StyleGregorova, J., Vychytilova-Faltejskova, P., & Sevcikova, S. (2021). Epigenetic Regulation of MicroRNA Clusters and Families during Tumor Development. Cancers, 13(6), 1333. https://doi.org/10.3390/cancers13061333