
Targeting KRAS in Cancer: Promising Therapeutic Strategies




Abstract
Simple Summary
Abstract
1. Introduction
2. Blood Cancers
3. Breast Cancer
4. Colorectal Cancer
5. Gynecological Cancers
6. Lung Cancer
7. Pancreatic Cancer
8. Prostate Cancer
9. Skin Cancer
10. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef]
- Saito, Y.; Koya, J.; Araki, M.; Kogure, Y.; Shingaki, S.; Tabata, M.; McClure, M.B.; Yoshifuji, K.; Matsumoto, S.; Isaka, Y.; et al. Landscape and function of multiple mutations within individual oncogenes. Nat. Cell Biol. 2020, 582, 95–99. [Google Scholar] [CrossRef]
- Del Re, M.; Rofi, E.; Restante, G.; Crucitta, S.; Arrigoni, E.; Fogli, S.; Di Maio, M.; Petrini, I.; Danesi, R. Implications of KRAS mutations in acquired resistance to treatment in NSCLC. Oncotarget 2018, 9, 6630–6643. [Google Scholar] [CrossRef]
- Mullard, A. Cracking KRAS. Nat. Rev. Drug Discov. 2019, 18, 887–891. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.F.; Braun, B.S.; Shannon, K.M. Targeting oncogenic Ras signaling in hematologic malignancies. Blood 2012, 120, 3397–3406. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, Y.; Sha, K.; Tang, Q.; Yang, X.; Yu, C.; Liu, Z.; Sun, W.; Cai, L.; Xu, C.; et al. KRAS (G12D) Cooperates with AML1/ETO to Initiate a Mouse Model Mimicking Human Acute Myeloid Leukemia. Cell. Physiol. Biochem. 2014, 33, 78–87. [Google Scholar] [CrossRef]
- Zhou, J.-D.; Yao, D.-M.; Li, X.-X.; Zhang, T.-J.; Zhang, W.; Ma, J.-C.; Guo, H.; Deng, Z.-Q.; Lin, J.; Qian, J. KRAS overexpression independent of RAS mutations confers an adverse prognosis in cytogenetically normal acute myeloid leukemia. Oncotarget 2017, 8, 66087–66097. [Google Scholar] [CrossRef]
- Vendramini, E.; Bomben, R.; Pozzo, F.; Benedetti, D.; Bittolo, T.; Rossi, F.M.; Bo, M.D.; Rabe, K.G.; Pozzato, G.; Zaja, F.; et al. KRAS, NRAS, and BRAF mutations are highly enriched in trisomy 12 chronic lymphocytic leukemia and are associated with shorter treatment-free survival. Leukemia 2019, 33, 2111–2115. [Google Scholar] [CrossRef] [PubMed]
- Jerchel, I.S.; Hoogkamer, A.Q.; Ariës, I.M.; Steeghs, E.M.P.; Boer, J.M.; Besselink, N.J.M.; Boeree, A.; Van De Ven, C.; De Groot-Kruseman, A.H.; De Haas, V.; et al. RAS pathway mutations as a predictive biomarker for treatment adaptation in pediatric B-cell precursor acute lymphoblastic leukemia. Leukemia 2017, 32, 931–940. [Google Scholar] [CrossRef]
- Hou, H.-A.; Tien, H.-F. Genomic landscape in acute myeloid leukemia and its implications in risk classification and targeted therapies. J. Biomed. Sci. 2020, 27, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, A.; Maharry, K.; Mrózek, K.; Thiede, C.; Marcucci, G.; Paschka, P.; Mayer, R.J.; Larson, R.A.; Liu, E.T.; Bloomfield, C.D. Patients with Acute Myeloid Leukemia and RAS Mutations Benefit Most from Postremission High-Dose Cytarabine: A Cancer and Leukemia Group B Study. J. Clin. Oncol. 2008, 26, 4603–4609. [Google Scholar] [CrossRef] [PubMed]
- Burgess, M.R.; Hwang, E.; Mroue, R.; Bielski, C.M.; Wandler, A.M.; Huang, B.J.; Firestone, A.J.; Young, A.; Lacap, J.A.; Crocker, L.; et al. KRAS Allelic Imbalance Enhances Fitness and Modulates MAP Kinase Dependence in Cancer. Cell 2017, 168, 817–829.e15. [Google Scholar] [CrossRef]
- Weisberg, E.; Meng, C.; Case, A.; Sattler, M.; Tiv, H.L.; Gokhale, P.C.; Buhrlage, S.; Wang, J.; Gray, N.; Stone, R.; et al. Evaluation of ERK as a therapeutic target in acute myelogenous leukemia. Leukemia 2020, 34, 625–629. [Google Scholar] [CrossRef] [PubMed]
- Steelman, L.S.; Franklin, R.A.; Abrams, S.L.; Chappell, W.; Kempf, C.R.; Bäsecke, J.; Stivala, F.; Donia, M.; Fagone, P.; Nicoletti, F.; et al. Roles of the Ras/Raf/MEK/ERK pathway in leukemia therapy. Leukemia 2011, 25, 1080–1094. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.H.; Gmachl, M.; Ramharter, J.; Savarese, F.; Gerlach, D.; Marszalek, J.R.; Sanderson, M.P.; Kessler, D.; Trapani, F.; Arnhof, H.; et al. BI-3406, a Potent and Selective SOS1–KRAS Interaction Inhibitor, Is Effective in KRAS-Driven Cancers through Combined MEK Inhibition. Cancer Discov. 2021, 11, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Choi, P.S.; Gelbard, M.; Meyerson, M. Identification and Characterization of Oncogenic SOS1 Mutations in Lung Adenocarcinoma. Mol. Cancer Res. 2019, 17, 1002–1012. [Google Scholar] [CrossRef]
- Koschut, D.; Ray, D.; Li, Z.; Giarin, E.; Groet, J.; Alić, I.; Kham, S.K.-Y.; Chng, W.J.; Ariffin, H.; Weinstock, D.M.; et al. RAS-protein activation but not mutation status is an outcome predictor and unifying therapeutic target for high-risk acute lymphoblastic leukemia. Oncogene 2021, 40, 746–762. [Google Scholar] [CrossRef]
- Galiè, M. RAS as Supporting Actor in Breast Cancer. Front. Oncol. 2019, 9, 1199. [Google Scholar] [CrossRef] [PubMed]
- Adnane, J.; Jackson, R.J.; Nicosia, S.V.; Cantor, A.B.; Pledger, W.J.; Sebti, S.M. Loss of p21WAF1/CIP1 accelerates Ras oncogenesis in a transgenic/knockout mammary cancer model. Oncogene 2000, 19, 5338–5347. [Google Scholar] [CrossRef][Green Version]
- Ricoult, S.J.H.; Yecies, J.L.; Ben-Sahra, I.; Manning, B.D. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene 2016, 35, 1250–1260. [Google Scholar] [CrossRef]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.-Y.; Hsu, R.-J.; Liu, J.-M.; Chen, S.-C.; Liao, G.-S.; Gao, H.-W.; Yu, C.-P. MicroRNA-382-5p aggravates breast cancer progression by regulating the RERG/Ras/ERK signaling axis. Oncotarget 2017, 8, 22443–22459. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Hao, Y.; Feig, L.A. The R-Ras GTPase Mediates Cross Talk between Estrogen and Insulin Signaling in Breast Cancer Cells. Mol. Cell. Biol. 2006, 26, 6372–6380. [Google Scholar] [CrossRef]
- Feng, M.; Bao, Y.; Li, Z.; Li, J.; Gong, M.; Lam, S.; Wang, J.; Marzese, D.M.; Donovan, N.; Tan, E.Y.; et al. RASAL2 activates RAC1 to promote triple-negative breast cancer progression. J. Clin. Investig. 2014, 124, 5291–5304. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.V.; Pellacani, D.; Lefort, S.; Kannan, N.; Osako, T.; Makarem, M.; Cox, C.L.; Kennedy, W.; Beer, P.A.; Carles, A.; et al. Barcoding reveals complex clonal dynamics of de novo transformed human mammary cells. Nat. Cell Biol. 2015, 528, 267–271. [Google Scholar] [CrossRef]
- Loi, S.; Dushyanthen, S.S.; Beavis, P.P.; Salgado, R.; Denkert, C.; Savas, P.P.; Combs, S.S.; Rimm, D.D.; Giltnane, J.J.; Estrada, M.V.M.; et al. RAS/MAPK Activation Is Associated with Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation Between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clin. Cancer Res. 2016, 22, 1499–1509. [Google Scholar] [CrossRef]
- Adeyinka, A.; Nui, Y.; Cherlet, T.; Snell, L.; Watson, P.H.; Murphy, L.C. Activated mitogen-activated protein kinase expression during human breast tumorigenesis and breast cancer progression. Clin. Cancer Res. 2002, 8, 1747–1753. [Google Scholar] [PubMed]
- Hoeflich, K.P.; O’Brien, C.; Boyd, Z.; Cavet, G.; Guerrero, S.; Jung, K.; Januario, T.; Savage, H.; Punnoose, E.; Truong, T.; et al. In vivo Antitumor Activity of MEK and Phosphatidylinositol 3-Kinase Inhibitors in Basal-Like Breast Cancer Models. Clin. Cancer Res. 2009, 15, 4649–4664. [Google Scholar] [CrossRef]
- Zhao, Y.; Ge, C.C.; Wang, J.; Wu, X.X.; Li, X.M.; Li, W.; Wang, S.S.; Liu, T.; Hou, J.Z.; Sun, H.; et al. MEK inhibitor, PD98059, promotes breast cancer cell migration by inducing beta-catenin nuclear accumulation. Oncol. Rep. 2017, 38, 3055–3063. [Google Scholar] [CrossRef]
- Di Simone, D.; Galimberti, S.; Basolo, F.; Ciardiello, F.; Petrini, M.; Scheper, R.J. c-Ha-ras transfection and expression of MDR-related genes in MCF-10A human breast cell line. Anticancer Res. 1997, 17, 3587–3592. [Google Scholar]
- Zoppoli, G.; Moran, E.; Soncini, D.; Cea, M.; Garuti, A.; Rocco, I.; Cirmena, G.; Grillo, V.; Bagnasco, L.; Icardi, G.; et al. Ras-induced resistance to lapatinib is overcome by MEK inhibition. Curr. Cancer Drug Targets 2010, 10, 168–175. [Google Scholar] [CrossRef] [PubMed]
- McGlynn, L.M.; Kirkegaard, T.; Edwards, J.; Tovey, S.; Cameron, D.; Twelves, C.; Bartlett, J.M.; Cooke, T.G. Ras/Raf-1/MAPK Pathway Mediates Response to Tamoxifen but not Chemotherapy in Breast Cancer Patients. Clin. Cancer Res. 2009, 15, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Porru, M.; Pompili, L.; Caruso, C.; Biroccio, A.; Leonetti, C. Targeting KRAS in metastatic colorectal cancer: Current strategies and emerging opportunities. J. Exp. Clin. Cancer Res. 2018, 37, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; Van Krieken, J.H.; Aderka, D.; Aguilar, E.A.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef] [PubMed]
- Parseghian, C.; Loree, J.; Morris, V.; Liu, X.; Clifton, K.; Napolitano, S.; Henry, J.; Pereira, A.; Vilar, E.; Johnson, B.; et al. Anti-EGFR-resistant clones decay exponentially after progression: Implications for anti-EGFR re-challenge. Ann. Oncol. 2019, 30, 243–249. [Google Scholar] [CrossRef]
- Siravegna, G.; Bardelli, A. Failure is not final: ctDNA-guided rechallenge therapy in colorectal cancer. Ann. Oncol. 2019, 30, 1671. [Google Scholar] [CrossRef] [PubMed]
- Allegra, C.J.; Jessup, J.M.; Somerfield, M.R.; Hamilton, S.R.; Hammond, E.H.; Hayes, D.F.; McAllister, P.K.; Morton, R.F.; Schilsky, R.L. American Society of Clinical Oncology Provisional Clinical Opinion: Testing for KRAS Gene Mutations in Patients with Metastatic Colorectal Carcinoma to Predict Response to Anti–Epidermal Growth Factor Receptor Monoclonal Antibody Therapy. J. Clin. Oncol. 2009, 27, 2091–2096. [Google Scholar] [CrossRef]
- Karnoub, A.E.; Weinberg, R.A. Ras oncogenes: Split personalities. Nat. Rev. Mol. Cell Biol. 2008, 9, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Papke, B.; Murarka, S.; Vogel, H.A.; Martin-Gago, P.; Kovacevic, M.; Truxius, D.C.; Fansa, E.K.; Ismail, S.; Zimmermann, G.; Heinelt, K.; et al. Identification of pyrazolopyridazinones as PDEdelta inhibitors. Nat. Commun. 2016, 7, 11360. [Google Scholar] [CrossRef]
- Porru, M.; Artuso, S.; Salvati, E.; Bianco, A.; Franceschin, M.; Diodoro, M.G.; Passeri, D.; Orlandi, A.; Savorani, F.; D’Incalci, M.; et al. Targeting G-Quadruplex DNA Structures by EMICORON Has a Strong Antitumor Efficacy against Advanced Models of Human Colon Cancer. Mol. Cancer Ther. 2015, 14, 2541–2551. [Google Scholar] [CrossRef] [PubMed]
- Shima, F.; Yoshikawa, Y.; Ye, M.; Araki, M.; Matsumoto, S.; Liao, J.; Hu, L.; Sugimoto, T.; Ijiri, Y.; Takeda, A.; et al. In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc. Natl. Acad. Sci. USA 2013, 110, 8182–8187. [Google Scholar] [CrossRef]
- Hiraoka, K.; Inoue, T.; Taylor, R.D.; Watanabe, T.; Koshikawa, N.; Yoda, H.; Shinohara, K.-I.; Takatori, A.; Sugimoto, H.; Maru, Y.; et al. Inhibition of KRAS codon 12 mutants using a novel DNA-alkylating pyrrole–imidazole polyamide conjugate. Nat. Commun. 2015, 6, 6706. [Google Scholar] [CrossRef]
- Tutuka, C.S.A.; Andrews, M.C.; Mariadason, J.M.; Ioannidis, P.; Hudson, C.; Cebon, J.; Behren, A. PLX8394, a new generation BRAF inhibitor, selectively inhibits BRAF in colonic adenocarcinoma cells and prevents paradoxical MAPK pathway activation. Mol. Cancer 2017, 16, 112. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Guo, X.; Zhang, H.; Xiang, Y.; Chen, J.; Yin, Y.; Cai, X.; Wang, K.; Wang, G.; Ba, Y.; et al. Role of miR-143 targeting KRAS in colorectal tumorigenesis. Oncogene 2009, 28, 1385–1392. [Google Scholar] [CrossRef]
- Gillson, J.; Ramaswamy, Y.; Singh, G.; Gorfe, A.A.; Pavlakis, N.; Samra, J.; Mittal, A.; Sahni, S. Small Molecule KRAS Inhibitors: The Future for Targeted Pancreatic Cancer Therapy? Cancers 2020, 12, 1341. [Google Scholar] [CrossRef]
- Samalin, E.; Bouché, O.; Thézénas, S.; François, E.; Adenis, A.; Bennouna, J.; Taieb, J.; Desseigne, F.; Seitz, J.F.; Conroy, T.; et al. Sorafenib and irinotecan (NEXIRI) as second- or later-line treatment for patients with metastatic colorectal cancer and KRAS-mutated tumours: A multicentre Phase I/II trial. Br. J. Cancer 2014, 110, 1148–1154. [Google Scholar] [CrossRef]
- Samalin, E.; de la Fouchardière, C.; Thézenas, S.; Boige, V.; Senellart, H.; Guimbaud, R.; Taïeb, J.; François, E.; Galais, M.-P.; Lièvre, A.; et al. Sorafenib Plus Irinotecan Combination in Patients with RAS-mutated Metastatic Colorectal Cancer Refractory to Standard Combined Chemotherapies: A Multicenter, Randomized Phase 2 Trial (NEXIRI-2/PRODIGE 27). Clin. Colorectal Cancer 2020, 19, 301–310.e1. [Google Scholar] [CrossRef]
- Torre, L.A.; Islami, F.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global Cancer in Women: Burden and Trends. Cancer Epidemiol. Biomark. Prev. 2017, 26, 444–457. [Google Scholar] [CrossRef]
- Spaans, V.M.; Trietsch, M.D.; Crobach, S.; Stelloo, E.; Kremer, D.; Osse, E.M.; Haar, N.T.T.; Van Eijk, R.; Muller, S.; Van Wezel, T.; et al. Designing a High-Throughput Somatic Mutation Profiling Panel Specifically for Gynaecological Cancers. PLoS ONE 2014, 9, e93451. [Google Scholar] [CrossRef]
- Kurman, R.J.; Shih Ie, M. Pathogenesis of ovarian cancer: Lessons from morphology and molecular biology and their clinical implications. Int. J. Gynecol. Pathol. 2008, 27, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Xiang, L.; Pei, X.; He, T.; Shen, X.; Wu, X.; Yang, H. Mutational analysis of KRAS and its clinical implications in cervical cancer patients. J. Gynecol. Oncol. 2018, 29, e4. [Google Scholar] [CrossRef]
- Sideris, M.; Emin, E.I.; Abdullah, Z.; Hanrahan, J.; Stefatou, K.M.; Sevas, V.; Emin, E.; Hollingworth, T.; Odejinmi, F.; Papagrigoriadis, S.; et al. The Role of KRAS in Endometrial Cancer: A Mini-Review. Anticancer Res. 2019, 39, 533–539. [Google Scholar] [CrossRef]
- Ring, K.L.; Yates, M.S.; Schmandt, R.; Onstad, M.; Zhang, Q.; Celestino, J.; Kwan, S.-Y.; Lu, K.H. Endometrial Cancers with Activating KRas Mutations Have Activated Estrogen Signaling and Paradoxical Response to MEK Inhibition. Int. J. Gynecol. Cancer 2017, 27, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.M.; Kipp, B.R.; Halling, K.C.; Kerr, S.E.; Smith, D.I.; Distad, T.J.; Clayton, A.C.; Medeiros, F. Molecular characterization of endometrial cancer: A correlative study assessing microsatellite instability, MLH1 hypermethylation, DNA mismatch repair protein expression, and PTEN, PIK3CA, KRAS, and BRAF mutation analysis. Int. J. Gynecol. Pathol. 2012, 31, 195–205. [Google Scholar] [CrossRef]
- Sasaki, H.; Nishii, H.; Takahashi, H.; Tada, A.; Furusato, M.; Terashima, Y.; Siegal, G.P.; Parker, S.L.; Kohler, M.F.; Berchuck, A. Mutation of the Ki-ras protooncogene in human endometrial hyperplasia and carcinoma. Cancer Res. 1993, 53, 1906–1910. [Google Scholar] [PubMed]
- Tsuda, H.; Jiko, K.; Yajima, M.; Yamada, T.; Tanemura, K.; Tsunematsu, R.; Ohmi, K.; Sonoda, T.; Hirohashi, S. Frequent Occurrence of c-Ki-ras Gene Mutations in Well Differentiated Endometrial Adenocarcinoma Showing Infiltrative Local Growth with Fibrosing Stromal Response. Int. J. Gynecol. Pathol. 1995, 14, 255–259. [Google Scholar] [CrossRef]
- Malpica, A.; Wong, K.-K. The molecular pathology of ovarian serous borderline tumors. Ann. Oncol. 2016, 27 (Suppl. 1), i16–i19. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Sill, M.W.; Thaker, P.H.; Bender, D.P.; Street, D.; McGuire, W.P.; Johnston, C.M.; Rotmensch, J. A phase II evaluation of selumetinib (AZD6244, ARRY-142886), a selective MEK-1/2 inhibitor in the treatment of recurrent or persistent endometrial cancer: An NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol. 2015, 138, 30–35. [Google Scholar] [CrossRef]
- Farley, J.; Brady, W.E.; Vathipadiekal, V.; Lankes, H.A.; Coleman, R.; Morgan, M.A.; Mannel, R.; Yamada, S.D.; Mutch, D.; Rodgers, W.H.; et al. Selumetinib in women with recurrent low-grade serous carcinoma of the ovary or peritoneum: An open-label, single-arm, phase 2 study. Lancet Oncol. 2013, 14, 134–140. [Google Scholar] [CrossRef]
- Adderley, H.; Blackhall, F.H.; Lindsay, C.R. KRAS-mutant non-small cell lung cancer: Converging small molecules and immune checkpoint inhibition. EBioMedicine 2019, 41, 711–716. [Google Scholar] [CrossRef]
- Mustachio, L.M.; Roszik, J. Current Targeted Therapies for the Fight against Non-Small Cell Lung Cancer. Pharmaceuticals 2020, 13, 374. [Google Scholar] [CrossRef]
- Janes, M.R.; Zhang, J.; Li, L.-S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589.e17. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.Y.; Zhao, Y.; Aronowitz, J.; Mai, T.T.; Vides, A.; Qeriqi, B.; Kim, D.; Li, C.; De Stanchina, E.; Mazutis, L.; et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nat. Cell Biol. 2020, 577, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Xue, J.Y.; Lito, P. Targeting KRAS(G12C): From Inhibitory Mechanism to Modulation of Antitumor Effects in Patients. Cell 2020, 183, 850–859. [Google Scholar] [CrossRef]
- McCarthy, M.J.; Pagba, C.V.; Prakash, P.; Naji, A.K.; Van Der Hoeven, D.; Liang, H.; Gupta, A.K.; Zhou, Y.; Cho, K.-J.; Hancock, J.F.; et al. Discovery of High-Affinity Noncovalent Allosteric KRAS Inhibitors That Disrupt Effector Binding. ACS Omega 2019, 4, 2921–2930. [Google Scholar] [CrossRef] [PubMed]
- Manchado, E.; Weissmueller, S.; Morris, J.P.; Chen, C.-C.; Wullenkord, R.; Lujambio, A.; De Stanchina, E.; Poirier, J.T.; Gainor, J.F.; Corcoran, R.B.; et al. A combinatorial strategy for treating KRAS-mutant lung cancer. Nat. Cell Biol. 2016, 534, 647–651. [Google Scholar] [CrossRef]
- Röth, S.; Macartney, T.J.; Konopacka, A.; Queisser, M.A.; Sapkota, G. Targeting Endogenous K-RAS for Degradation Through the Affinity-Directed Protein Missile System. Cell. Chem. Biol. 2019, 27, 1151–1163.e6. [Google Scholar] [CrossRef]
- Mustachio, L.M.; Lu, Y.; Tafe, L.J.; Memoli, V.; Rodriguez-Canales, J.; Mino, B.; Villalobos, P.A.; Wistuba, I.; Katayama, H.; Hanash, S.M.; et al. Deubiquitinase USP18 Loss Mislocalizes and Destabilizes KRAS in Lung Cancer. Mol. Cancer Res. 2017, 15, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Chung, V.; McDonough, S.; Philip, P.A.; Cardin, D.; Wang-Gillam, A.; Hui, L.; Tejani, M.A.; Seery, T.E.; Dy, I.A.; Al Baghdadi, T.; et al. Effect of Selumetinib and MK-2206 vs Oxaliplatin and Fluorouracil in Patients with Metastatic Pancreatic Cancer After Prior Therapy: SWOG S1115 Study Randomized Clinical Trial. JAMA Oncol. 2017, 3, 516–522. [Google Scholar] [CrossRef]
- Mahalingam, D.; Goel, S.; Aparo, S.; Patel Arora, S.; Noronha, N.; Tran, H.; Chakrabarty, R.; Selvaggi, G.; Gutierrez, A.; Coffey, M.; et al. A Phase II Study of Pelareorep (REOLYSIN®) in Combination with Gemcitabine for Patients with Advanced Pancreatic Adenocarcinoma. Cancers 2018, 10, 160. [Google Scholar] [CrossRef] [PubMed]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef]
- Weng, C.-C.; Ding, P.-Y.; Liu, Y.-H.; Hawse, J.R.; Subramaniam, M.; Wu, C.-C.; Lin, Y.-C.; Chen, C.-Y.; Hung, W.-C.; Cheng, K.-H. Mutant Kras-induced upregulation of CD24 enhances prostate cancer stemness and bone metastasis. Oncogene 2019, 38, 2005–2019. [Google Scholar] [CrossRef]
- Yang, Q.; Lang, C.; Wu, Z.; Dai, Y.; He, S.; Guo, W.; Huang, S.; Du, H.; Ren, D.; Peng, X. MAZ promotes prostate cancer bone metastasis through transcriptionally activating the KRas-dependent RalGEFs pathway. J. Exp. Clin. Cancer Res. 2019, 38, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Merrick, B.A.; Phadke, D.P.; Bostrom, M.A.; Shah, R.R.; Wright, G.M.; Wang, X.; Gordon, O.; Pelch, K.E.; Auerbach, S.S.; Paules, R.S.; et al. Arsenite malignantly transforms human prostate epithelial cells in vitro by gene amplification of mutated KRAS. PLoS ONE 2019, 14, e0215504. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-S.; Shankar, S.; Dhanasekaran, S.M.; Ateeq, B.; Sasaki, A.T.; Jing, X.; Robinson, D.; Cao, Q.; Prensner, J.R.; Yocum, A.K.; et al. Characterization of KRAS Rearrangements in Metastatic Prostate Cancer. Cancer Discov. 2011, 1, 35–43. [Google Scholar] [CrossRef]
- Kharmate, G.; Hosseini-Beheshti, E.; Caradec, J.; Chin, M.Y.; Tomlinson Guns, E.S. Epidermal Growth Factor Receptor in Prostate Cancer Derived Exosomes. PLoS ONE 2016, 11, e0154967. [Google Scholar]
- Kharmate, G.; Hosseini-Beheshti, E.; Caradec, J.; Chin, M.Y.; Tomlinson Guns, E.S. Correction: Epidermal Growth Factor Receptor in Prostate Cancer Derived Exosomes. PLoS ONE 2016, 11, e0157392. [Google Scholar] [CrossRef] [PubMed]
- Nickols, N.G.; Nazarian, R.; Zhao, S.G.; Tan, V.; Uzunangelov, V.; Xia, Z.; Baertsch, R.; Neeman, E.; Gao, A.C.; Thomas, G.V.; et al. MEK-ERK signaling is a therapeutic target in metastatic castration resistant prostate cancer. Prostate Cancer Prostatic Dis. 2019, 22, 531–538. [Google Scholar] [CrossRef]
- Vanni, I.; Tanda, E.T.; Dalmasso, B.; Pastorino, L.; Andreotti, V.; Bruno, W.; Boutros, A.; Spagnolo, F.; Ghiorzo, P. Non-BRAF Mutant Melanoma: Molecular Features and Therapeutical Implications. Front. Mol. Biosci. 2020, 7, 172. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, P.; Kuphal, S.; Spruss, T.; Hellerbrand, C.; Bosserhoff, A.K. Wild-type KRAS is a novel therapeutic target for melanoma contributing to primary and acquired resistance to BRAF inhibition. Oncogene 2017, 37, 897–911. [Google Scholar] [CrossRef] [PubMed]
- Missero, C. The genetic evolution of skin squamous cell carcinoma: Tumor suppressor identity matters. Exp. Dermatol. 2016, 25, 863–864. [Google Scholar] [CrossRef] [PubMed]


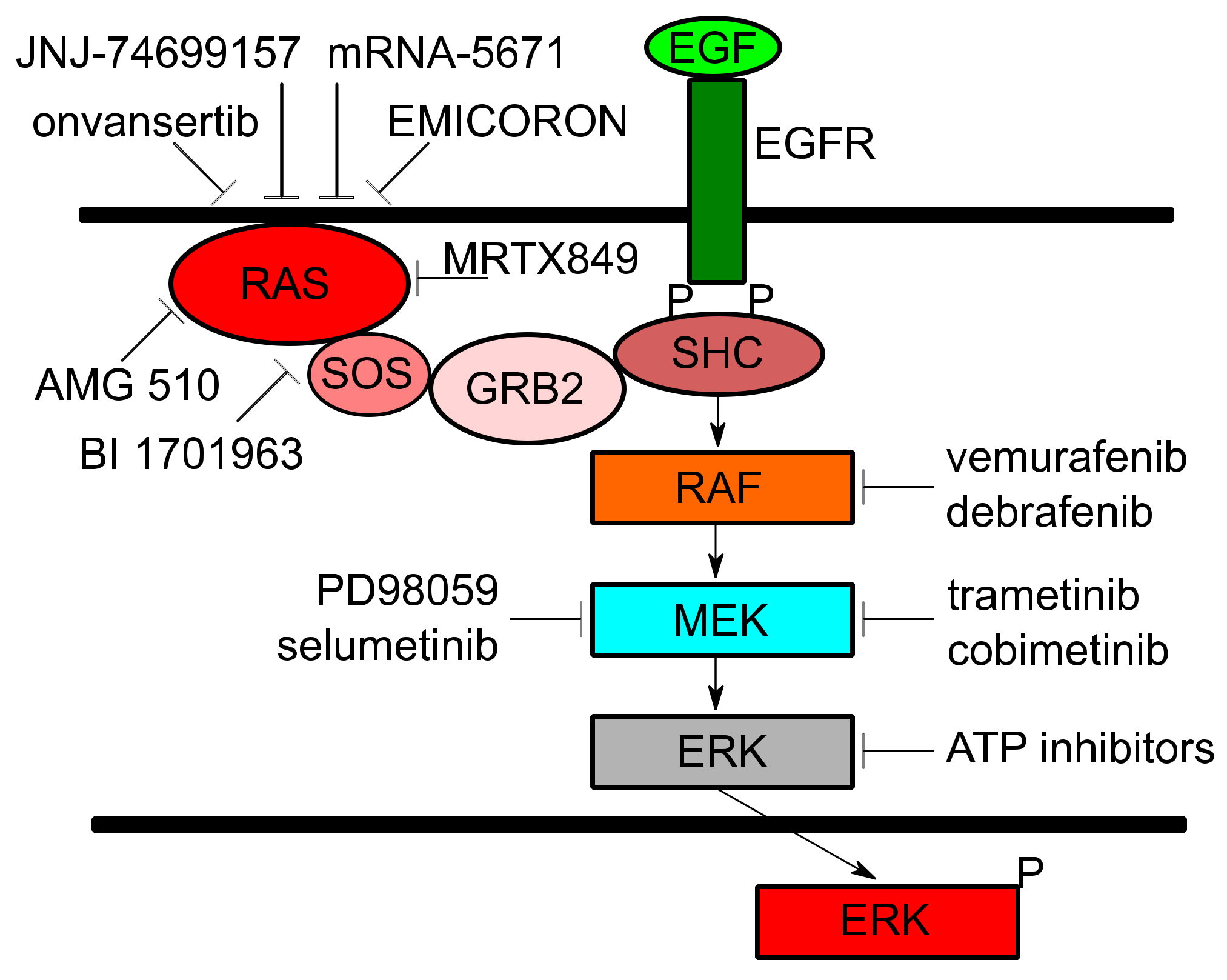{kind=link}
| Trial ID | Trial Title | Target(s) | Cancer(s) | Mechanism |
|---|---|---|---|---|
| NCT01274624 | Study of REOLYSIN in Combination With FOLFIRI and Bevacizumab in FOLFIRI Naïve Patients With KRAS Mutant Metastatic Colorectal Cancer | Activated RAS | CRC | Stimulates lysis of tumor cells and induces antitumor immunity |
| NCT03808558 | Phase 2 Study of TVB-2640 in KRAS Non-Small Cell Lung Carcinomas | FASN | NSCLC | Disrupts tumor lipid rafts and RAS localization |
| NCT03600883 | A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of AMG 510 in Subjects With Solid Tumors With a Specific KRAS Mutation (CodeBreak 100) | KRAS G12C | NSCLC, solid tumors | Binds to cysteine residue in KRAS G12C mutations holding protein in inactive form and prevents downstream signaling |
| NCT03785249 | Phase 1/2 Study of MRTX849 in Patients With Cancer Having a KRAS G12C Mutation KRYSTAL-1 | KRAS G12C | NSCLC, CRC, other solid tumors | Modifies mutant cysteine 12 in a GDP-bound state and inhibits KRAS dependent signaling |
| NCT04006301 | First-in-Human Study of JNJ-74699157 in Participants With Tumors Harboring the KRAS G12C Mutation | KRAS G12C | NSCLC, solid tumors | Binds to KRAS G23C and holds the protein in inactive form, preventing downstream signaling |
| NCT03948763 | A Study of mRNA-5671/V941 as Monotherapy and in Combination With Pembrolizumab (V941-001) | KRAS G12D, G12V, G13D, and G12C | NSCLC, CRC, pancreatic | Induces T-cell dependent immune responses to destroy tumors presenting KRAS mutations |
| NCT04132505 | Binimetinib and Hydroxychloroquine in Treating Patients With KRAS Mutant Metastatic Pancreatic Cancer | MEK, Autophagy | pancreatic | Binimetinib: Noncompetitive ATP inhibitor preventing MEK signaling. Hydroxychloroquine: Inhibits autophagy to avoid metabolism of Binimetinib |
| NCT03040986 | Selumetinib Sulfate in Treating Patients With Locally Advanced or Metastatic Pancreatic Cancer With KRAS G12R Mutations | MEK | pancreatic | Potent and selective inhibitor against MEK1/2 and KRAS dependent signaling |
| NCT02642042 | Trametinib and Docetaxel in Treating Patients With Recurrent or Stage IV KRAS Mutation Positive Non-small Cell Lung Cancer | MEK | NSCLC | Trametinib: Inhibits MEK signaling downstream of KRAS. Docetaxel: Chemotherapeutic |
| NCT02101788 | Trametinib in Treating Patients With Recurrent or Progressive Low-Grade Ovarian Cancer or Peritoneal Cavity Cancer | MEK | ovarian, peritoneal | Inhibits MEK signaling downstream of KRAS |
| NCT02613650 | A Trial of mFOLFIRI With MEK162 in Patients With Advanced RAS (HRAS, NRAS, or KRAS) Positive Metastatic Colorectal Cancers | MEK | CRC | MEK162: Uncompetitive ATP inhibitor suppresses the activity of MEK1/2 and KRAS downstream signaling. mFOLFIRI: Chemotherapeutic |
| NCT03704688 | Trial of Trametinib and Ponatinib in Patients With KRAS Mutant Advanced Non-Small Cell Lung Cancer | MEK, Multi-tyrosine Kinase | NSCLC | Trametinib: Inhibits MEK signaling downstream of KRAS. Ponatinib: Targets multiple tyrosine kinases and inhibits signaling |
| NCT03829410 | Onvansertib in Combination With FOLFIRI and Bevacizumab for Second Line Treatment of Metastatic Colorectal Cancer Patients With a KRAS Mutation | PLK1, VEGF | CRC | Onvansertib: Selectively inhibits PLK1 causing mitotic arrest and cell death. Bevacizumab: Binds to VEGF and reduces blood supply to tumors. FOLFIRI: Chemotherapeutic |
| NCT04111458 | A Study to Test Different Doses of BI 1701963 Alone and Combined With Trametinib in Patients With Different Types of Advanced Cancer (Solid Tumours With KRAS Mutation) | SOS1, MEK | NSCLC, solid tumors | BI 1701963: Inhibits KRAS binding to SOS1, leading to KRAS inactivation. Trametinib: Inhibits MEK signaling downstream of KRAS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mustachio, L.M.; Chelariu-Raicu, A.; Szekvolgyi, L.; Roszik, J. Targeting KRAS in Cancer: Promising Therapeutic Strategies. Cancers 2021, 13, 1204. https://doi.org/10.3390/cancers13061204
Mustachio LM, Chelariu-Raicu A, Szekvolgyi L, Roszik J. Targeting KRAS in Cancer: Promising Therapeutic Strategies. Cancers. 2021; 13(6):1204. https://doi.org/10.3390/cancers13061204
Chicago/Turabian StyleMustachio, Lisa Maria, Anca Chelariu-Raicu, Lorant Szekvolgyi, and Jason Roszik. 2021. "Targeting KRAS in Cancer: Promising Therapeutic Strategies" Cancers 13, no. 6: 1204. https://doi.org/10.3390/cancers13061204
APA StyleMustachio, L. M., Chelariu-Raicu, A., Szekvolgyi, L., & Roszik, J. (2021). Targeting KRAS in Cancer: Promising Therapeutic Strategies. Cancers, 13(6), 1204. https://doi.org/10.3390/cancers13061204