
ANO1 Expression Orchestrates p27Kip1/MCL1-Mediated Signaling in Head and Neck Squamous Cell Carcinoma

 ,
,  , , ,
, , ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
2.1. ANO1 Expression Promotes Cell Proliferation and EMT of Primary Cancer Cells Derived from HNSCC
2.2. ANO1 Knockdown Leads to Downregulation of Pro-Survival Protein MCL1 Conferring Apoptosis
2.3. ANO1 Exists in the Mitochondria of UT-SCC Cells with High ANO1 Expression
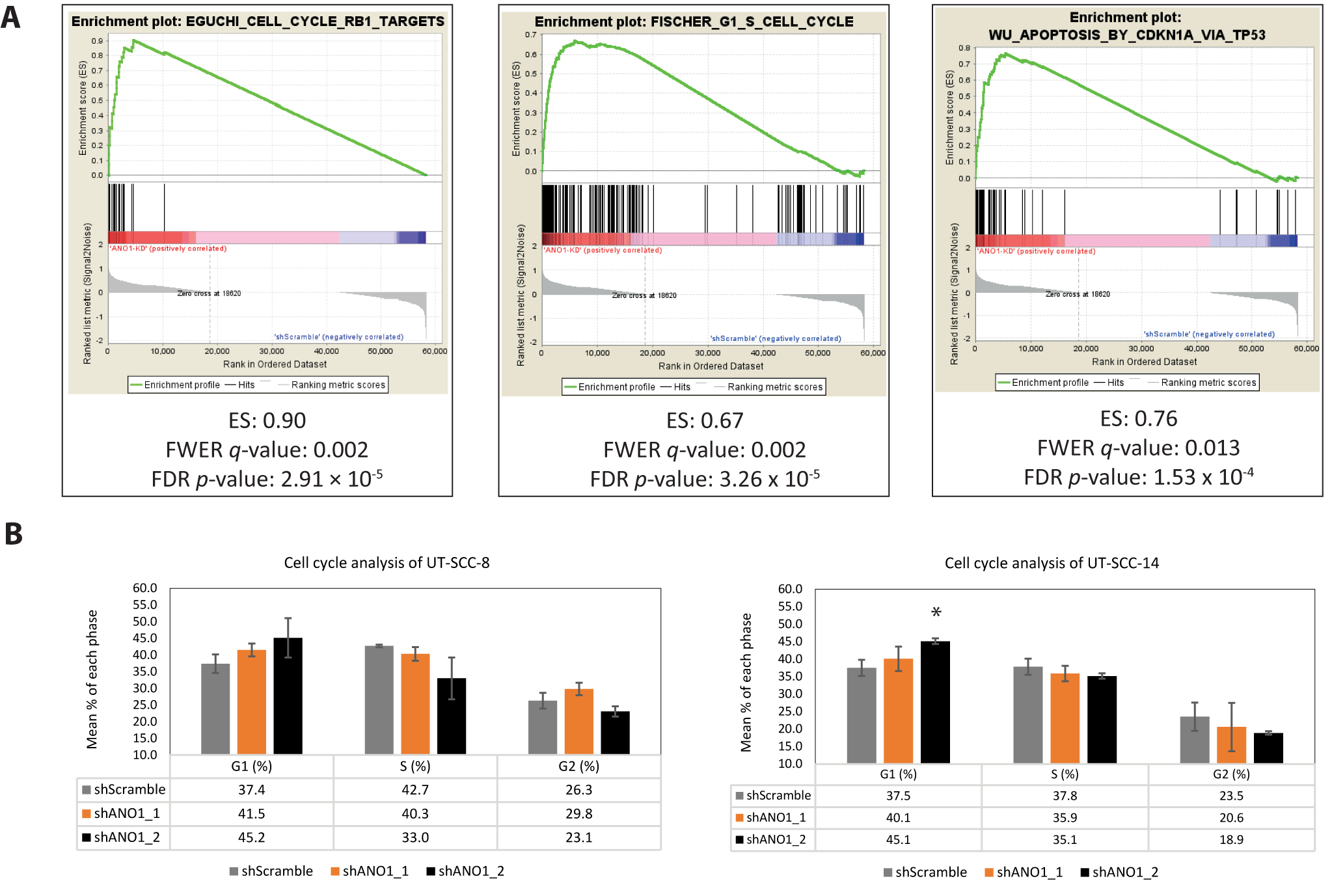
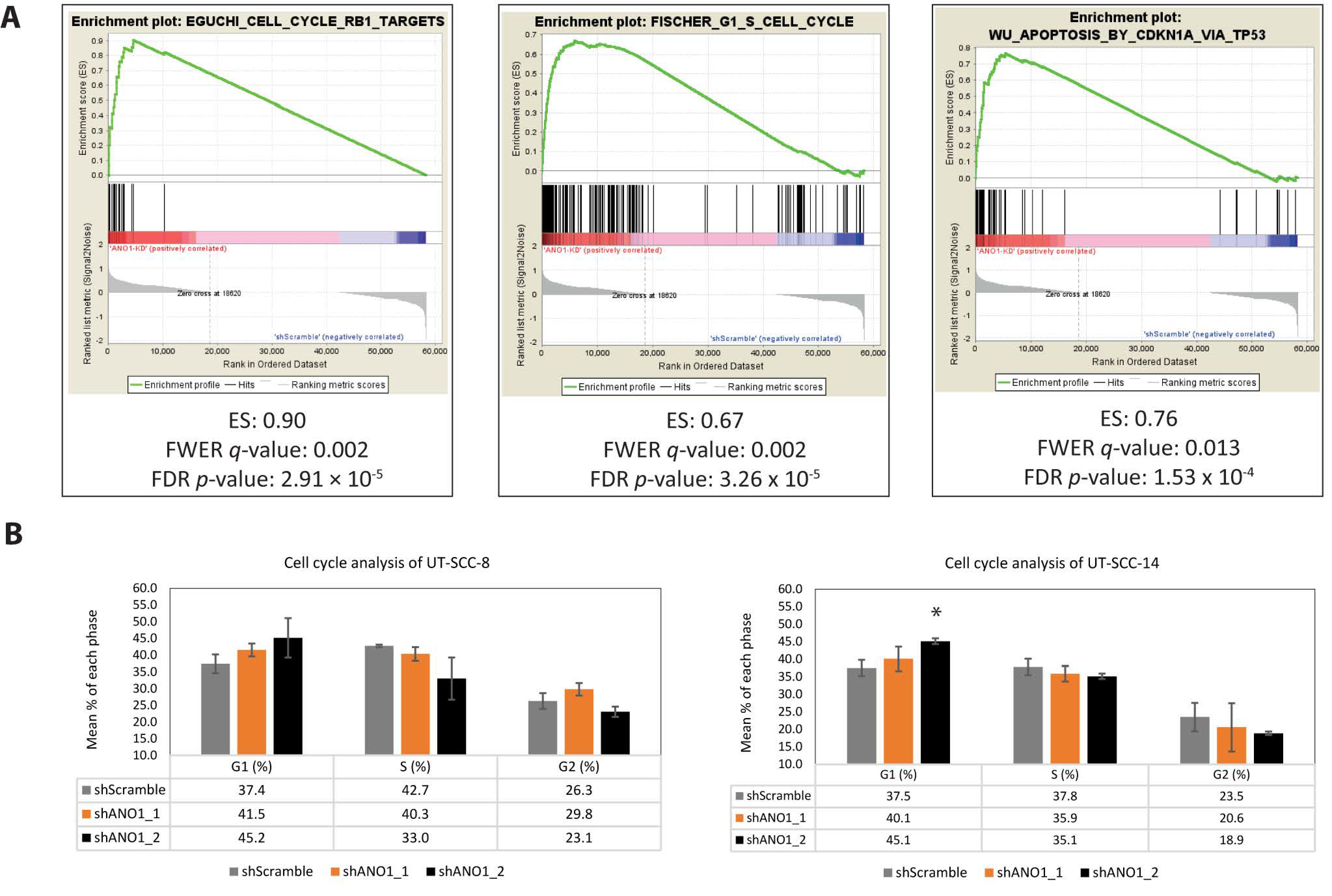
2.4. Pathway Analysis Revealed ANO1 Involvement in Oncogenic Signaling, Including Apoptosis and Cell Cycle Progression
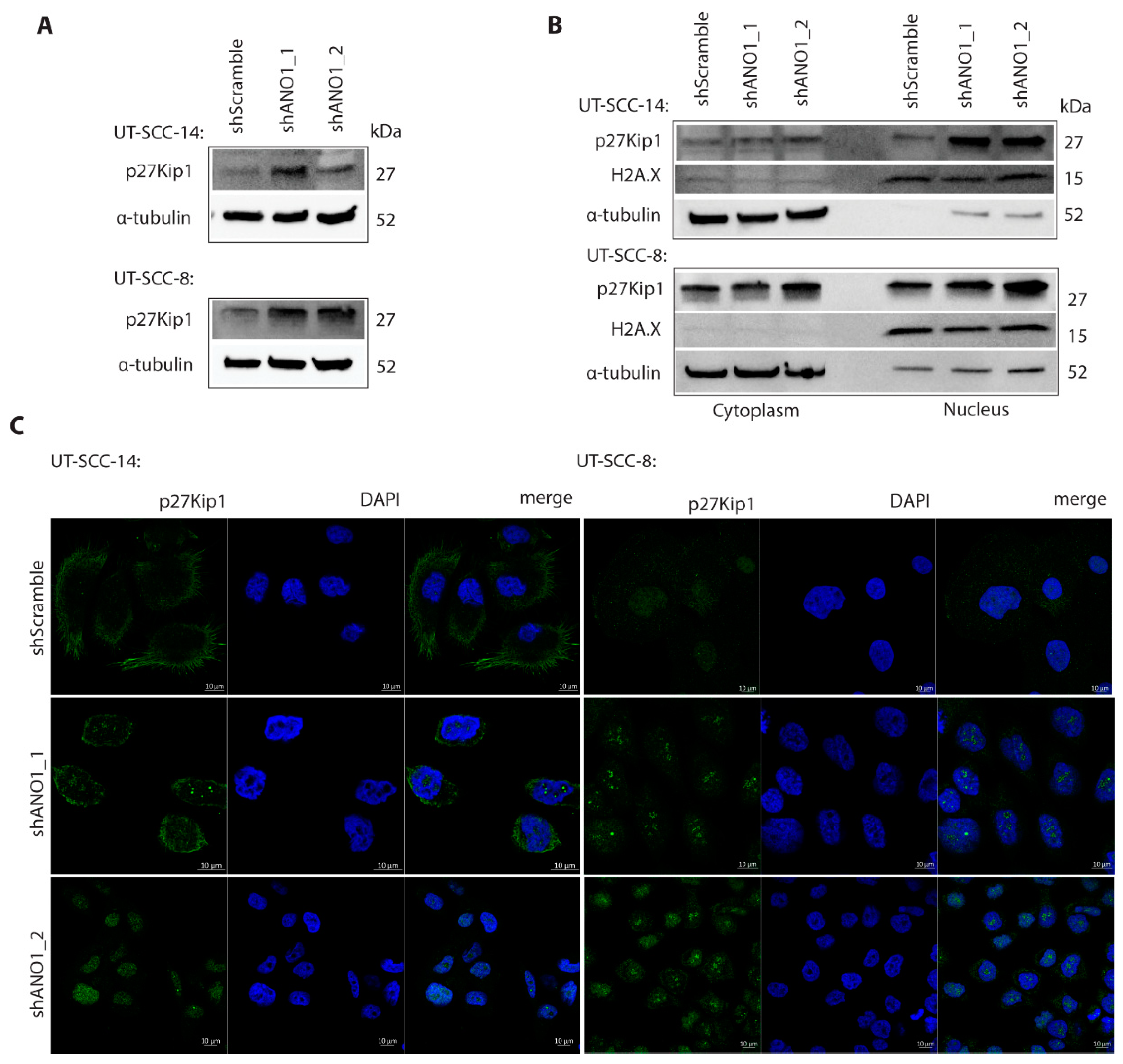
2.5. ANO1 Regulates p27Kip1 Stability and Subcellular Localization
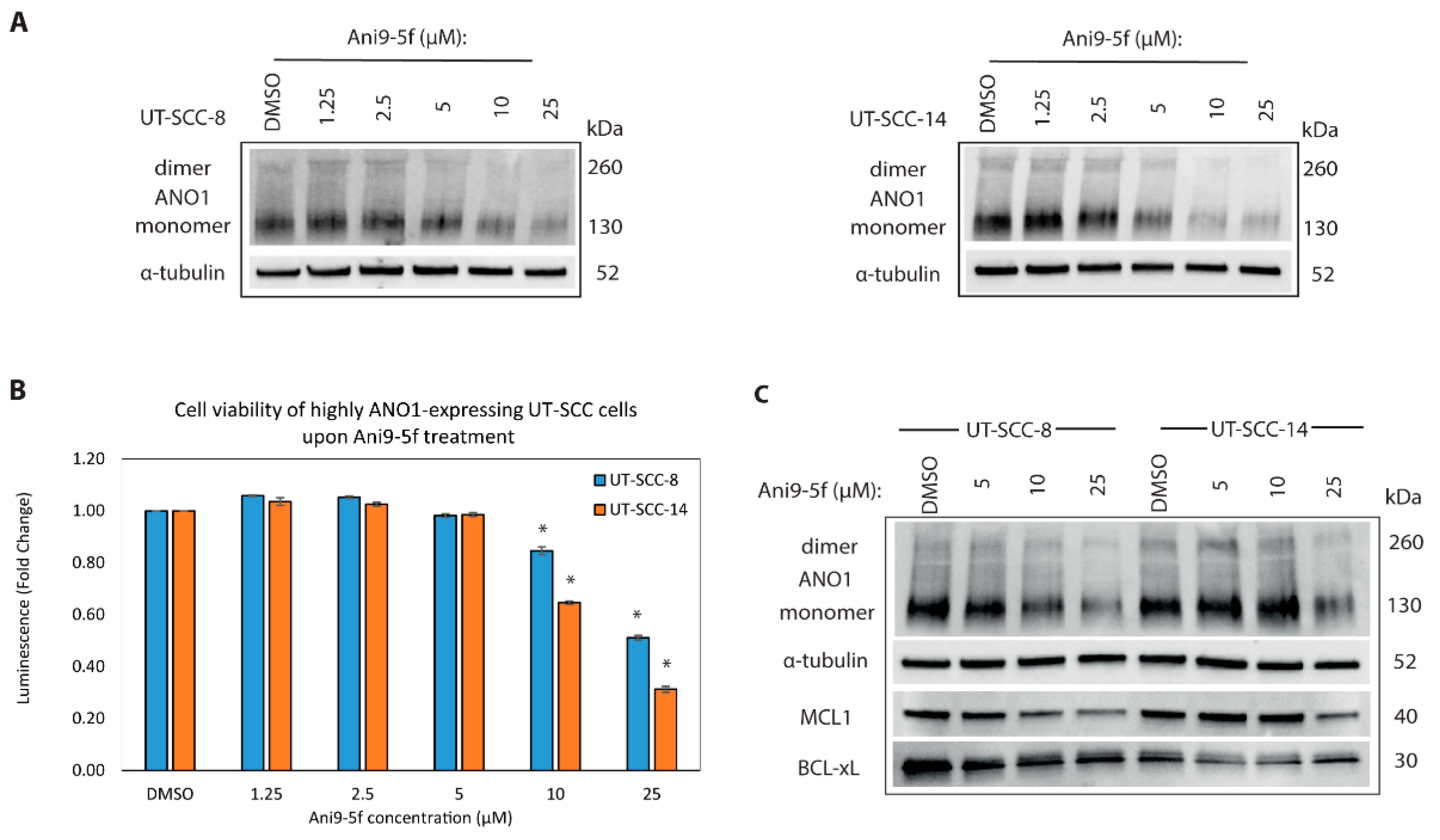
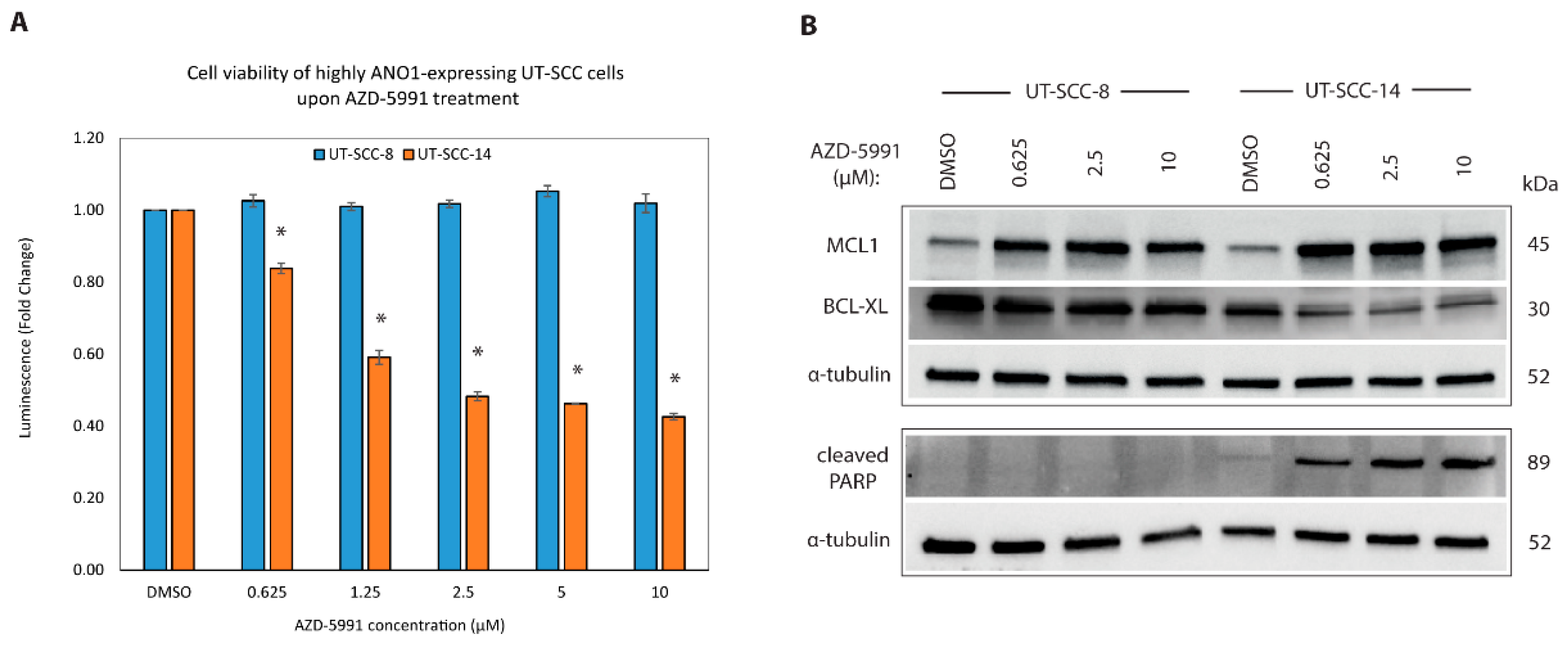
2.6. New ANO1 Small-Molecule Inhibitor Efficiently Decreases Cell Viability of ANO1HIGH HNSCC Cell Lines
2.7. Targeting MCL1 Induces Apoptosis with Concomitant Reduced Expression of BCL-XL
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Antibodies, Reagents and Chemicals
4.3. Generation of the ArrayCGH Profiles
4.4. Generation of Stable ANO1-Knockdown UT-SCC Cells
4.5. Sample Preparation for RNA Sequencing
4.6. RNA Extraction and Purification
4.7. RNA Sequencing and Data Analysis
4.8. Gene Set Enrichment Analysis (GSEA)
4.9. Protein Extraction, Immunoblot and Immunoprecipitation
4.10. Cell Proliferation Assay (MTT)
4.11. Cell Viability, Apoptosis, and Drug Treatment Assays
4.12. Cell Cycle Analysis
4.13. Immunofluorescence Staining and Confocal Microscopy
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, R.; Agrawal, N.; Fakhry, C. Anatomical Sites and Subsites of Head and Neck Cancer. Tumours Skull Base Paranasal Sinuses 2015, 2015, 1–11. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Blot, W.J.; McLaughlin, J.K.; Winn, D.M.; Austin, D.F.; Greenberg, R.S.; Preston-Martin, S.; Bernstein, L.; Schoenberg, J.B.; Stemhagen, A.; Fraumeni, J.F. Smoking and drinking in relation to oral and pharyngeal cancer. Cancer Res. 1988, 48, 3282–3287. [Google Scholar] [PubMed]
- Chaturvedi, A.K.; Engels, E.A.; Pfeiffer, R.M.; Hernandez, B.Y.; Xiao, W.; Kim, E.; Jiang, B.; Goodman, M.T.; Sibug-Saber, M.; Cozen, W.; et al. Human Papillomavirus and Rising Oropharyngeal Cancer Incidence in the United States. J. Clin. Oncol. 2011, 29, 4294–4301. [Google Scholar] [CrossRef] [PubMed]
- Leemans, C.R.; Snijders, P.J.F.; Brakenhoff, R.H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 2018, 18, 269–282. [Google Scholar] [CrossRef]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Prim. 2020, 6, 1–22. [Google Scholar] [CrossRef]
- Zibelman, M.; Mehra, R. Overview of Current Treatment Options and Investigational Targeted Therapies for Locally Advanced Squamous Cell Carcinoma of the Head and Neck. Am. J. Clin. Oncol. 2016, 39, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Jin, Y.; Gisselsson, D.; Wennerberg, J.; Wah, T.; Strömbäck, B.; Kwong, Y.-L.; Mertens, F. Molecular cytogenetic characterization of the 11q13 amplicon in head and neck squamous cell carcinoma. Cytogenet. Genome Res. 2006, 115, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.; Bill, A.; Godse, N.R.; Khan, N.I.; Kass, J.I.; Steehler, K.; Kemp, C.; Davis, K.; Bertrand, C.A.; Vyas, A.R.; et al. TMEM16A/ANO1 suppression improves response to antibody-mediated targeted therapy of EGFR and HER2/ERBB2. Genes Chromosom. Cancer 2017, 56, 460–471. [Google Scholar] [CrossRef]
- Britschgi, A.; Bill, A.; Brinkhaus, H.; Rothwell, C.; Clay, I.; Duss, S.; Rebhan, M.; Raman, P.; Guy, C.T.; Wetzel, K.; et al. Calcium-activated chloride channel ANO1 promotes breast cancer progression by activating EGFR and CAMK signaling. Proc. Natl. Acad. Sci. USA 2013, 110, E1026–E1034. [Google Scholar] [CrossRef]
- Liu, F.; Cao, Q.-H.; Lü, D.-J.; Luo, B.; Lu, X.-F.; Luo, R.-C.; Wang, X.-G. TMEM16A overexpression contributes to tumor invasion and poor prognosis of human gastric cancer through TGF-β signaling. Oncotarget 2015, 6, 11585–11599. [Google Scholar] [CrossRef]
- Berglund, E.; Akçakaya, P.; Berglund, D.; Karlsson, F.; Vukojević, V.; Lee, L.; Bogdanović, D.; Lui, W.-O.; Larsson, C.; Zedenius, J.; et al. Functional role of the Ca2+-activated Cl− channel DOG1/TMEM16A in gastrointestinal stromal tumor cells. Exp. Cell Res. 2014, 326, 315–325. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, S.; Hou, F.; Zhang, C.; Gao, J.; Wang, K. Inhibition of Ca2+-activated chloride channel ANO1 suppresses ovarian cancer through inactivating PI3K/Akt signaling. Int. J. Cancer 2019, 144, 2215–2226. [Google Scholar] [CrossRef]
- Liu, W.; Lu, M.; Liu, B.; Huang, Y.; Wang, K. Inhibition of Ca2+-activated Cl− channel ANO1/TMEM16A expression suppresses tumor growth and invasiveness in human prostate carcinoma. Cancer Lett. 2012, 326, 41–51. [Google Scholar] [CrossRef]
- Song, Y.; Gao, J.; Guan, L.; Chen, X.; Gao, J.; Wang, K. Inhibition of ANO1/TMEM16A induces apoptosis in human prostate carcinoma cells by activating TNF-α signaling. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Lee, Y.-S.; Lee, J.K.; Bae, Y.; Lee, B.-S.; Kim, E.; Cho, C.-H.; Ryoo, K.; Yoo, J.; Kim, C.-H.; Yi, G.-S.; et al. Suppression of 14-3-3γ-mediated surface expression of ANO1 inhibits cancer progression of glioblastoma cells. Sci. Rep. 2016, 6, 26413. [Google Scholar] [CrossRef]
- Ayoub, C.M.; Wasylyk, C.; Li, Y.; A Thomas, E.; Marisa, L.; Robe, A.; Roux, M.; Abecassis, J.; De Reyniès, A.; Wasylyk, B. ANO1 amplification and expression in HNSCC with a high propensity for future distant metastasis and its functions in HNSCC cell lines. Br. J. Cancer 2010, 103, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, C.; Martins, J.R.; Rudin, F.; Schneider, S.; Dietsche, T.; Fischer, C.A.; Tornillo, L.; Terracciano, L.M.; Schreiber, R.; Bubendorf, L.; et al. Enhanced Expression of ANO1 in Head and Neck Squamous Cell Carcinoma Causes Cell Migration and Correlates with Poor Prognosis. PLoS ONE 2012, 7, e43265. [Google Scholar] [CrossRef]
- Duvvuri, U.; Shiwarski, D.J.; Xiao, D.; Bertrand, C.; Huang, X.; Edinger, R.S.; Rock, J.R.; Harfe, B.D.; Henson, B.J.; Kunzelmann, K.; et al. TMEM16A Induces MAPK and Contributes Directly to Tumorigenesis and Cancer Progression. Cancer Res. 2012, 72, 3270–3281. [Google Scholar] [CrossRef] [PubMed]
- Shiwarski, D.J.; Shao, C.; Bill, A.; Kim, J.; Xiao, D.; Bertrand, C.A.; Seethala, R.S.; Sano, D.; Myers, J.N.; Ha, P.; et al. To “Grow” or “Go”: TMEM16A Expression as a Switch between Tumor Growth and Metastasis in SCCHN. Clin. Cancer Res. 2014, 20, 4673–4688. [Google Scholar] [CrossRef] [PubMed]
- Stanich, J.E.; Gibbons, S.J.; Eisenman, S.T.; Bardsley, M.R.; Rock, J.R.; Harfe, B.D.; Ordog, T.; Farrugia, G. Ano1 as a regulator of proliferation. Am. J. Physiol. Liver Physiol. 2011, 301, G1044–G1051. [Google Scholar] [CrossRef] [PubMed]
- Guan, L.; Song, Y.; Gao, J.; Gao, J.; Wang, K. Inhibition of calcium-activated chloride channel ANO1 suppresses proliferation and induces apoptosis of epithelium originated cancer cells. Oncotarget 2016, 7, 78619–78630. [Google Scholar] [CrossRef] [PubMed]
- Allawzi, A.M.; Vang, A.; Clements, R.T.; Jhun, B.S.; Kue, N.R.; Mancini, T.J.; Landi, A.K.; Terentyev, D.; O-Uchi, J.; Comhair, S.A.; et al. Activation of Anoctamin-1 Limits Pulmonary Endothelial Cell Proliferation via p38–Mitogen-activated Protein Kinase–Dependent Apoptosis. Am. J. Respir. Cell Mol. Biol. 2018, 58, 658–667. [Google Scholar] [CrossRef]
- Bill, A.; Gutierrez, A.; Kulkarni, S.; Kemp, C.; Bonenfant, D.; Voshol, H.; Duvvuri, U.; Gaither, L.A. ANO1/TMEM16A interacts with EGFR and correlates with sensitivity to EGFR-targeting therapy in head and neck cancer. Oncotarget 2015, 6, 9173–9188. [Google Scholar] [CrossRef] [PubMed]
- Perez-Cornejo, P.; Gokhale, A.; Duran, C.; Cui, Y.; Xiao, Q.; Hartzell, H.C.; Faundez, V. Anoctamin 1 (Tmem16A) Ca2+-activated chloride channel stoichiometrically interacts with an ezrin-radixin-moesin network. Proc. Natl. Acad. Sci. USA 2012, 109, 10376–10381. [Google Scholar] [CrossRef]
- Sayáns, M.P.; Petronacci, C.M.C.; Pouso, A.I.L.; Iruegas, E.P.; Carrión, A.B.; Peñaranda, J.M.S.; García, A.G. Comprehensive Genomic Review of TCGA Head and Neck Squamous Cell Carcinomas (HNSCC). J. Clin. Med. 2019, 8, 1896. [Google Scholar] [CrossRef]
- Bill, A.; Hall, M.L.; Borawski, J.; Hodgson, C.; Jenkins, J.; Piechon, P.; Popa, M.O.; Rothwell, C.; Tranter, P.; Tria, S.; et al. Small molecule facilitated degradation of ANO1—A new targeting approach for anticancer therapeutics. J. Biol. Chem. 2014, 289, e11029-41. [Google Scholar] [CrossRef] [PubMed]
- Boedtkjer, D.M.B.; Kim, S.; Jensen, A.B.; Matchkov, V.M.; E Andersson, K. New selective inhibitors of calcium-activated chloride channels—T16Ainh-A01, CaCCinh-A01 and MONNA—What do they inhibit? Br. J. Pharmacol. 2015, 172, 4158–4172. [Google Scholar] [CrossRef]
- Seo, Y.; Lee, H.K.; Park, J.; Jeon, D.-K.; Jo, S.; Jo, M.; Namkung, W. Ani9, A Novel Potent Small-Molecule ANO1 Inhibitor with Negligible Effect on ANO2. PLoS ONE 2016, 11, e0155771. [Google Scholar] [CrossRef]
- Seo, Y.; Kim, J.; Chang, J.; Kim, S.S.; Namkung, W.; Kim, I. Synthesis and biological evaluation of novel Ani9 derivatives as potent and selective ANO1 inhibitors. Eur. J. Med. Chem. 2018, 160, 245–255. [Google Scholar] [CrossRef]
- Fujise, K.; Zhang, D.; Liu, J.-L.; Yeh, E.T.H. Regulation of Apoptosis and Cell Cycle Progression by MCL1. J. Biol. Chem. 2000, 275, 39458–39465. [Google Scholar] [CrossRef] [PubMed]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Carter, R.J.; Milani, M.; Butterworth, M.; Alotibi, A.; Harper, N.; Yedida, G.; Greaves, G.; Al-Zebeeby, A.; Jorgensen, A.L.; Schache, A.G.; et al. Exploring the potential of BH3 mimetic therapy in squamous cell carcinoma of the head and neck. Cell Death Dis. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Senichkin, V.V.; Streletskaia, A.Y.; Zhivotovsky, B.; Kopeina, G.S. Molecular Comprehension of Mcl-1: From Gene Structure to Cancer Therapy. Trends Cell Biol. 2019, 29, 549–562. [Google Scholar] [CrossRef]
- Perciavalle, R.M.; Stewart, D.P.; Koss, B.; Lynch, J.; Milasta, S.; Bathina, M.; Temirov, J.; Cleland, M.M.; Pelletier, S.; Schuetz, J.D.; et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat. Cell Biol. 2012, 14, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Fairbrother, A.A.W.J.; Leverson, J.D.; Souers, J.D.L.A.J. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat. Rev. Drug Discov. 2017, 16, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Tron, A.E.; Belmonte, M.A.; Adam, A.; Aquila, B.M.; Boise, L.H.; Chiarparin, E.; Cidado, J.; Embrey, K.J.; Gangl, E.; Gibbons, F.D.; et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, T.; Hesabi, B.; Moons, D.S.; Pandolfi, P.P.; Hansel, K.S.; Koff, A.; Kiyokawa, H. Targeted Disruption of CDK4 Delays Cell Cycle Entry with Enhanced p27Kip1 Activity. Mol. Cell. Biol. 1999, 19, 7011–7019. [Google Scholar] [CrossRef]
- Chu, I.M.; Hengst, L.; Slingerland, J.M. The Cdk inhibitor p27 in human cancer: Prognostic potential and relevance to anticancer therapy. Nat. Rev. Cancer 2008, 8, 253–267. [Google Scholar] [CrossRef]
- Lepikhova, T.; Karhemo, P.-R.; Louhimo, R.; Yadav, B.; Murumägi, A.; Kulesskiy, E.; Kivento, M.; Sihto, H.; Grénman, R.; Syrjänen, S.M.; et al. Drug-Sensitivity Screening and Genomic Characterization of 45 HPV-Negative Head and Neck Carcinoma Cell Lines for Novel Biomarkers of Drug Efficacy. Mol. Cancer Ther. 2018, 17, 2060–2071. [Google Scholar] [CrossRef]
- Loh, C.-Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef]
- Nguyen, P.T.; Kudo, Y.; Yoshida, M.; Kamata, N.; Ogawa, I.; Takata, T. N-cadherin expression is involved in malignant behavior of head and neck cancer in relation to epithelial-mesenchymal transition. Histol. Histopathol. 2011, 26, 147–156. [Google Scholar] [PubMed]
- Yang, M.-H.; Hsu, D.S.-S.; Wang, H.-W.; Wang, H.-J.; Lan, H.-Y.; Yang, W.-H.; Huang, C.-H.; Kao, S.-Y.; Tzeng, C.-H.; Tai, S.-K.; et al. Bmi1 is essential in Twist1-induced epithelial–mesenchymal transition. Nat. Cell Biol. 2010, 12, 982–992. [Google Scholar] [CrossRef]
- Pehkonen, H.; Von Nandelstadh, P.; Karhemo, P.-R.; Lepikhova, T.; Grenman, R.; Lehti, K.; Monni, O. Liprin-α1 is a regulator of vimentin intermediate filament network in the cancer cell adhesion machinery. Sci. Rep. 2016, 6, 24486. [Google Scholar] [CrossRef] [PubMed]
- Godse, N.R.; Khan, N.; Yochum, Z.A.; Gomez-Casal, R.; Kemp, C.; Shiwarski, D.J.; Seethala, R.S.; Kulich, S.; Seshadri, M.; Burns, T.F.; et al. TMEM16A/ANO1 Inhibits Apoptosis via Downregulation of Bim Expression. Clin. Cancer Res. 2017, 23, 7324–7332. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.J.; A Yun, J.; Jabeen, S.; Jeon, E.K.; Won, H.S.; Ko, Y.H.; Kim, S.Y. Prognostic significance of TMEM16A, PPFIA1, and FADD expression in invasive ductal carcinoma of the breast. World J. Surg. Oncol. 2014, 12, 137. [Google Scholar] [CrossRef]
- Uchi, J.; Vang, A.; Cypress, M.W.; Fernandez-Nicolas, A.; Mancini, T.; Jhun, B.S.; Clements, R.T.; Choudhary, G. Role of Mitochondrial Expression of the Calcium-Activated Chloride Channel Anoctamin-1 in Pulmonary Artery Endothelial Cells. Biophys. J. 2020, 118, 448a. [Google Scholar] [CrossRef]
- Weeden, C.E.; Ah-Cann, C.; Holik, A.Z.; Pasquet, J.; Garnier, J.-M.; Merino, D.; Lessene, G.; Asselin-Labat, M.-L. Dual inhibition of BCL-XL and MCL-1 is required to induce tumour regression in lung squamous cell carcinomas sensitive to FGFR inhibition. Oncogene 2018, 37, 4475–4488. [Google Scholar] [CrossRef] [PubMed]
- Greaves, G.; Milani, M.; Butterworth, M.; Carter, R.J.; Byrne, M.P.; Eyers, P.A.; Luo, X.; Cohen, G.M.; Varadarajan, S. BH3-only proteins are dispensable for apoptosis induced by pharmacological inhibition of both MCL-1 and BCL-XL. Cell Death Differ. 2018, 26, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.S.; Park, J.Y.; Park, S.-H.; Ha, S.H.; An, A.R.; Noh, S.J.; Kwon, K.S.; Jung, S.H.; Park, H.S.; Kang, M.J.; et al. Expression of ANO1/DOG1 is associated with shorter survival and progression of breast carcinomas. Oncotarget 2017, 9, 607–621. [Google Scholar] [CrossRef] [PubMed]
- Sabile, A.; Meyer, A.M.; Wirbelauer, C.; Hess, D.; Kogel, U.; Scheffner, M.; Krek, W. Regulation of p27 Degradation and S-Phase Progression by Ro52 RING Finger Protein. Mol. Cell. Biol. 2006, 26, 5994–6004. [Google Scholar] [CrossRef]
- Rahman, S.F.A.; Muniandy, K.; Soo, Y.K.; Tiew, E.Y.H.; Tan, K.X.; Bates, T.E.; Mohana-Kumaran, N. Co-inhibition of BCL-XL and MCL-1 with selective BCL-2 family inhibitors enhances cytotoxicity of cervical cancer cell lines. Biochem. Biophys. Rep. 2020, 22, 100756. [Google Scholar] [CrossRef] [PubMed]
- Charoenrat, P.; Modjtahedi, H.; Rhys-Evans, P.; Court, W.J.; Box, G.M.; A Eccles, S. Epidermal growth factor-like ligands differentially up-regulate matrix metalloproteinase 9 in head and neck squamous carcinoma cells. Cancer Res. 2000, 60, 1121–1128. [Google Scholar]
- Hara, T.; Kamura, T.; Nakayama, K.; Oshikawa, K.; Hatakeyama, S.; Nakayama, K.-I. Degradation of p27 at the G0-G1 Transition Mediated by a Skp2-independent Ubiquitination Pathway. J. Biol. Chem. 2001, 276, 48937–48943. [Google Scholar] [CrossRef]
- Kamura, T.; Hara, T.; Matsumoto, M.; Ishida, N.; Okumura, F.; Hatakeyama, S.; Yoshida, M.; Nakayama, K.; Nakayama, K.I. Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27Kip1 at G1 phase. Nat. Cell Biol. 2004, 6, 1229–1235. [Google Scholar] [CrossRef]
- Moser, R.; Xu, C.; Kao, M.; Annis, J.; Lerma, L.A.; Schaupp, C.M.; Gurley, K.E.; Jang, I.S.; Biktasova, A.; Yarbrough, W.G.; et al. Functional Kinomics Identifies Candidate Therapeutic Targets in Head and Neck Cancer. Clin. Cancer Res. 2014, 20, 4274–4288. [Google Scholar] [CrossRef] [PubMed]
- Jamil, S.; Mojtabavi, S.; Hojabrpour, P.; Cheah, S.; Duronio, V. An Essential Role for MCL-1 in ATR-mediated CHK1 Phosphorylation. Mol. Biol. Cell 2008, 19, 3212–3220. [Google Scholar] [CrossRef]
- Thomas, R.L.; Roberts, D.J.; Kubli, D.A.; Lee, Y.; Quinsay, M.N.; Owens, J.B.; Fischer, K.M.; Sussman, M.A.; Miyamoto, S.; Gustafsson, Å.B. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev. 2013, 27, 1365–1377. [Google Scholar] [CrossRef]
- Stojanovski, D.; Koutsopoulos, O.S.; Okamoto, K.; Ryan, M.T. Levels of human Fis1 at the mitochondrial outer membrane regulate mitochondrial morphology. J. Cell Sci. 2004, 117, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Grenman, R.; Carey, T.E.; Mcclatchey, K.D.; Wagner, J.G.; Pekkolaheino, K.; Schwartz, D.R.; Wolf, G.T.; Lacivita, L.P.; Ho, L.; Baker, S.R.; et al. Invitro Radiation-Resistance among Cell-Lines Established from Patients with Squamous-Cell Carcinoma of the Head and Neck. Cancer 1991, 67, 2741–2747. [Google Scholar] [CrossRef]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar]
- Hubbard, T.J.P.; Aken, B.L.; Beal, K.; Ballester, B.; Caccamo, M.; Chen, Y.; Clarke, L.; Coates, G.; Cunningham, F.; Cutts, T.; et al. Ensembl 2007. Nucleic Acids Res. 2006, 35, D610–D617. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Log2FC | padj | Description (Source HGNC) |
|---|---|---|---|
| MCL1 | −0.78 | 4.79 × 10−13 | Myeloid cell leukemia 1; BCL2 family member |
| USP43 | −0.75 | 5.12 × 10−7 | Ubiquitin specific peptidase 43 |
| CDKN1B | +0.60 | 3.01 × 10−5 | Cyclin dependent kinase inhibitor 1B |
| TGFA | −0.77 | 0.0001 | Transforming growth factor alpha |
| FIS1 | −0.40 | 0.0002 | Fission, mitochondrial 1 |
| E2F8 | +1.87 | 0.0004 | E2F transcription factor 8 |
| DNAJB4 | +0.74 | 0.0008 | DnaJ heat shock protein family (Hsp40) member B4 |
| HLA-A | −0.89 | 0.0029 | Major histocompatibility complex, class I, A |
| COX7C | +0.70 | 0.0031 | Cytochrome c oxidase subunit 7C |
| PINK1 | −0.58 | 0.0066 | PTEN induced putative kinase 1 |
| TRIM21 | −0.66 | 0.0077 | Tripartite motif containing 21 |
| RB1 | +0.50 | 0.0077 | RB transcriptional corepressor 1 |
| STUB1 | −0.33 | 0.0088 | STIP1 homology and U-box containing protein 1 |
| PSMD5 | +0.27 | 0.0093 | Proteasome 26S subunit, non-ATPase 5 |
| FADD | −0.49 | 0.0102 | Fas associated via death domain |
| NDUFA13 | −0.40 | 0.0102 | NADH: ubiquinone oxidoreductase subunit A13 |
| BCL-XL | −0.54 | 0.0127 | BCL2 like1; protein phosphatase 1, regulatory subunit 52 |
| WEE1 | +0.74 | 0.0188 | WEE1 G2 checkpoint kinase |
| PSMB10 | −0.63 | 0.0236 | Proteasome subunit beta 10 |
| CDKN2C | +1.27 | 0.0257 | Cyclin dependent kinase inhibitor 2C |
| FURIN | −0.55 | 0.0266 | Furin, paired basic amino acid cleaving enzyme |
| PSEN2 | −0.50 | 0.0323 | Presenilin 2 |
| HDAC1 | −0.36 | 0.0328 | Histone deacetylase 1 |
| USP37 | +0.48 | 0.0334 | Ubiquitin specific peptidase 37 |
| UBE2L6 | −1.16 | 0.0406 | Ubiquitin conjugating enzyme E2 L6 |
| UBE2E3 | +0.34 | 0.0428 | Ubiquitin conjugating enzyme E2 E3 |
| USP48 | +0.19 | 0.0481 | Ubiquitin specific peptidase 48 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filippou, A.; Pehkonen, H.; Karhemo, P.-R.; Väänänen, J.; Nieminen, A.I.; Klefström, J.; Grénman, R.; Mäkitie, A.A.; Joensuu, H.; Monni, O. ANO1 Expression Orchestrates p27Kip1/MCL1-Mediated Signaling in Head and Neck Squamous Cell Carcinoma. Cancers 2021, 13, 1170. https://doi.org/10.3390/cancers13051170
Filippou A, Pehkonen H, Karhemo P-R, Väänänen J, Nieminen AI, Klefström J, Grénman R, Mäkitie AA, Joensuu H, Monni O. ANO1 Expression Orchestrates p27Kip1/MCL1-Mediated Signaling in Head and Neck Squamous Cell Carcinoma. Cancers. 2021; 13(5):1170. https://doi.org/10.3390/cancers13051170
Chicago/Turabian StyleFilippou, Artemis, Henna Pehkonen, Piia-Riitta Karhemo, Juho Väänänen, Anni I. Nieminen, Juha Klefström, Reidar Grénman, Antti A. Mäkitie, Heikki Joensuu, and Outi Monni. 2021. "ANO1 Expression Orchestrates p27Kip1/MCL1-Mediated Signaling in Head and Neck Squamous Cell Carcinoma" Cancers 13, no. 5: 1170. https://doi.org/10.3390/cancers13051170
APA StyleFilippou, A., Pehkonen, H., Karhemo, P.-R., Väänänen, J., Nieminen, A. I., Klefström, J., Grénman, R., Mäkitie, A. A., Joensuu, H., & Monni, O. (2021). ANO1 Expression Orchestrates p27Kip1/MCL1-Mediated Signaling in Head and Neck Squamous Cell Carcinoma. Cancers, 13(5), 1170. https://doi.org/10.3390/cancers13051170