
Transitioning the Molecular Tumor Board from Proof of Concept to Clinical Routine: A German Single-Center Analysis

 , , , , , , , , , , , , , , ,
, , , , , , , , , , , , , , ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. MTB Organization and Patients
2.2. Molecular Diagnostics
3. Results
3.1. Patient Characteristics
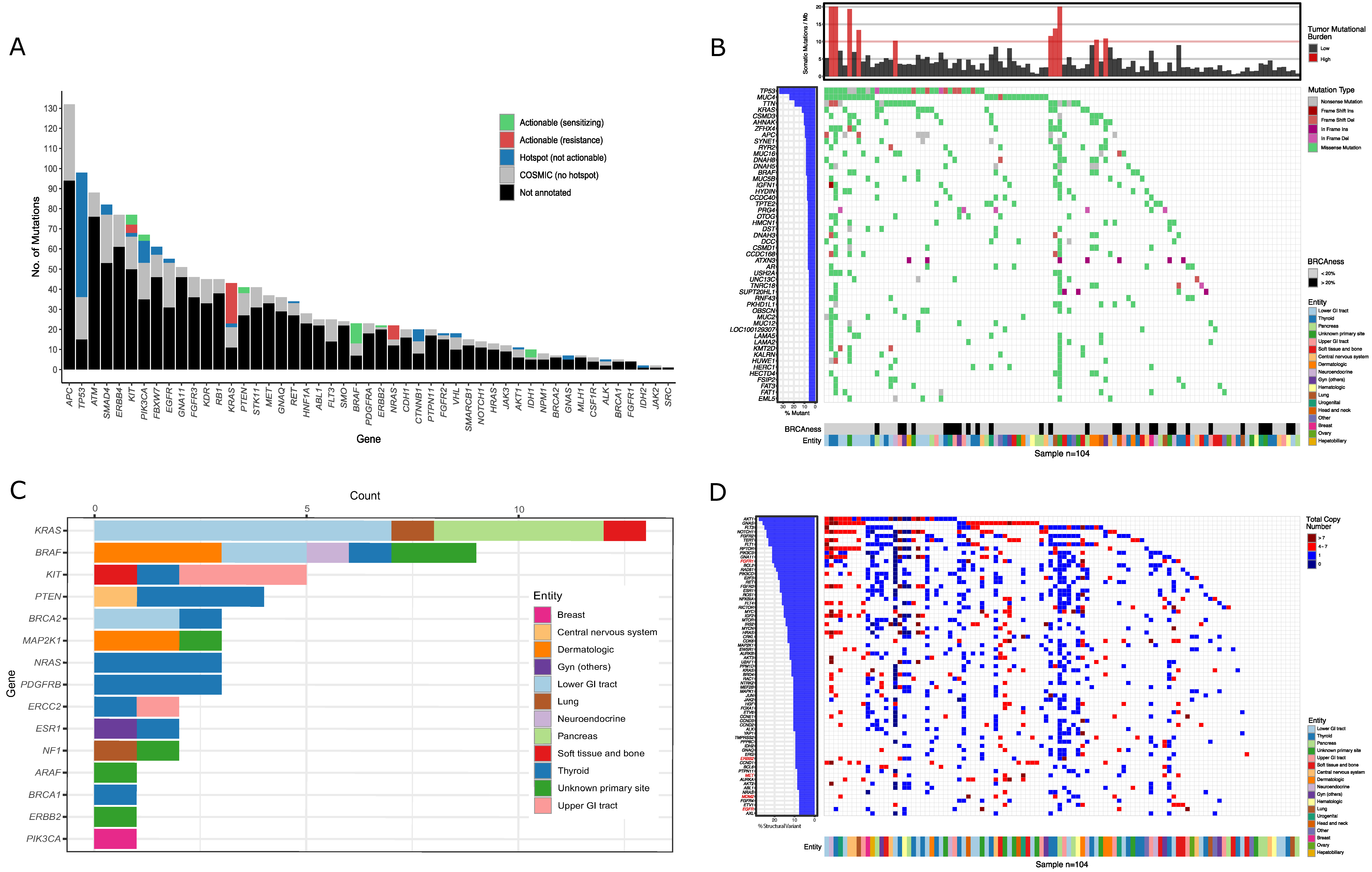
3.2. Molecular Diagnostic Testing
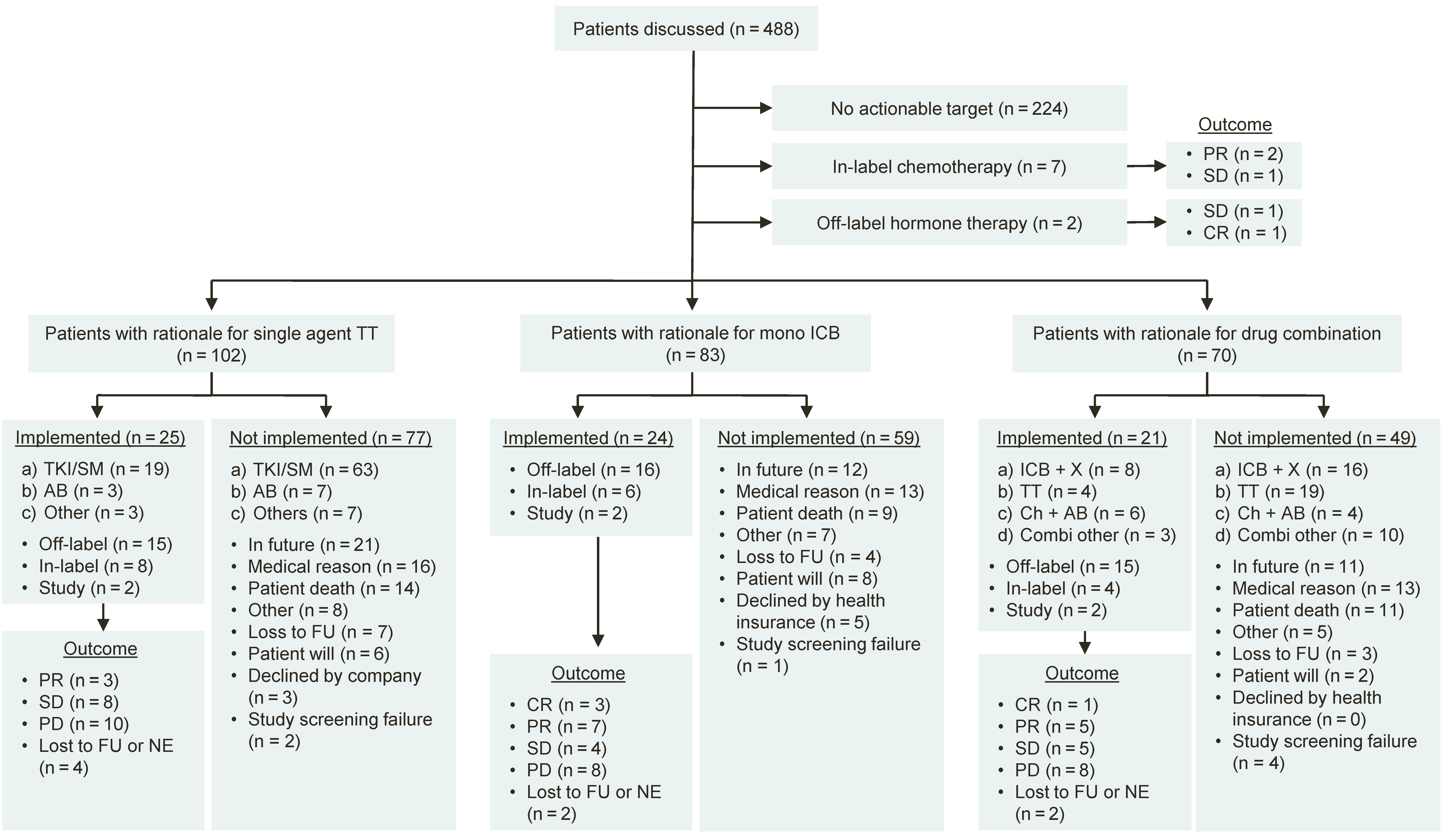
3.3. Treatment Recommendations
3.4. Clinical Outcome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mullard, A. NEWS & AnaLYSIS 2018 FDA Drug Approvals. Nat. Rev. Drug Discov. 2019, 18, 85–89. [Google Scholar] [PubMed]
- Mullard, A. 2019 FDA Drug Approvals. Nat. Rev. Drug Discov. 2020, 19, 79–84. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration (FDA). Grants Accelerated Approval to Pembrolizumab for First Tissue/Site Agnostic Indication; FDA: Rockwell, MD, USA, 2017.
- U.S. Food and Drug Administration (FDA). Approves Pembrolizumab for the First-Line Treatment of MSI-H/DMMR Colorectal Cancer. Available online: https://www.ascopost.com/issues/july-10-2020/fda-approves-pembrolizumab-for-the-first-line-treatment-of-msi-hdmmr-colorectal-cancer/ (accessed on 10 January 2021).
- U.S. Food and Drug Administration (FDA). Approves Pembrolizumab for Adults and Children with TMB-H Solid Tumors. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-pembrolizumab-adults-and-children-tmb-h-solid-tumors (accessed on 10 January 2021).
- U.S. Food and Drug Administration (FDA). Approves Larotrectinib for Solid Tumors with NTRK Gene Fusions. Available online: https://www.fda.gov/drugs/fda-approves-larotrectinib-solid-tumors-ntrk-gene-fusions (accessed on 10 January 2021).
- Hyman, D.M.; Taylor, B.S.; Baselga, J. Implementing Genome-Driven Oncology. Cell 2017, 168, 584–599. [Google Scholar] [CrossRef]
- Le Tourneau, C.; Delord, J.-P.P.; Gonçalves, A.; Gavoille, C.; Dubot, C.; Isambert, N.; Campone, M.; Trédan, O.; Massiani, M.-A.A.; Mauborgne, C.; et al. Molecularly Targeted Therapy Based on Tumour Molecular Profiling versus Conventional Therapy for Advanced Cancer (SHIVA): A Multicentre, Open-Label, Proof-of-Concept, Randomised, Controlled Phase 2 Trial. Lancet Oncol. 2015, 16, 1324–1334. [Google Scholar] [CrossRef]
- Prasad, V. The Precision-Oncology Illusion. Nat. Outlook 2016, 537, S63. [Google Scholar]
- Tannock, I.F.; Hickman, J.A. Limits to Personalized Cancer Medicine. N. Engl. J. Med. 2016, 375, 1289–1294. [Google Scholar] [CrossRef] [PubMed]
- Hainsworth, J.D.; Meric-Bernstam, F.; Swanton, C.; Hurwitz, H.; Spigel, D.R.; Sweeney, C.; Burris, H.; Bose, R.; Yoo, B.; Stein, A.; et al. Targeted Therapy for Advanced Solid Tumors on the Basis of Molecular Profiles: Results from Mypathway, an Open-Label, Phase IIA Multiple Basket Study. J. Clin. Oncol. 2018, 36, 536–542. [Google Scholar] [CrossRef]
- Hyman, D.M.; Piha-Paul, S.A.; Won, H.; Rodon, J.; Saura, C.; Shapiro, G.I.; Juric, D.; Quinn, D.I.; Moreno, V.; Doger, B.; et al. HER Kinase Inhibition in Patients with HER2-and HER3-Mutant Cancers. Nature 2018, 554, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Stockley, T.L.; Oza, A.M.; Berman, H.K.; Leighl, N.B.; Knox, J.J.; Shepherd, F.A.; Chen, E.X.; Krzyzanowska, M.K.; Dhani, N.; Joshua, A.M.; et al. Molecular Profiling of Advanced Solid Tumors and Patient Outcomes with Genotype-Matched Clinical Trials: The Princess Margaret IMPACT/COMPACT Trial. Genome Med. 2016, 8, 109. [Google Scholar] [CrossRef]
- Dalton, W.B.; Forde, P.M.; Kang, H.; Connolly, R.M.; Stearns, V.; Gocke, C.D.; Eshleman, J.R.; Axilbund, J.; Petry, D.; Geoghegan, C.; et al. Personalized Medicine in the Oncology Clinic: Implementation and Outcomes of the Johns Hopkins Molecular Tumor Board. JCO Precis. Oncol. 2017, 2017, PO.16.00046. [Google Scholar] [CrossRef]
- Sohal, D.P.S.; Rini, B.I.; Khorana, A.A.; Dreicer, R.; Abraham, J.; Procop, G.W.; Saunthararajah, Y.; Pennell, N.A.; Stevenson, J.P.; Pelley, R.; et al. Prospective Clinical Study of Precision Oncology in Solid Tumors. J. Natl. Cancer Inst. 2016, 108, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Kieler, M.; Unseld, M.; Bianconi, D.; Waneck, F.; Mader, R.; Wrba, F.; Fuereder, T.; Marosi, C.; Raderer, M.; Staber, P.; et al. Interim Analysis of a Real-World Precision Medicine Platform for Molecular Profiling of Metastatic or Advanced Cancers: MONDTI. ESMO Open 2019, 4, e000538. [Google Scholar] [CrossRef]
- Belin, L.; Kamal, M.; Mauborgne, C.; Plancher, C.; Mulot, F.; Delord, J.P.; Gonçalves, A.; Gavoille, C.; Dubot, C.; Isambert, N.; et al. Randomized Phase II Trial Comparing Molecularly Targeted Therapy Based on Tumor Molecular Profiling versus Conventional Therapy in Patients with Refractory Cancer: Cross-over Analysis from the SHIVA Trial. Ann. Oncol. 2017, 28, 590–596. [Google Scholar] [CrossRef]
- World Health Organization. Latest Global Cancer Data: Cancer Burden Rises to 18.1 Million New Cases and 9.6 Million Cancer Deaths in 2018; WHO: Geneva, Switzerland, 2018. [Google Scholar]
- Hoefflin, R.; Geißler, A.-L.; Fritsch, R.; Claus, R.; Wehrle, J.; Metzger, P.; Reiser, M.; Mehmed, L.; Fauth, L.; Heiland, D.H.; et al. Personalized Clinical Decision Making Through Implementation of a Molecular Tumor Board: A German Single-Center Experience. JCO Precis. Oncol. 2018, 2018, PO.18.00105. [Google Scholar] [CrossRef]
- Schildhaus, H.-U. Der Prädiktive Wert Der PD-L1-Diagnostik. Pathologe 2018, 39, 498–519. [Google Scholar] [CrossRef]
- Boland, C.R.; Thibodeau, S.N.; Hamilton, S.R.; Sidransky, D.; Eshleman, J.R.; Burt, R.W.; Meltzer, S.J.; Rodriguez-Bigas, M.A.; Fodde, R.; Ranzani, G.N.; et al. A National Cancer Institute Workshop on Microsatellite Instability for Cancer Detection and Familial Predisposition: Development of International Criteria for the Determination of Microsatellite Instability in Colorectal Cancer. Cancer Res. 1998, 58, 5248–5257. [Google Scholar]
- Geißler, A.-L.; Geißler, M.; Kottmann, D.; Lutz, L.; Fichter, C.D.; Fritsch, R.; Weddeling, B.; Makowiec, F.; Werner, M.; Lassmann, S. ATM Mutations and E-Cadherin Expression Define Sensitivity to EGFR-Targeted Therapy in Colorectal Cancer. Oncotarget 2017, 8, 17164. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.M.; Zhang, S.; Geiger, T.; Hafez, M.J.; Bacher, J.; Berg, K.D.; Eshleman, J.R. Comparison of the Microsatellite Instability Analysis System and the Bethesda Panel for the Determination of Microsatellite Instability in Colorectal Cancers. J. Mol. Diagn. 2006, 8, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic Mutation and Copy Number Alteration Discovery in Cancer by Exome Sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Larson, D.E.; Wilson, R.K. Using VarScan 2 for Germline Variant Calling and Somatic Mutation Detection. In Current Protocols in Bioinformatics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 15.4.1–15.4.17. ISBN 0471250953. [Google Scholar]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public Archive of Relationships among Sequence Variation and Human Phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef]
- Li, Q.; Wang, K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am. J. Hum. Genet. 2017, 100, 267–280. [Google Scholar] [CrossRef]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic Cancer Genetics at High-Resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef] [PubMed]
- Sherry, S.T.; Ward, M.; Sirotkin, K. DbSNP-Database for Single Nucleotide Polymorphisms and Other Classes of Minor Genetic Variation. Genome Res. 1999, 9, 677–679. [Google Scholar]
- Chang, M.T.; Bhattarai, T.S.; Schram, A.M.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; Chakravarty, D.; Phillips, S.; Kandoth, C.; Penson, A.; et al. Accelerating Discovery of Functional Mutant Alleles in Cancer. Cancer Discov. 2018, 8, 174–183. [Google Scholar] [CrossRef]
- Chang, M.T.; Asthana, S.; Gao, S.P.; Lee, B.H.; Chapman, J.S.; Kandoth, C.; Gao, J.J.; Socci, N.D.; Solit, D.B.; Olshen, A.B.; et al. Identifying Recurrent Mutations in Cancer Reveals Widespread Lineage Diversity and Mutational Specificity. Nat. Biotechnol. 2016, 34, 155–163. [Google Scholar] [CrossRef]
- Cotto, K.C.; Wagner, A.H.; Feng, Y.-Y.; Kiwala, S.; Coffman, A.C.; Spies, G.; Wollam, A.; Spies, N.C.; Griffith, O.L.; Griffith, M. DGIdb 3.0: A Redesign and Expansion of the Drug–Gene Interaction Database. Nucleic Acids Res. 2017, 46, 1068–1073. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. DbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jian, X.; Boerwinkle, E. DbNSFP: A Lightweight Database of Human Nonsynonymous SNPs and Their Functional Predictions. Hum. Mutat. 2011, 32, 894–899. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2008; ISBN 3-900051-07-0. [Google Scholar]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open Software Development for Computational Biology and Bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed]
- Hübschmann, D.; Jopp-Saile, L.; Andresen, C.; Krämer, S.; Gu, Z.; Heilig, C.E.; Kreutzfeldt, S.; Teleanu, V.; Fröhling, S.; Eils, R.; et al. Analysis of Mutational Signatures with yet Another Package for Signature Analysis. Genes Chromosom. Cancer 2020, 1–18. [Google Scholar] [CrossRef]
- Boeva, V.; Zinovyev, A.; Bleakley, K.; Vert, J.P.; Janoueix-Lerosey, I.; Delattre, O.; Barillot, E. Control-Free Calling of Copy Number Alterations in Deep-Sequencing Data Using GC-Content Normalization. Bioinformatics 2011, 27, 268–269. [Google Scholar] [CrossRef]
- Boeva, V.; Popova, T.; Bleakley, K.; Chiche, P.; Cappo, J.; Schleiermacher, G.; Janoueix-Lerosey, I.; Delattre, O.; Barillot, E. Control-FREEC: A Tool for Assessing Copy Number and Allelic Content Using next-Generation Sequencing Data. Bioinformatics 2012, 28, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Nicorici, D.; Satalan, M.; Edgren, H.; Kangaspeska, S.; Murumagi, A.; Kallioniemi, O.; Virtanen, S.; Kilkku, O. FusionCatcher-a Tool for Finding Somatic Fusion Genes in Paired-End RNA-Sequencing Data. BioRxiv 2014, 011650. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Kovaleva, V.; Geissler, A.-L.; Lutz, L.; Fritsch, R.; Makowiec, F.; Wiesemann, S.; Hopt, U.T.; Passlick, B.; Werner, M.; Lassmann, S. Spatio-Temporal Mutation Profiles of Case-Matched Colorectal Carcinomas and Their Metastases Reveal Unique de Novo Mutations in Metachronous Lung Metastases by Targeted next Generation Sequencing. Mol. Cancer 2016, 15, 63. [Google Scholar] [CrossRef]
- González-Pérez, A.; López-Bigas, N. Improving the Assessment of the Outcome of Nonsynonymous SNVs with a Consensus Deleteriousness Score, Condel. Am. J. Hum. Genet. 2011, 88, 440–449. [Google Scholar] [CrossRef]
- Chakravarty, D.; Gao, J.; Phillips, S.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 2017, PO.17.00011. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L. Signatures of Mutational Processes in Human Cancer. Nature 2013, 50, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The Repertoire of Mutational Signatures in Human Cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Polak, P.; Kim, J.; Braunstein, L.Z.; Karlic, R.; Haradhavala, N.J.; Tiao, G.; Rosebrock, D.; Livitz, D.; Kübler, K.; Mouw, K.W.; et al. A Mutational Signature Reveals Alterations Underlying Deficient Homologous Recombination Repair in Breast Cancer. Nat. Genet. 2017, 49, 1476–1486. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A. Olaparib Monotherapy in Patients with Advanced Cancer and a Germline BRCA1/2 Mutation. J. Clin. Oncol. 2015, 33, 244. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Porta, N.; Bianchini, D.; McGovern, U.; Elliott, T.; Jones, R.; Syndikus, I.; Ralph, C.; Jain, S.; Varughese, M.; et al. Olaparib in Patients with Metastatic Castration-Resistant Prostate Cancer with DNA Repair Gene Aberrations (TOPARP-B): A Multicentre, Open-Label, Randomised, Phase 2 Trial. Lancet Oncol. 2020, 21, 162–174. [Google Scholar] [CrossRef]
- Gröschel, S.; Hübschmann, D.; Raimondi, F.; Horak, P.; Warsow, G.; Fröhlich, M.; Klink, B.; Gieldon, L.; Hutter, B.; Kleinheinz, K.; et al. Defective Homologous Recombination DNA Repair as Therapeutic Target in Advanced Chordoma. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Leichsenring, J.; Horak, P.; Kreutzfeldt, S.; Heining, C.; Christopoulos, P.; Volckmar, A.; Neumann, O.; Kirchner, M.; Ploeger, C.; Budczies, J. Variant Classification in Precision Oncology. Int. J. Cancer 2019, 145, 2996–3010. [Google Scholar] [CrossRef]
- Watson, S.; Menis, J.; Baldini, C.; Martin-Romano, P.; Michot, J.M.; Hollebecque, A.; Armand, J.P.; Massard, C.; Soria, J.C.; Postel-Vinay, S.; et al. Time to Progression Ratio in Cancer Patients Enrolled in Early Phase Clinical Trials: Time for New Guidelines? Br. J. Cancer 2018, 119, 937–939. [Google Scholar] [CrossRef]
- Radovich, M.; Kiel, P.J.; Nance, S.M.; Niland, E.E.; Parsley, M.E.; Ferguson, M.E.; Jiang, G.; Ammakkanavar, N.R.; Einhorn, L.H.; Cheng, L.; et al. Clinical Benefit of a Precision Medicine Based Approach for Guiding Treatment of Refractory Cancers. Oncotarget 2016, 7, 56491–56500. [Google Scholar] [CrossRef]
- Seeber, A.; Gastl, G.; Ensinger, C.; Spizzo, G.; Willenbacher, W.; Kocher, F.; Leitner, C.; Willenbacher, E.; Amann, A.; Steiner, N.; et al. Treatment of Patients with Refractory Metastatic Cancer According to Molecular Profiling on Tumor Tissue in the Clinical Routine: An Interim-Analysis of the ONCO-T-PROFILE Project. Genes and Cancer 2016, 7, 301–308. [Google Scholar] [CrossRef]
- Birendra, K.C.; Afzal, M.Z.; Sochaki, A.; Wentland, K.A.; Chang, R.; Singh, S.; O’Rourke, T. Tumor Molecular Profiling in the Treatment of Refractory Cancers. J. Exp. Ther. Oncol. 2015, 11, 27–32. [Google Scholar]
- Cirkel, G.A.; Weeber, F.; Bins, S.; Gadellaa-van Hooijdonk, C.G.M.; Van Werkhoven, E.; Willems, S.M.; Van Stralen, M.; Veldhuis, W.B.; Ubink, I.; Steeghs, N.; et al. The Time to Progression Ratio: A New Individualized Volumetric Parameter for the Early Detection of Clinical Benefit of Targeted Therapies. Ann. Oncol. 2016, 27, 1638–1643. [Google Scholar] [CrossRef]
- Mock, A.; Heilig, C.E.; Kreutzfeldt, S.; Huebschmann, D.; Heining, C.; Schröck, E.; Brors, B.; Stenzinger, A.; Jäger, D.; Schlenk, R.; et al. Community-Driven Development of a Modified Progression-Free Survival Ratio for Precision Oncology. ESMO Open 2019, 4, e000583. [Google Scholar] [CrossRef] [PubMed]
- Migliorini, D.; Aguiar, D.; Vargas, M.-I.; Lobrinus, A.; Dietrich, P.-Y. BRAF/MEK Double Blockade in Refractory Anaplastic Pleomorphic Xanthoastrocytoma. Neurology 2017, 88, 1291–1293. [Google Scholar] [CrossRef]
- Chamberlain, M.C. Salvage Therapy with BRAF Inhibitors for Recurrent Pleomorphic Xanthoastrocytoma: A Retrospective Case Series. J. Neurooncol. 2013, 114, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Rivalland, G.; Kao, S.C.-H.; Pavlakis, N.; Hughes, B.G.M.; Thapa, B.; Pal, A.; Walkiewicz, M.; Zimet, A.S.; White, S.; O’Byrne, K.; et al. Outcomes of Anti-PD-1 Therapy in Mesothelioma and Correlation with PD-L1 Expression. J. Clin. Oncol. 2017, 35, 8514. [Google Scholar] [CrossRef]
- Massard, C.; Michiels, S.; Ferté, C.; Le Deley, M.C.; Lacroix, L.; Hollebecque, A.; Verlingue, L.; Ileana, E.; Rosellini, S.; Ammari, S.; et al. High-Throughput Genomics and Clinical Outcome in Hard-to-Treat Advanced Cancers: Results of the MOSCATO 01 Trial. Cancer Discov. 2017, 7, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Van der Velden, D.L.; Van Herpen, C.M.L.; Van Laarhoven, H.W.M.; Smit, E.F.; Groen, H.J.M.; Willems, S.M.; Nederlof, P.M.; Langenberg, M.H.G.; Cuppen, E.; Sleijfer, S.; et al. Molecular Tumor Boards: Current Practice and Future Needs. Ann. Oncol. 2017, 28, 3070–3075. [Google Scholar] [CrossRef]
- Vis, D.J.; Lewin, J.; Liao, R.G.; Mao, M.; Andre, F.; Ward, R.L.; Calvo, F.; Teh, B.T.; Camargo, A.A.; Knoppers, B.M.; et al. Towards a Global Cancer Knowledge Network: Dissecting the Current International Cancer Genomic Sequencing Landscape. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 1145–1151. [Google Scholar] [CrossRef]
- Rieke, D.T.; Lamping, M.; Schuh, M.; Le Tourneau, C.; Basté, N.; Burkard, M.E.; Metzeler, K.H.; Leyvraz, S.; Keilholz, U. Comparison of Treatment Recommendations by Molecular Tumor Boards Worldwide. JCO Precis. Oncol. 2018, 2018, po.18.00098. [Google Scholar] [CrossRef]
- Ray, T. CMS-Proposed Coverage of NGS Cancer Tests Could Lead to Off-Label Scripts, Oncologists Worry. Precision Oncology News. 2018. Available online: https://www.precisiononcologynews.com/molecular-diagnostics/cms-proposed-coverage-ngs-cancer-tests-could-lead-label-scripts-oncologists#.YEJcAcpKiUl (accessed on 10 January 2021).
- Basse, C.; Morel, C.; Alt, M.; Sablin, M.P.; Franck, C.; Pierron, G.; Callens, C.; Melaabi, S.; Masliah-Planchon, J.; Bataillon, G.; et al. Relevance of a Molecular Tumour Board (MTB) for Patients’ Enrolment in Clinical Trials: Experience of the Institut Curie. ESMO Open 2018, 3, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.A.; Kushnir, M.; Mak, G.; Winter, H.; Curiel, T.; Voskoboynik, M.; Moschetta, M.; Rozumna-Martynyuk, N.; Balbi, K.; Bennett, P.; et al. Prospective Analysis of 895 Patients on a UK Genomics Review Board. ESMO Open 2019, 4, e000469. [Google Scholar] [CrossRef]
- Davis, A.A.; Patel, V.G. The Role of PD-L1 Expression as a Predictive Biomarker: An Analysis of All US Food and Drug Administration (FDA) Approvals of Immune Checkpoint Inhibitors. J. Immunother. Cancer 2019, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/ Mismatch Repair–Deficient Cancer: Results from the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S. Mismatch-Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science 2017, 357, eaan6733. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Fakih, M.G.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.; et al. Association of Tumour Mutational Burden with Outcomes in Patients with Select Advanced Solid Tumours Treated with Pembrolizumab in KEYNOTE-158. Ann. Oncol. 2019, 30, v477–v478. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Callahan, M.K.; Awad, M.M.; Calvo, E.; Ascierto, P.A.; Atmaca, A.; Rizvi, N.A.; Hirsch, F.R.; Selvaggi, G.; Szustakowski, J.D.; et al. Tumor Mutational Burden and Efficacy of Nivolumab Monotherapy and in Combination with Ipilimumab in Small-Cell Lung Cancer. Cancer Cell 2018, 33, 853–861.e4. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration (FDA). Grants Regular Approval to Pertuzumab for Adjuvant Treatment of HER2-Positive Breast Cancer; FDA: Rockwell, MD, USA, 2012.



{kind=link}
{kind=link}
{kind=link}
| Characteristic | No. | (%) |
|---|---|---|
| Total | 488 | |
| Sex | ||
| Female | 230 | (47.1) |
| Male | 258 | (52.9) |
| Median Age | 54 | range (1–88) |
| Patients with Solid Tumors: Stage at Presentation | 470 | (96.5) |
| Metastatic Disease | 383 | (81.5) |
| Localized Disease | 86 | (18.3) |
| Complete Remission | 1 | (0.2) |
| Tumor type | ||
| Lower GI tract | 68 | (13.9) |
| Pancreas | 50 | (10.2) |
| Upper GI tract | 42 | (8.6) |
| Central nervous system | 45 | (9.2) |
| Unknown Primary Site | 37 | (7.6) |
| Hepatobiliary | 30 | (6.1) |
| Thyroid | 30 | (6.1) |
| Soft tissue and bone | 37 | (7.6) |
| Gyn (others) | 18 | (3.7) |
| Head and neck | 19 | (3.9) |
| Breast | 21 | (4.3) |
| Urogenital | 12 | (2.5) |
| Ovary | 12 | (2.5) |
| Dermatologic | 18 | (3.7) |
| Hematologic | 17 | (3.5) |
| Lung | 16 | (3.3) |
| Neuroendocrine | 10 | (2.0) |
| Other | 6 | (1.2) |
| Previous Lines of Therapy | 2.05 | (0–12) |
| 0 | 66 | (13.6) |
| 1 | 152 | (31.2) |
| 2 to 3 | 192 | (39.2) |
| >3 | 79 | (16.2) |
| Unknown | 1 | (0.2) |
| Reason for Referral | ||
| Progression to standard of care treatment | 381 | (78.1) |
| Rare Tumor | 51 | (10.5) |
| Young Age | 29 | (5.9) |
| Unknown Primary Site | 20 | (4.1) |
| Other | 7 | (1.4) |
| Recommendations | No. | (%) |
|---|---|---|
| Meetings | 95 | |
| Case Discussions (per patient average) | 499 | (2.5) |
| Recommendations | 1411 | |
| Diagnostic | 762 | (54.0) |
| Treatment | 367 | (26.0) |
| No treatment recommendation | 224 | (15.9) |
| Conditional treatment recommendation | 58 | (4.1) |
| Patients with diagnostic recommendations | 487 | (99.8) |
| Routine molecular analysis | 485 | (99.4) |
| Extended genetic analysis | 183 | (37.5) |
| Rebiopsy | 40 | (5.2) |
| Other | 14 | (1.8) |
| Patients with Treatment recommendations | 264 | |
| Not implemented | 188 | (71.2) |
| Implemented | 76 | (28.8) |
| Stable disease (off-label) | 19 (13) | (25.0) |
| Partial response (off-label) | 17 (12) | (22.4) |
| Complete remission (off-label) | 5 (5) | (6.6) |
| Disease control rate (off-label) | 41 (30) | (8.4) |
| Cancer Type | Rational for Treatment Recommendation | Board Recommendation | EL | L | R | PFS2 (Week) | PFS1 (Week) | PFSr | Outcome |
|---|---|---|---|---|---|---|---|---|---|
| Adrenocortical Carcinoma | Positive IO-Panel (TPS 2%, CPS 3, IC-Score 0). Chromosomal instability (CNV in 16 oncogenes and 28 tumor suppressor genes) | Study: Nivolumab for rare cancers (NCT02832167) | m2C | off | SD | 36 | 37 | 1.0 | SD for 36 weeks |
| Tumor mutational burden high (11.07/Mb) | Pembrolizumab | m2C | off | SD | 53 | 3 | 17.5 | SD for 53 weeks | |
| CRC | Positive IO-Panel (TPS 5%, CPS 10, IC-Score 1). | Combination of atezolizumab and cobimetinib | m2C | off | PR | 76 | 11 | 6.9 | PR for 76 weeks then switch to best supportive care |
| CUP | Signet ring cell carcinoma of unknown primary site with chromosomal Instability incl. a high copy number gain in EGFR (×338). | Combination of cetuximab and FOLFIRI | m2A | off | SD | 25 | 31 | 0.8 | SD for 25 weeks the PD with new ascites. Switch to Combination of paclitaxel and ramucirumab |
| Histiocytosis | Erdheim Chester disease with BRAF-V600E mutation. | BRAF-inhibition with vemurafenib or dabrafenib | m1B | off | PR | >133 | 51 | >2.6 | Initial treatment with vemurafenib. Due to toxicity switch to dabrafenib after 10 weeks. Very good PR in cMRI after 39 weeks therefore switch to maintenance therapy with peg-INF. PR still ongoing |
| Meningioma | Anaplastic meningioma. IHC shows strong staining for somatostatin on 100% of tumor cells | Octreotide | m3 | off | SD | 18 | n.a. | n.a. | SD for 18 weeks then PD with new resection. PFS1 n.a. since no prior systemic therapy received. Only surgery and radiation therapy |
| Mesothelioma | Despite negative IO-Panel (TPS < 1%, CPS 10, IC-Score 1) data show response to checkpoint-Inhibition [62] | Pembrolizumab | m1C | off | SD | 32 | 56 | 0.6 | SD for 32 weeks then patient death |
| PXA | Pleomorphic xanthoastrocytoma with BRAF V600E mutation | BRAF-Inhibition with combination of dabrafenib and trametinib | m1C | off | CR | >64 | n.a. | n.a. | CR for 64 weeks and ongoing. PFS1 n.a. since no prior systemic therapy received. Only surgery and radiation therapy. |
| Prostate | Deleterious BRCA2 mutation. | Platinum-based chemotherapy followed by olaparib | m2A | off | SD | 31 | 7 | 4.5 | SD for 31 weeks then PD |
| Salivary | IHC shows strong androgen receptor staining in 90% of tumor cells. | Combination of degarelix and bicalutamide | m1C | off | CR | 66 | n.a. | n.a. | CR for 66 weeks then PD with new metastasis. PFS1 n.a. after resection (R0) and adjuvant radiochemotherapy |
| Sarcoma | WES shows BRCAness of 29% | Combination of olaparib and trabectedine | m3 | off | SD | 6 | 4 | 1.3 | SD in first staging after 2 weeks. Due to pain initiation of palliative radiation therapy with consecutive esophagitis and pancytopenia resulting in death at 6 weeks |
| Thyroid (anaplastic) | Positive IO-Panel (TPS 5%, CPS 9, IC-Score 1). Study availability | Combination of pembrolizumab and lenvatinib | m2C | off | PR | >53 | 16 | >3.3 | PR for 53 weeks and ongoing |
| (anaplastic) | Positive IO-Panel (TPS > 80%, intratumoral TILs) | Pembrolizumab | m2C | off | CR | 25 | 7 | 3.0 | CR but died due to pulmonary bleeding at 21 weeks |
| (anaplastic) | Positive IO-Panel (TPS 5%, intratumoral TILs). Chromosomal instability with strong amplification of PDGFRA (×28) and PDGFB (×29) | Combination of pembrolizumab and lenvatinib | m2C | off | CR | 83 | 16 | 3.9 | CR for 83 weeks and ongoing. Lenvatinib was discontinued after 52 weeks. |
| (anaplastic) | RNA-Seq shows upregulation of FGFR3 signaling pathway | FGFR3-Inhibition with lenvatinib | m3 | off | SD | 11 | n.a. | n.a. | Good clinical response with regressive local relapse and significant clinical improvement. Therapy was discontinued after 14 weeks due to weight loss, then PD and death. |
| (medullary) | RET Met918Thr Mutation | Selpercatinib | m1A | off | PR | 35 | 15 | 2.3 | Radiologic PR. Serologic decrease of calcitonin from 8554 pg/mL to 12 pg/mL. Response ongoing. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoefflin, R.; Lazarou, A.; Hess, M.E.; Reiser, M.; Wehrle, J.; Metzger, P.; Frey, A.V.; Becker, H.; Aumann, K.; Berner, K.; et al. Transitioning the Molecular Tumor Board from Proof of Concept to Clinical Routine: A German Single-Center Analysis. Cancers 2021, 13, 1151. https://doi.org/10.3390/cancers13051151
Hoefflin R, Lazarou A, Hess ME, Reiser M, Wehrle J, Metzger P, Frey AV, Becker H, Aumann K, Berner K, et al. Transitioning the Molecular Tumor Board from Proof of Concept to Clinical Routine: A German Single-Center Analysis. Cancers. 2021; 13(5):1151. https://doi.org/10.3390/cancers13051151
Chicago/Turabian StyleHoefflin, Rouven, Adriana Lazarou, Maria Elena Hess, Meike Reiser, Julius Wehrle, Patrick Metzger, Anna Verena Frey, Heiko Becker, Konrad Aumann, Kai Berner, and et al. 2021. "Transitioning the Molecular Tumor Board from Proof of Concept to Clinical Routine: A German Single-Center Analysis" Cancers 13, no. 5: 1151. https://doi.org/10.3390/cancers13051151
APA StyleHoefflin, R., Lazarou, A., Hess, M. E., Reiser, M., Wehrle, J., Metzger, P., Frey, A. V., Becker, H., Aumann, K., Berner, K., Boeker, M., Buettner, N., Dierks, C., Duque-Afonso, J., Eisenblaetter, M., Erbes, T., Fritsch, R., Ge, I. X., Geißler, A.-L., ... von Bubnoff, N. (2021). Transitioning the Molecular Tumor Board from Proof of Concept to Clinical Routine: A German Single-Center Analysis. Cancers, 13(5), 1151. https://doi.org/10.3390/cancers13051151