
Application of Multilayer Evidence for Annotation of C-Terminal BRCA2 Variants

 , ,
, ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
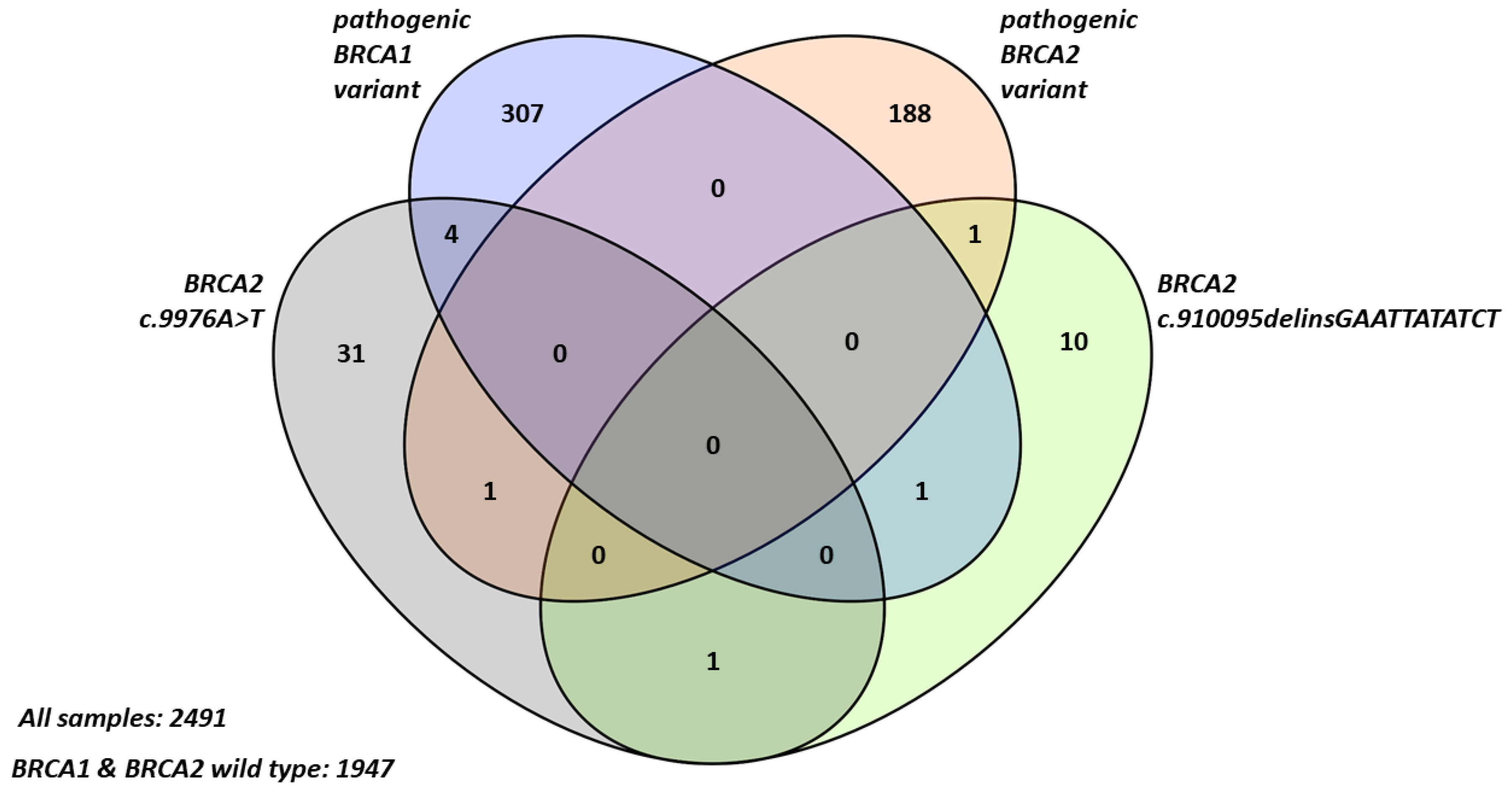
2.1. Frequency and Characteristics of BRCA2 Terminal Stop Codon Variants in Breast Cancer Patients
2.2. Familial Cancer Prevalence in Probands with BRCA2 Terminal Stop Codon Variants
2.2.1. Hereditary Breast and Ovarian Cancer (HBOC) Syndrome-Related Cancers
2.2.2. Prevalence of Other Cancers
2.3. Functional Evaluation of the Potential Pathogenicity of BRCA2 C-Terminal Stop Codon Variants (Loss Of Heterozygosity, Allelic Imbalance, Minor Allele Frequency)
2.4. Re-Analysis of BRCA2 c.9976A>T and c.10095delinsGAATTATATCT Variants by Re-Analysis of All Published Data Where These Variants Were Investigated
3. Discussion
4. Materials and Methods
4.1. Cases: Patients and Relatives
4.2. Nucleic Acid Extraction
4.3. Genetic Analysis (Sequence and Copy Number Analysis by Next Generation Sequencing (NGS) and Multiplex Ligation-Dependent Probe Amplification)
4.4. Sanger Validation and LOH Analysis
4.5. Transcript Allelic Imbalance
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Spurdle, A.B.; Healey, S.; Devereau, A.; Hogervorst, F.B.L.; Monteiro, A.N.A.; Nathanson, K.L.; Radice, P.; Stoppa-Lyonnet, D.; Tavtigian, S.; Wappenschmidt, B.; et al. ENIGMA-Evidence-based network for the interpretation of germline mutant alleles: An international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Hum. Mutat. 2011, 33, 2–7. [Google Scholar] [CrossRef]
- Fradet-Turcotte, A.; Sitz, J.; Grapton, D.; Orthwein, A. BRCA2 functions: From DNA repair to replication fork stabilization. Endocr. Relat. Cancer 2016, 23, T1–T17. [Google Scholar] [CrossRef]
- Davies, O.R.; Pellegrini, L. Interaction with the BRCA2 C terminus protects RAD51–DNA filaments from disassembly by BRC repeats. Nat. Struct. Mol. Biol. 2007, 14, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Esashi, F.; Christ, N.; Gannon, J.; Liu, Y.; Hunt, T.L.; Jasin, M.; West, S.C. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nat. Cell Biol. 2005, 434, 598–604. [Google Scholar] [CrossRef]
- Esashi, F.; Galkin, V.E.; Yu, X.; Egelman, E.H.; West, S.C. Stabilization of RAD51 nucleoprotein filaments by the C-terminal region of BRCA2. Nat. Struct. Mol. Biol. 2007, 14, 468–474. [Google Scholar] [CrossRef]
- Schlacher, K.; Wu, H.; Jasin, M. A Distinct Replication Fork Protection Pathway Connects Fanconi Anemia Tumor Suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef]
- Martin, S.T.; Matsubayashi, H.; Rogers, C.D.; Philips, J.; Couch, F.J.; Brune, K.; Yeo, C.J.; Kern, S.E.; Hruban, R.H.; Goggins, M. Increased prevalence of the BRCA2 polymorphic stop codon K3326X among individuals with familial pancreatic cancer. Oncogene 2005, 24, 3652–3656. [Google Scholar] [CrossRef]
- Thompson, E.R.; Gorringe, K.L.; Rowley, S.M.; Li, N.; McInerny, S.; Wong-Brown, M.W.; Devereux, L.; Li, J.; Trainer, A.H.; Mitchell, G.; et al. Reevaluation of the BRCA2 truncating allele c.9976A > T (p.Lys3326Ter) in a familial breast cancer context. Sci. Rep. 2015, 5, 14800. [Google Scholar] [CrossRef]
- Selvan, M.E.; Klein, R.J.; Gümüş, Z.H. Rare, Pathogenic Germline Variants in Fanconi Anemia Genes Increase Risk for Squamous Lung Cancer. Clin. Cancer Res. 2019, 25, 1517–1525. [Google Scholar] [CrossRef] [PubMed]
- Mazoyer, S.; Dunning, A.M.; Serova, O.; Dearden, J.; Puget, N.; Healey, C.S.; Gayther, S.A.; Mangion, J.; Stratton, M.R.; Lynch, H.T.; et al. A polymorphic stop codon in BRCA2. Nat. Genet. 1996, 14, 253–254. [Google Scholar] [CrossRef]
- Johnson, N.; Fletcher, O.; Palles, C.; Rudd, M.; Webb, E.; Sellick, G.; Silva, I.D.S.; McCormack, V.; Gibson, L.; Fraser, A.; et al. Counting potentially functional variants in BRCA1, BRCA2 and ATM predicts breast cancer susceptibility. Hum. Mol. Genet. 2007, 16, 1051–1057. [Google Scholar] [CrossRef]
- Higgs, J.E.; Harkness, E.F.; Bowers, N.L.; Howard, E.; Wallace, A.J.; Lalloo, F.; Newman, W.G.; Evans, D.G. The BRCA2 Polymorphic Stop Codon: Stuff or Nonsense? J. Med. Genet. 2015, 52, 642–645. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, K.; Hall, P.; Gonzalez-Neira, A.; Ghoussaini, M.; Dennis, J.; Milne, R.L.; Schmidt, M.K.; Chang-Claude, J.; Bojesen, S.E.; Bolla, M.K.; et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat. Genet. 2013, 45, 353–361. [Google Scholar] [CrossRef]
- Meeks, H.D.; Song, H.; Michailidou, K.; Bolla, M.K.; Dennis, J.; Wang, Q.; Barrowdale, D.; Frost, D.; McGuffog, L.; Ellis, S.; et al. BRCA2 Polymorphic Stop Codon K3326X and the Risk of Breast, Prostate, and Ovarian Cancers. J. Natl. Cancer Inst. 2015, 108. [Google Scholar] [CrossRef]
- Stafford, J.L.; Dyson, G.; Levin, N.K.; Chaudhry, S.; Rosati, R.; Kalpage, H.; Wernette, C.; Petrucelli, N.; Simon, M.S.; Tainsky, M.A. Reanalysis of BRCA1/2 negative high risk ovarian cancer patients reveals novel germline risk loci and insights into missing heritability. PLoS ONE 2017, 12, e0178450. [Google Scholar] [CrossRef]
- Delahaye-Sourdeix, M.; Anantharaman, D.; Timofeeva, M.N.; Gaborieau, V.; Chabrier, A.; Vallée, M.P.; Lagiou, P.; Holcátová, I.; Richiardi, L.; Kjaerheim, K.; et al. A Rare Truncating BRCA2 Variant and Genetic Susceptibility to Upper Aerodigestive Tract Cancer. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef]
- Ratajska, M.; Brozek, I.; Senkus-Konefka, E.; Jassem, J.; Stepnowska, M.; Palomba, G.; Pisano, M.; Casula, M.; Palmieri, G.; Borg, A.; et al. BRCA1 and BRCA2 point mutations and large rearrangements in breast and ovarian cancer families in Northern Poland. Oncol. Rep. 2008, 19, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Machackova, E.; Foretova, L.; Lukesova, M.; Vasickova, P.; Navratilova, M.; Coene, I.; Pavlu, H.; Kosinova, V.; Kuklova, J.; Claes, K. Spectrum and characterisation of BRCA1 and BRCA2 deleterious mutations in high-risk Czech patients with breast and/or ovarian cancer. BMC Cancer 2008, 8, 140. [Google Scholar] [CrossRef] [PubMed]
- German Consortium for Hereditary Breast and Ovarian Cancer Comprehensive analysis of 989 patients with breast or ovarian cancer provides BRCA1 and BRCA2 mutation profiles and frequencies for the German population. Int. J. Cancer 2002, 97, 472–480. [CrossRef]
- Cvok, M.L.; Sabol, M.; Musani, V.; Ozretić, P.; Levanat, S. New sequence variants in BRCA1 and BRCA2 genes detected by high-resolution melting analysis in an elderly healthy female population in Croatia. Clin. Chem. Lab. Med. 2008, 46, 1376–1383. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.A.; Greenhalf, B.; Ellis, I.; Sina-Frey, M.; Rieder, H.; Korte, B.; Gerdes, B.; Kress, R.; Ziegler, A.; Raeburn, J.A.; et al. BRCA2 Germline Mutations in Familial Pancreatic Carcinoma. J. Natl. Cancer Inst. 2003, 95, 214–221. [Google Scholar] [CrossRef]
- Koczkowska, M.; Zuk, M.; Gorczynski, A.; Ratajska, M.; Lewandowska, M.; Biernat, W.; Limon, J.; Wasag, B. Detection of somatic BRCA 1/2 mutations in ovarian cancer—Next-generation sequencing analysis of 100 cases. Cancer Med. 2016, 5, 1640–1646. [Google Scholar] [CrossRef]
- Caminsky, N.G.; Mucaki, E.J.; Perri, A.M.; Lu, R.; Knoll, J.H.M.; Rogan, P.K. Prioritizing Variants in Complete Hereditary Breast and Ovarian Cancer Genes in Patients Lacking Known BRCA Mutations. Hum. Mutat. 2016, 37, 640–652. [Google Scholar] [CrossRef] [PubMed]
- Thomassen, M.; Hansen, T.V.O.; Borg, A.; Lianee, H.T.; Wikman, F.; Pedersen, I.S.; Bisgaard, M.L.; Nielsen, F.C.; Kruse, T.A.; Gerdes, A.-M. BRCA1 and BRCA2 mutations in Danish families with hereditary breast and/or ovarian cancer. Acta Oncol. 2008, 47, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Meisel, C.; Sadowski, C.E.; Kohlstedt, D.; Keller, K.; Stäritz, F.; Grübling, N.; Becker, K.; Mackenroth, L.; Rump, A.; Schröck, E.; et al. Spectrum of genetic variants of BRCA1 and BRCA2 in a German single center study. Arch. Gynecol. Obstet. 2017, 295, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Akbari, M.R.; Malekzadeh, R.; Nasrollahzadeh, D.; Amanian, D.; Islami, F.; Li, S.; Zandvakili, I.; Shakeri, R.; Sotoudeh, M.; Aghcheli, K.; et al. Germline BRCA2 mutations and the risk of esophageal squamous cell carcinoma. Oncogene 2007, 27, 1290–1296. [Google Scholar] [CrossRef][Green Version]
- Borg, Å.; Haile, R.W.; Malone, K.E.; Capanu, M.; Diep, A.; Törngren, T.; Teraoka, S.; Begg, C.B.; Thomas, D.C.; Concannon, P.; et al. Characterization of BRCA1 and BRCA2 deleterious mutations and variants of unknown clinical significance in unilateral and bilateral breast cancer: The WECARE study. Hum. Mutat. 2010, 31, E1200–E1240. [Google Scholar] [CrossRef]
- Krainer, M.; Silva-Arrieta, S.; Fitzgerald, M.G.; Shimada, A.; Ishioka, C.; Kanamaru, R.; Macdonald, D.J.; Unsal, H.; Finkelstein, D.M.; Bowcock, A.; et al. Differential Contributions of BRCA1 and BRCA2 to Early-Onset Breast Cancer. N. Engl. J. Med. 1997, 336, 1416–1422. [Google Scholar] [CrossRef]
- Malone, K.E.; Daling, J.R.; Neal, C.; Suter, N.M.; O’Brien, C.; Cushing-Haugen, K.; Jonasdottir, T.J.; Thompson, J.D.; Ostrander, E.A. Frequency of BRCA1/BRCA2 mutations in a population-based sample of young breast carcinoma cases. Cancer 2000, 88, 1393–1402. [Google Scholar] [CrossRef]
- Bergthorsson, J.; Ejlertsen, B.; Olsen, J.; Borg, Å.; Nielsen, K.; Barkardottir, R.; Klausen, S.; Mouridsen, H.; Winther, K.; Fenger, K.; et al. BRCA1 and BRCA2 mutation status and cancer family history of Danish women affected with multifocal or bilateral breast cancer at a young age. J. Med. Genet. 2001, 38, 361–368. [Google Scholar] [CrossRef][Green Version]
- Hamann, U.; Liu, X.; Bungardt, N.; Ulmer, H.U.; Bastert, G.; Sinn, H.-P. Similar contributions of BRCA1 and BRCA2 germline mutations to early-onset breast cancer in Germany. Eur. J. Hum. Genet. 2003, 11, 464–467. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Musolino, A.; Bella, M.A.; Bortesi, B.; Michiara, M.; Naldi, N.; Zanelli, P.; Capelletti, M.; Pezzuolo, D.; Camisa, R.; Savi, M.; et al. BRCA mutations, molecular markers, and clinical variables in early-onset breast cancer: A population-based study. Breast 2007, 16, 280–292. [Google Scholar] [CrossRef]
- Juwle, A.; Saranath, D. BRCA1/BRCA2 gene mutations/SNPs and BRCA1 haplotypes in early-onset breast cancer patients of Indian ethnicity. Med. Oncol. 2012, 29, 3272–3281. [Google Scholar] [CrossRef]
- Claes, K.; Poppe, B.; Machackova, E.; Coene, I.; Foretova, L.; de Paepe, A.; Messiaen, L. Differentiating pathogenic mutations from polymorphic alterations in the splice sites of BRCA1 and BRCA2. Genes Chromosom. Cancer 2003, 37, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Hadjisavvas, A.; Charalambous, E.; Adamou, A.; Christodoulou, C.G.; Kyriacou, K. BRCA2 germline mutations in Cypriot patients with familial breast/ovarian cancer. Hum. Mutat. 2003, 21, 171. [Google Scholar] [CrossRef]
- Giannini, G.; Capalbo, C.; Ristori, E.; Ricevuto, E.; Sidoni, T.; Buffone, A.; Cortesi, E.; Marchetti, P.; Scambia, G.; Tomao, S.; et al. Novel BRCA1 and BRCA2 germline mutations and assessment of mutation spectrum and prevalence in Italian breast and/or ovarian cancer families. Breast Cancer Res. Treat. 2006, 100, 83–91. [Google Scholar] [CrossRef]
- Simard, J.-C.; Dumont, M.; Moisan, A.-M.; Gaborieau, V.; Vézina, H.; Durocher, F.; Chiquette, J.; Plante, M.; Avard, D.; Bessette, P.; et al. Evaluation of BRCA1 and BRCA2 mutation prevalence, risk prediction models and a multistep testing approach in French-Canadian families with high risk of breast and ovarian cancer. J. Med. Genet. 2006, 44, 107–121. [Google Scholar] [CrossRef]
- Beristain, E.; Martínez-Bouzas, C.; Guerra, I.; Viguera, N.; Moreno, J.; Ibáñez, E.; Diez, J.; Rodríguez, F.; Mallabiabarrena, G.; Luján, S.; et al. Differences in the frequency and distribution of BRCA1 and BRCA2 mutations in breast/ovarian cancer cases from the Basque country with respect to the Spanish population: Implications for genetic counselling. Breast Cancer Res. Treat. 2007, 106, 255–262. [Google Scholar] [CrossRef]
- Kuusisto, K.M.; Bebel, A.; Vihinen, M.; Schleutker, J.; Sallinen, S.-L. Screening for BRCA1, BRCA2, CHEK2, PALB2, BRIP1, RAD50, and CDH1 mutations in high-risk Finnish BRCA1/2-founder mutation-negative breast and/or ovarian cancer individuals. Breast Cancer Res. 2011, 13, R20. [Google Scholar] [CrossRef]
- Cherbal, F.; Salhi, N.; Bakour, R.; Adane, S.; Boualga, K.; Maillet, P. BRCA1 and BRCA2 Unclassified Variants and Missense Polymorphisms in Algerian Breast/Ovarian Cancer Families. Dis. Markers 2012, 32, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Jalkh, N.; Nassar-Slaba, J.; Chouery, E.; Salem, N.; Uhrchammer, N.; Golmard, L.; Stoppa-Lyonnet, D.; Bignon, Y.-J.; Mégarbané, A. Prevalance of BRCA1 and BRCA2 mutations in familial breast cancer patients in Lebanon. Hered. Cancer Clin. Pr. 2012, 10, 7. [Google Scholar] [CrossRef]
- Dobričić, J.; Krivokuća, A.; Brotto, K.; Mališić, E.; Radulović, S.; Branković-Magić, M. Serbian high-risk families: Extensive results on BRCA mutation spectra and frequency. J. Hum. Genet. 2013, 58, 501–507. [Google Scholar] [CrossRef][Green Version]
- Hilton, J.L.; Geisler, J.P.; Rathe, J.A.; Hattermann-Zogg, M.A.; Deyoung, B.; Buller, R.E. Inactivation of BRCA1 and BRCA2 in Ovarian Cancer. J. Natl. Cancer Inst. 2002, 94, 1396–1406. [Google Scholar] [CrossRef]
- Haraldsson, K.; Loman, N.; Zhang, Q.X.; Johannsson, O.; Olsson, H.; Borg, A. BRCA2 germ-line mutations are frequent in male breast cancer patients without a family history of the disease. Cancer Res. 1998, 58, 1367–1371. [Google Scholar] [PubMed]
- Ding, Y.C.; Steele, L.; Kuan, C.-J.; Greilac, S.; Neuhausen, S.L. Mutations in BRCA2 and PALB2 in male breast cancer cases from the United States. Breast Cancer Res. Treat. 2010, 126, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.G.R.; Bulman, M.; Young, K.; Howard, E.; Bayliss, S.; Wallace, A.; Lalloo, F. BRCA1/2 mutation analysis in male breast cancer families from North West England. Fam. Cancer 2007, 7, 113–117. [Google Scholar] [CrossRef]
- Obazee, O.; Archibugi, L.; Andriulli, A.; Soucek, P.; Małecka-Panas, E.; Ivanauskas, A.; Johnson, T.; Gazouli, M.; Pausch, T.; Lawlor, R.T.; et al. Germline BRCA2 K3326X and CHEK2 I157T mutations increase risk for sporadic pancreatic ductal adenocarcinoma. Int. J. Cancer 2019, 145, 686–693. [Google Scholar] [CrossRef]
- Wang, Y.; McKay, J.D.; Rafnar, T.; Wang, Z.; Timofeeva, M.N.; Broderick, P.; Zong, X.; Laplana, M.; Wei, Y.; Han, Y.; et al. Rare variants of large effect in BRCA2 and CHEK2 affect risk of lung cancer. Nat. Genet. 2014, 46, 736–741. [Google Scholar] [CrossRef]
- Rudd, M.F.; Webb, E.L.; Matakidou, A.; Sellick, G.S.; Williams, R.D.; Bridle, H.; Eisen, T.; Houlston, R.S. Variants in the GH-IGF axis confer susceptibilityto lung cancer. Genome Res. 2006, 16, 693–701. [Google Scholar] [CrossRef]
- Rafnar, T.; Sigurjonsdottir, G.R.; Stacey, S.N.; Halldorsson, G.; Sulem, P.; Pardo, L.M.; Helgason, H.; Sigurdsson, S.T.; Gudjonsson, T.; Tryggvadottir, L.; et al. Association of BRCA2 K3326* With Small Cell Lung Cancer and Squamous Cell Cancer of the Skin. J. Natl. Cancer Inst. 2018, 110, 967–974. [Google Scholar] [CrossRef]
- Ge, Y.; Wang, Y.; Shao, W.; Jin, J.; Du, M.; Ma, G.; Chu, H.; Wang, M.; Zhang, Z. Rare variants in BRCA2 and CHEK2 are associated with the risk of urinary tract cancers. Sci. Rep. 2016, 6, 33542. [Google Scholar] [CrossRef]
- Tuominen, R.; Engström, P.G.; Helgadottir, H.; Eriksson, H.; Unneberg, P.; Kjellqvist, S.; Yang, M.; Lindén, D.; Edsgärd, D.; Hansson, J.; et al. The role of germline alterations in the DNA damage response genes BRIP1 and BRCA2 in melanoma susceptibility. Genes Chromosom. Cancer 2016, 55, 601–611. [Google Scholar] [CrossRef]
- Palmirotta, R.; Lovero, D.; Stucci, L.S.; Silvestris, E.; Quaresmini, D.; Cardascia, A.; Silvestris, F. Double Heterozygosity for BRCA1 Pathogenic Variant and BRCA2 Polymorphic Stop Codon K3326X: A Case Report in a Southern Italian Family. Int. J. Mol. Sci. 2018, 19, 285. [Google Scholar] [CrossRef]
- Heidemann, S.; Fischer, C.; Engel, C.; Fischer, B.; Harder, L.; Schlegelberger, B.; Niederacher, D.; Goecke, T.O.; Doelken, S.C.; Dikow, N.; et al. Double heterozygosity for mutations in BRCA1 and BRCA2 in German breast cancer patients: Implications on test strategies and clinical management. Breast Cancer Res. Treat. 2012, 134, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Hinson, S.R.; Ohashi, A.; Farrugia, D.; Wendt, P.; Tavtigian, S.V.; Deffenbaugh, A.; Goldgar, D.; Couch, F.J. Functional evaluation and cancer risk assessment of BRCA2 unclassified variants. Cancer Res. 2005, 65, 417–426. [Google Scholar] [PubMed]
- Kuznetsov, S.G.; Liu, P.; Sharan, S.K. Mouse embryonic stem cell–based functional assay to evaluate mutations in BRCA2. Nat. Med. 2008, 14, 875–881. [Google Scholar] [CrossRef]
- Easton, D.F. Breast Cancer Linkage Consortium Cancer Risks in BRCA2 Mutation Carriers. J. Natl. Cancer Inst. 1999, 91, 1310–1316. [Google Scholar] [CrossRef]
- Van Asperen, C.J.; Brohet, R.M.; Meijers-Heijboer, E.J.; Hoogerbrugge, N.; Verhoef, S.; Vasen, H.F.A.; Ausems, M.G.E.M.; Menko, F.H.; Garcia, E.B.G.; Klijn, J.G.M.; et al. Cancer risks in BRCA2 families: Estimates for sites other than breast and ovary. J. Med. Genet. 2005, 42, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, A.C.; Sinilnikova, O.M.; Simard, J.; Léoné, M.; Dumont, M.; Neuhausen, S.L.; Struewing, J.P.; Stoppa-Lyonnet, D.; Barjhoux, L.; Hughes, D.J.; et al. RAD51 135G→C Modifies Breast Cancer Risk among BRCA2 Mutation Carriers: Results from a Combined Analysis of 19 Studies. Am. J. Hum. Genet. 2007, 81, 1186–1200. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, T.R.; Friebel, T.M.; Friedman, E.; Hamann, U.; Huo, D.; Kwong, A.; Olah, E.; Olopade, O.I.; Solano, A.R.; Teo, S.-H.; et al. Mutational spectrum in a worldwide study of 29,700 families with BRCA 1or BRCA2 mutations. Hum. Mutat. 2018, 39, 593–620. [Google Scholar] [CrossRef] [PubMed]
- Bozsik, A.; Pócza, T.; Papp, J.; Vaszkó, T.; Butz, H.; Patócs, A.; Oláh, E. Complex Characterization of Germline Large Genomic Rearrangements of the BRCA1 and BRCA2 Genes in High-Risk Breast Cancer Patients—Novel Variants from a Large National Center. Int. J. Mol. Sci. 2020, 21, 4650. [Google Scholar] [CrossRef] [PubMed]
- Baughan, S.; Tainsky, M. K3326X and Other C-Terminal BRCA2 Variants Implicated in Hereditary Cancer Syndromes: A Review. Cancers 2021, 13, 447. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Proband | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| # | Gender | BRCA2 C-Terminal Variant | Pathogenic BRCA1/BRCA2 Variant | Disease: | 1st Breast Cancer | 2nd Breast Cancer | Ovarian Cancer | ||||||
| Age of onset | ER | PR | HER2 | Ki67 (%) | Hist | Age | Age | Hist | |||||
| 1 | F | c.9976A>T | — | sol | 43 | pos | pos | neg | 25 | DUC | - | - | - |
| 2 | F | c.9976A>T | — | sol | 55 | pos | neg | neg | n.a. | DUC | - | - | - |
| 3 | F | c.9976A>T | — | sol | 48 | pos | pos | neg | n.a. | DUC | - | - | - |
| 4 | F | c.9976A>T | — | sol | 47 | pos | pos | neg | 5 | DUC | - | - | - |
| 5 | F | c.9976A>T | — | sol | 31 | pos | pos | neg | 40 | DUC | - | - | - |
| 6 | F | c.9976A>T | — | sol | 68 | pos | pos | neg | 1 | DUC | - | - | - |
| 7 | F | c.9976A>T | — | sol | 57 | pos | pos | neg | 10 | DUC | - | - | - |
| 8 | F | c.9976A>T | — | sol | 36 | neg | neg | neg | 40 | DUC | - | - | - |
| 9 | F | c.9976A>T | — | sol | 42 | pos | pos | neg | 70 | LOB | - | - | - |
| 10 | F | c.9976A>T | — | sol | 46 | pos | pos | neg | n.a. | DUC | - | - | - |
| 11 | F | c.9976A>T | — | sol | 41 | pos | pos | pos | 10 | DUC | - | - | - |
| 12 | F | c.9976A>T | — | sol | 34 | pos | pos | pos | 67 | DUC | - | - | - |
| 13 | F | c.9976A>T | — | sol | 39 | neg | neg | neg | 20 | DUC | - | - | - |
| 14 | F | c.9976A>T | — | sol | 46 | pos | pos | neg | 5 | DUC | - | - | - |
| 15 | F | c.9976A>T | — | sol | 41 | pos | neg | neg | 25 | DUC | - | - | - |
| 16 | F | c.9976A>T | — | sol | 46 | pos | pos | neg | n.a. | DUC | - | - | - |
| 17 | F | c.9976A>T | — | sol | 44 | pos | pos | neg | n.a. | DUC | - | - | - |
| 18 | F | c.9976A>T | — | sol | 38 | pos | pos | neg | 25 | DUC | - | - | - |
| 19 | F | c.9976A>T | — | multi | 33 | pos | pos | neg | 50 | DUC | - | 33 | adenocar-cinoma |
| 20 | F | c.9976A>T | — | sol | 33 | pos | pos | neg | 25 | DUC | - | - | - |
| 21 | F | c.9976A>T | — | sol | 61 | neg | neg | neg | 25 | DUC | - | - | - |
| 22 | F | c.9976A>T | — | sol | 46 | neg | neg | poz | 10 | DUC | - | - | - |
| 23 | F | c.9976A>T | — | sol | 40 | pos | neg | neg | 70 | DUC | - | - | - |
| 24 | F | c.9976A>T | — | multi | 48 | pos | n.a. | neg | n.a. | DUC | 48 | - | - |
| 25 | F | c.9976A>T | — | sol | 26 | pos | pos | pos | 35 | DUC | - | - | - |
| 26 | F | c.9976A>T | — | sol | 41 | pos | pos | neg | n.a. | DUC | - | - | - |
| 27 | F | c.9976A>T | — | sol | 44 | pos | pos | neg | 25 | DUC | - | - | - |
| 28 | F | c.9976A>T | — | sol | 33 | pos | pos | neg | 35 | DUC | - | - | - |
| 29 | F | c.9976A>T | — | sol | 43 | pos | pos | neg | 7 | DUC | - | - | - |
| 30 | M | c.9976A>T | — | sol | 27 | n.a. | n.a. | n.a. | n.a. | DUC | - | - | - |
| 31 | F | c.9976A>T | — | sol | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | - | 45 | cystadenoc arcinoma mucinosum |
| 32 | F | c.10095delinsGAATTATATCT | — | sol | 44 | pos | pos | neg | 10 | DUC | - | - | - |
| 33 | F | c.10095delinsGAATTATATCT | — | sol | 36 | neg | neg | neg | 30 | DUC | - | - | - |
| 34 | F | c.10095delinsGAATTATATCT | — | sol | 55 | neg | neg | neg | 50 | DUC | - | - | - |
| 35 | F | c.10095delinsGAATTATATCT | — | sol | 55 | n.a. | n.a. | n.a. | n.a. | n.a. | - | - | - |
| 36 | F | c.10095delinsGAATTATATCT | — | sol | 40 | pos | pos | neg | 15 | DUC | - | - | - |
| 37 | F | c.10095delinsGAATTATATCT | — | sol | 27 | pos | pos | neg | n.a. | LOB | - | - | - |
| 38 | F | c.10095delinsGAATTATATCT | — | sol | 39 | pos | pos | neg | 10 | LOB | - | - | - |
| 39 | F | c.10095delinsGAATTATATCT | — | sol | 45 | pos | pos | neg | 1 | DUC | - | - | - |
| 40 | F | c.10095delinsGAATTATATCT | — | sol | 38 | pos | pos | neg | 2 | DUC | - | - | - |
| 41 | F | c.10095delinsGAATTATATCT | — | multi | 55 | pos | pos | neg | 20 | LOB | 58 | - | - |
| 42 | F | c.9976A>T & c.10095delinsGAATTATATCT | — | sol | 41 | pos | pos | neg | n.a. | DUC | - | - | - |
| 43 | F | c.10095delinsGAATTATATCT | BRCA1 c.5251C>T (p.Arg1751*) | sol | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | - | 36 | high grade serosus carcinoma |
| 44 | F | c.9976A>T | BRCA1 c.1687C>T (p.Gln563*) | sol | 40 | pos | pos | pos | 50 | DUC | - | - | - |
| 45 | F | c.9976A>T | BRCA1 c.68_69delAG (p.Glu23Valfs*) | sol | 54 | neg | neg | neg | 85 | DUC | - | - | - |
| 46 | F | c.9976A>T | BRCA1 c.3018_3021del4 (p.His1006Glnfs*17) | sol | 38 | n.a. | n.a. | n.a. | n.a. | n.a. | - | - | - |
| 47 | F | c.9976A>T | BRCA1 c.181T>G (p.Cys61Gly) | sol | 49 | neg | neg | neg | 90 | DUC | - | - | - |
| 48 | M | c.9976A>T | BRCA2 c.8378G>A (p.Gly2793Glu) | sol | 79 | pos | pos | neg | 25 | DUC | - | - | - |
| 49 | F | c.10095delinsGAATTATATCT | BRCA2 c.7595_7596insTT (p.Ala2534Leufs*18) | sol | 38 | poz | poz | n.a. | 5 | DUC | - | - | - |
| Clinicopathological Parameter | BRCA1/2 Wild Type | Pathogenic BRCA1 Variant | Pathogenic BRCA2 Variant | BRCA2 c.9976A>T | BRCA2 c.10095delins GAATTATATCT | Pathogenic BRCA1 + BRCA2 c.9976A>T |
|---|---|---|---|---|---|---|
| Number of probands (n) | 1947 | 307 | 188 | 31 | 10 | 4 |
| Age at disease onset (years): | ||||||
| Breast cancer (mean ± SD) | 43.38 ± 9.33 | 39.48 ± 8.93 a | 41.93 ± 8.63 b | 43.1 ± 9.10 b | 43.4 ± 9.37 | 42.25 ± 7.54 |
| Ovarian cancer (mean ± SD) | 49.07 ± 13.85 | 48.41 ± 7.99 | 55.3 ± 10.03 | 45.00 | — | — |
| Male breast cancer (mean ± SD) | 59.74 ± 11.08 | 47.00 | 62.87 ± 8.82 | 27.00 | — | — |
| Multiplex tumors from all patients (proportion (mpx cases/all)) | 0.08 (156/1947) | 0.2 (60/307) a | 0.18 (34/188) a | 0.06 (2/31) | 0.10 (1/10) | 0.00 (0/4) |
| Tumor characteristics: | ||||||
| Ki67 of breast cancer (mean ± SD) | 30 ± 25 | 58 ± 24 a | 30 ± 24 | 28 ± 21 | 17 ± 16 | 75 ± 22 |
| ER pos proportion (95%CI) | 0.70 (0.68–0.72) | 0.19 (0.15–0.24) a | 0.78 (0.72–0.84) b | 0.86 (0.69–0.95) b | 0.77 (0.44–0.95) b | 0.33 (0.05–0.79) |
| PR pos proportion (95%CI) | 0.64 (0.62–0.66) | 0.15 (0.11–0.20) a | 0.67 (0.60–0.74) b | 0.75 (0.56–0.87) | 0.77 (0.44–0.94) | 0.33 (0.05–0.79) |
| HER2 pos proportion (95%CI) | 0.24 (0.22–0.26) | 0.06 (0.04–0.11) a | 0.10 (0.06–0.16) a | 0.13 (0.05–0.31) | 0 (0.00–0.34) | 0.33 (0.05–0.79) |
| TNBC proportion (95%CI) | 0.20 (0.19–0.23) | 0.75 (0.70–0.80) a | 0.19 (0.14–0.26) b | 0.10 (0.03–0.27) b | 0.22 (0.05–0.55) | 0.66 (0.20–0.94) |
| Tumor prevalence in families (proportion (95%CI)) | ||||||
| Breast cancer <50 years of age in the family | 0.10 (0.09–0.11) | 0.21 (0.16–0.25) a | 0.24 (0.18–0.30) a | 0.16 (0.06–0.33) | 0.10 (0.00–0.42) | 0 (0.00–0.54) |
| Breast cancer at any age in the family | 0.42 (0.40–0.44) | 0.61 (0.56–0.66) a | 0.60 (0.53–0.67) a | 0.38 (0.24–0.56) b,c | 0.5 (0.24–0.76) | 0.25 (0.03–0.71) |
| Ovarian cancer at any age in the family | 0.06 (0.05–0.08) | 0.21 (0.17–0.26) a | 0.08 (0.05–0.13) b | 0.06 (0.01–0.21) | 0 (0.00–0.32) | 0.25 (0.03–0.71) |
| Breast and/or ovarian cancer at any age in the family | 0.47 (0.44–0.49) | 0.69 (0.63–0.74) a | 0.65 (0.58–0.72) a | 0.45 (0.29–0.62) b,c | 0.5 (0.23–0.76) | 0.5 (0.15–0.85) |
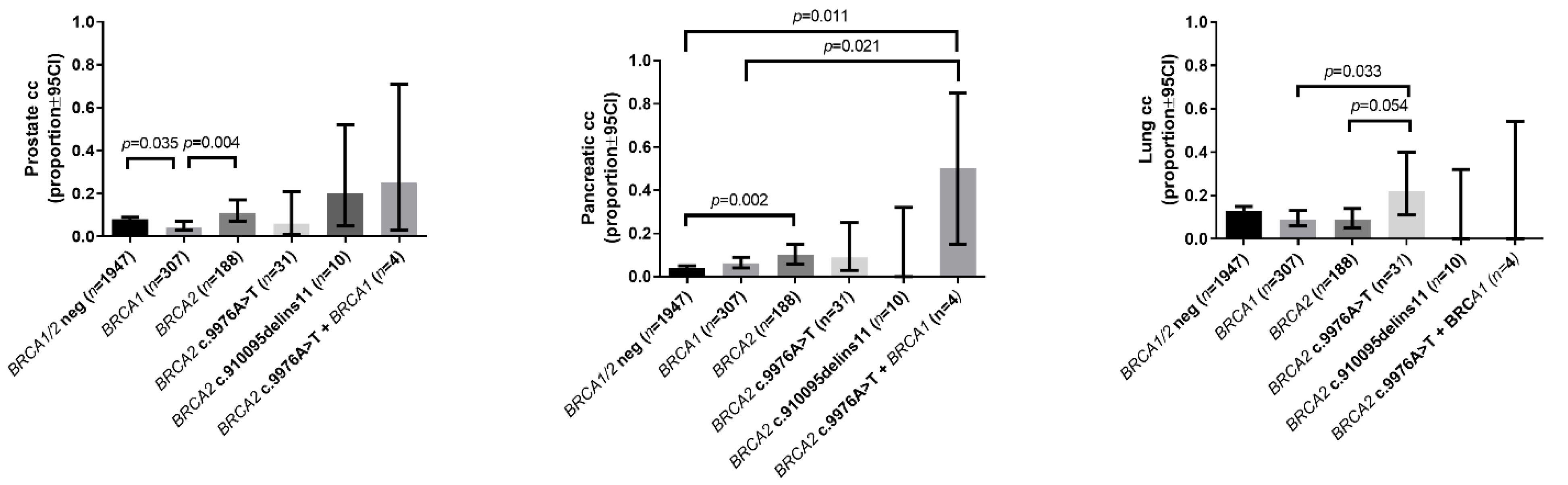
| Prostate cancer in the family | 0.08 (0.07–0.09) | 0.04 (0.03–0.07) a | 0.11 (0.07–0.17) b | 0.06 (0.01–0.21) | 0.20 (0.05–0.52) | 0.25 (0.03–0.71) |
| Pancreatic cancer in the family | 0.04 (0.03–0.05) | 0.06 (0.04–0.09) | 0.10 (0.06–0.15) a | 0.09 (0.03–0.25) | 0 (0.00–0.32) | 0.5 (0.15–0.85) a,b |
| Lung cancer in the family | 0.13 (0.12–0.15) | 0.09 (0.06–0.13) | 0.09 (0.05–0.14) | 0.22 (0.11–0.40) b,c | 0 (0.00–0.32) | 0 (0.00–0.54) |
| Skin cancer in the family | 0.04 (0.03–0.05) | 0.03 (0.02–0.06) | 0.03 (0.01–0.07) | 0.09 (0.02–0.25) | 0.10 (0.00–0.42) | 0 (0.00–0.54) |
| Head and neck cancer in the family | 0.05 (0.04–0.06) | 0.05 (0.03–0.08) | 0.08 (0.05–0.13) | 0 (0.00–0.13) | 0 (0.00–0.32) | 0 (0.00–0.54) |
| Hepatobiliary cancer in the family | 0.03 (0.02–0.04) | 0.03 (0.01–0.05) | 0.04 (0.02–0.08) | 0 (0.00–0.13) | 0 (0.00–0.32) | 0.25 (0.03–0.71) |
| Gastric cancer in the family | 0.08 (0.07–0.09) | 0.08 (0.06–0.12) | 0.03 (0.01–0.06) | 0.09 (0.02–0.25) | 0.10 (0.00–0.42) | 0 (0.00–0.54) |
| Cancer Type | Reference | Number of Probands Screened (Germline) | Number of Patients Carrying BRCA2 c.10095delinsGAATTATATCT Variant | Allelic Frequency | Clinical Interpretation |
|---|---|---|---|---|---|
| breast, ovarian cancer | Meindl et al. 2002 [20] | 989 | 3 | 0.00303 | VUS |
| breast, ovarian cancer | Ratajska et al. 2008 [18] | 64 | 2 | 0.03125 | VUS |
| breast, ovarian cancer | Machackova et al. 2008 [19] | 1010 | 1 | 0.00099 | VUS |
| breast, ovarian cancer | Cvok et al. 2008 [21] | 115 | 1 | 0.00869 | clinically not important |
| breast, ovarian cancer | Thomassen et al. 2008 ** [25] | na | 1 | na | not interpreted |
| breast, ovarian cancer | Meisel et al. 2017 **,† [26] | 523 | 3 | 0.00573 | VUS |
| ovarian cancer | Koczkowska et al. 2016 [23] | 22* | 1 | na * | benign |
| pancreatic cancer | Hahn et al. 2013 [22] | 26 | 1 | 0.03846 | VUS |
| Cancer Type | Reference | Number of Probands Screened | Number of Patients Carrying BRCA2 c.9976A>T Variant | Allelic Frequency | Odds Ratio (OR) (Patients vs. Controls) (Confidence Intervals) |
|---|---|---|---|---|---|
| breast, ovarian cancer | current study | 2138 | 46 | 0.01485 | na |
| breast cancer | Mazoyer et al. 1996 [11] | 513 | 11 | 0.01267 | OR: 1.01 (0.41–2.48) |
| breast cancer | Johnson et al. 2007 [12] | 473 | 11 | 0.011628 | OR: 1.16 (0.79–1.63) |
| breast cancer | Borg et al. 2010 [28] | 2103 | 40 | 0.00951 | na |
| breast cancer | Michailidou et al. 2013 [14] | 10052 | 80 | 0.008 | RR: 1.39 1.39 (1.13–1.71) |
| breast cancer | Thompson et al. 2015 [9] | 2634 | 66 | 0.01252 | OR: 1.53 (1.00–2.34); (p = 0.047) |
| breast cancer | Meeks et al. 2016 [15] | 41081 | 852 | 0.01036 | OR: 1.28 (1.17–1.40); (p = 5.86 × 10−6) |
| breast cancer, early onset | Krainer et al.l. 1997 [29] | 73 | 1 | 0.00684 | na |
| breast cancer, early onset | Malone et al. 2000 [30] | 386 | 2 | 0.00259 | na |
| breast cancer, early onset | Bergthorsson et al. 2001 [31] | 119 | 1 | 0.00420 | na |
| breast cancer, early onset | Hamann et al. 2003 [32] | 91 | 1 | 0.00549 | na |
| breast cancer, early onset | Musolino et al. 2007 [33] | 66 | 3 | 0.02272 | na |
| breast cancer, early onset | Juwle et al. 2012 [34] | 50 | 2 | 0.01 | na |
| breast cancer, early onset | Juwle et al. 2012 [34] | 50 | 2 | 0.02 | na |
| breast, ovarian cancer | Claes et al. 2003 [35] | 249 | 8 | 0.01606 | na |
| breast, ovarian cancer | Hadjisavvas et al. 2003 [36] | 26 | 1 | 0.01923 | na |
| breast, ovarian cancer | Giannini et al. 2006 [37] | 73 | 1 | 0.00684 | na |
| breast, ovarian cancer | Simard et al. 2007 [38] | 143 | 2 | 0.00699 | na |
| breast, ovarian cancer | Beristain et al. 2007 [39] | 236 | 1 | 0.00211 | na |
| breast, ovarian cancer | Ratajska et al. 2008 [18] | 64 | 0 | 0 | na |
| breast, ovarian cancer | Kuusisto et al. 2011 [40] | 82 | 1 | 0.012 | OR: 0.41 (0.05–3.24); (p = 0.702) |
| breast, ovarian cancer | Cherbal et al. 2012 [41] | 79 | 1 | 0.00632 | na |
| breast, ovarian cancer | Jalkh et al. 2012 [42] | 72 | 1 | 0.00694 | na |
| breast, ovarian cancer | Dobričić et al. 2013 [43] | 71 | 1 | 0.00704 | na |
| breast, ovarian cancer | Higgs et al. 2015-cohort 1 [13] | 1850 | 23 | 0.00621 | |
| breast, ovarian cancer | Higgs et al. 2015-cohort 2 [13] | 1576 | not reported | na | na |
| breast, ovarian cancer | Higgs et al. 2015-cohort 3 [13] | 1395 | 43 | 0.01541 | |
| ovarian cancer | Mazoyer et al. 1996 [11] | 361 | 7 | 0.00969 | na |
| ovarian cancer | Hilton et al. 2002 [44] | 92 | 1 | 0.00543 | na |
| ovarian cancer | Meeks et al. 2016 [15] | 14514 | 311 | 0.01071 | OR: 1.26 (1.10–1.43); (p = 3.84 ×10−3) |
| ovarian cancer | Stafford et al. 2017 [16] | 48 | 4* | 0.00416 | OR: 4.95 (p = 0.01) |
| male breast | Haraldsson et al. 1998 [45] | 34 | 1 | 0.01470 | na |
| male breast | Ding et al. 2011 [46] | 115 | 2 | 0.00869 | na |
| male breast | Evans et al. 2008 [47] | 64 | 1 | 0.00781 | na |
| familial pancreatic cancer | Martin et al. 2005 [8] | 144 | 8 | 0.02777 | OR: 4.24 (p < 0.05) |
| sporadic pancreatic cancer | Obazee et al. 2019 [48] | 2835 | 69 | 0.0123 | OR: 1.78 (1.26–2.52); (p = 0.00119) |
| lung cancer | Wang et al. 2014 [49] | 21435 | 298 | 0.01434 | OR: 1.83 (OR) = 2.47; (p = 4.74 × 10−20) |
| lung cancer | Rudd et al. 2006 [50] | 1526 | 14 | 0.009 | OR: 1.72 (0.15–2.57); (p = 0.0075) |
| lung cancer | Rafnar et al. 2018 [51] | 4 461 | na | na | OR: 1.54 (1.23–1.91); (p = 0.00012) |
| incl: small cell lung cancer | 800 | na | na | OR: 2.06 (1.35–3.16) | |
| incl: squamous cell lung carcinoma (SQLC) | 901 | na | na | OR: 1.71 (1.10–2.67); (p = 0.02) | |
| lung squamous cell carcinoma | Esai Selvan et al. 2019 [10] | 318 | na | na | OR: 3.0 (1.4–6.4); (p = 0.0053) |
| esophageal squamous cell carcinoma | Akbari et al. 2008 [27] | 197 | 9 | 0.02284 | OR: 6.0 (1.3–28);(p = 0.01) |
| UADT squamous cell carcinoma | Delahaye-Sourdeix et al. 2015 [17] | 5942 | 149 | 0.01253 | OR: 2.53 (1.89–3.38); (p = 3 × 10−10) |
| bladder cancer | Ge et al. 2016 [52] | 3591 | 41 | 0.0096 | OR: 1.70 (1.19–2.42); (p = 0.0036) |
| renal cell carcinoma | Ge et al. 2016 [52] | 1322 | 13 | 0.0125 | OR: 1.60 (0.91–2.82); (p = 0.103) |
| prostate cancer | Ge et al. 2016 [52] | 1151 | 8 | 0.0076 | OR: 0.85 (0.41–1.74); (p = 0.647) |
| squamous cell carcinoma of the skin | Rafnar et al. 2018 [51] | OR: 1.69 (1.26–2.26) | |||
| melanoma | Tuominen et al. 2016 [53] | 452 | 12 | 0.01304 | OR: 2.80 (1.04–7.58), (p = 0.035) |
| Study | Note | Number of Cases Screened | Number of Cases with BRCA2 c.9976A>T Variant Alone | Number of Cases Carrying BRCA2 c.9976A>T Variant WITH BRCA2 c.6275_6276delTT Variant | ||
|---|---|---|---|---|---|---|
| (#) | (%) | |||||
| Current study | breast, ovarian cancer | 2138 | 46 | 0 | 0 | |
| Higgs et al. 2015 [13] | High-risk breast/ovarian cancer families, Manchester region of North West England | breast, ovarian cancer | 1850 | 23 | 18 | 0.0097 |
| Research study: familial breast/ovarian cancer cases, North West | breast, ovarian cancer | 1576 | not reported | 25 | 0.0159 | |
| Samples from Liverpool (UK), Irish Republic, Finland and Germany | breast, ovarian cancer | 1395 | 43 | 4 | 0.0029 | |
| Mazoyer et al. 1996 [11] | breast cancer | 513 | 11 | 2 | 0.0039 | |
| Mazoyer et al. 1996 [11] | ovarian cancer | 361 | 7 | 0 | 0 | |
| Martin et al. 2005 [8] | familial pancreatic cancer | 144 | 8 | 0 | 0 | |
| Akbari et al. 2008 [27] | esophageal squamous cell carcinoma | 197 | 9 | 0 | 0 | |
| Wang et al. 2014 [49] | Meta-analysis of 4 lung cancer GWAS studies | lung cancer | 21,435 | 298 | 0/70 | 0 |
| Rafnar et al. 2018 [51] | Analysis of 3 studies | lung cancer | 4461 | na | 0 | 0 |
| incl: small cell lung cancer | 800 | na | 0 | 0 | ||
| incl: squamous cell lung carcinoma (SQLC) | 901 | na | 0 | 0 | ||
| Meeks et al. 2016 [15] | breast cancer | 41,081 | 852 | 233/306 | 0.7614 | |
| Haraldsson et al. 1998 [45] | male breast cancer | 34 | 0 | 1 | 0.0294 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butz, H.; Papp, J.; Bozsik, A.; Krokker, L.; Pócza, T.; Oláh, E.; Patócs, A. Application of Multilayer Evidence for Annotation of C-Terminal BRCA2 Variants. Cancers 2021, 13, 881. https://doi.org/10.3390/cancers13040881
Butz H, Papp J, Bozsik A, Krokker L, Pócza T, Oláh E, Patócs A. Application of Multilayer Evidence for Annotation of C-Terminal BRCA2 Variants. Cancers. 2021; 13(4):881. https://doi.org/10.3390/cancers13040881
Chicago/Turabian StyleButz, Henriett, János Papp, Anikó Bozsik, Lilla Krokker, Tímea Pócza, Edit Oláh, and Attila Patócs. 2021. "Application of Multilayer Evidence for Annotation of C-Terminal BRCA2 Variants" Cancers 13, no. 4: 881. https://doi.org/10.3390/cancers13040881
APA StyleButz, H., Papp, J., Bozsik, A., Krokker, L., Pócza, T., Oláh, E., & Patócs, A. (2021). Application of Multilayer Evidence for Annotation of C-Terminal BRCA2 Variants. Cancers, 13(4), 881. https://doi.org/10.3390/cancers13040881