
The Role of BRCA1/2-Mutated Tumor Microenvironment in Breast Cancer

 , , ,
, , ,
Abstract
Simple Summary
Abstract
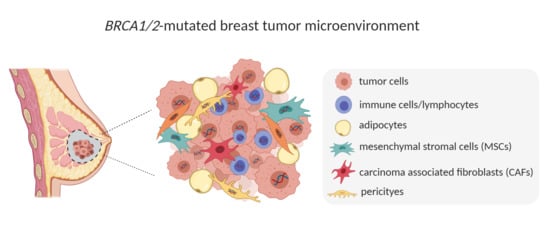
1. Introduction
2. Breast Tumor Microenvironment: Modulator of Tumor Initiation, Progression, Metastasis and Therapy Response
3. BRCA1/2-Deficient Tumor Microenvironment
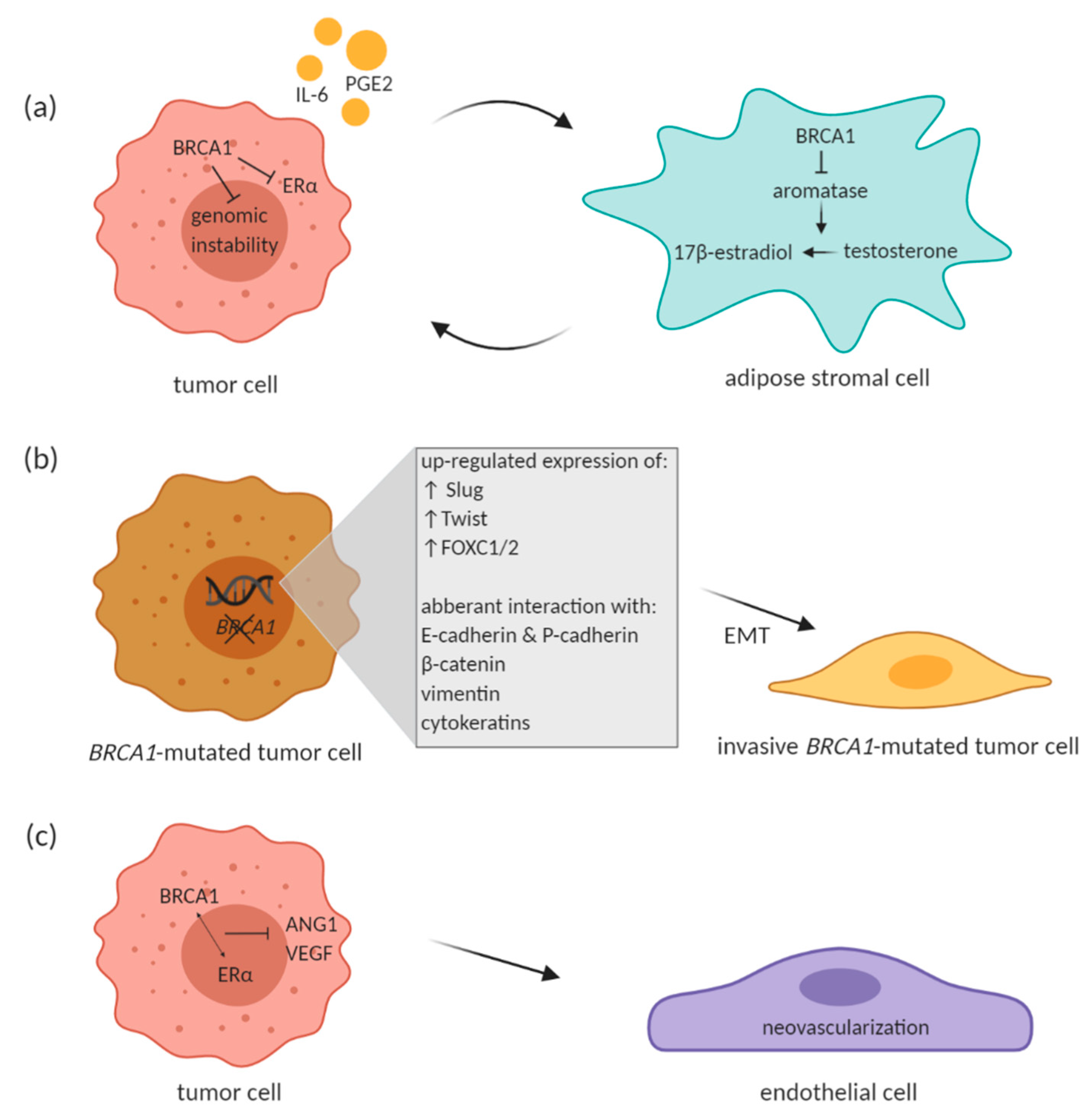
3.1. EMT Process in BRCA-Deficient Tumors
3.2. Impact of BRCA Deficiency on Tumor Neovascularization
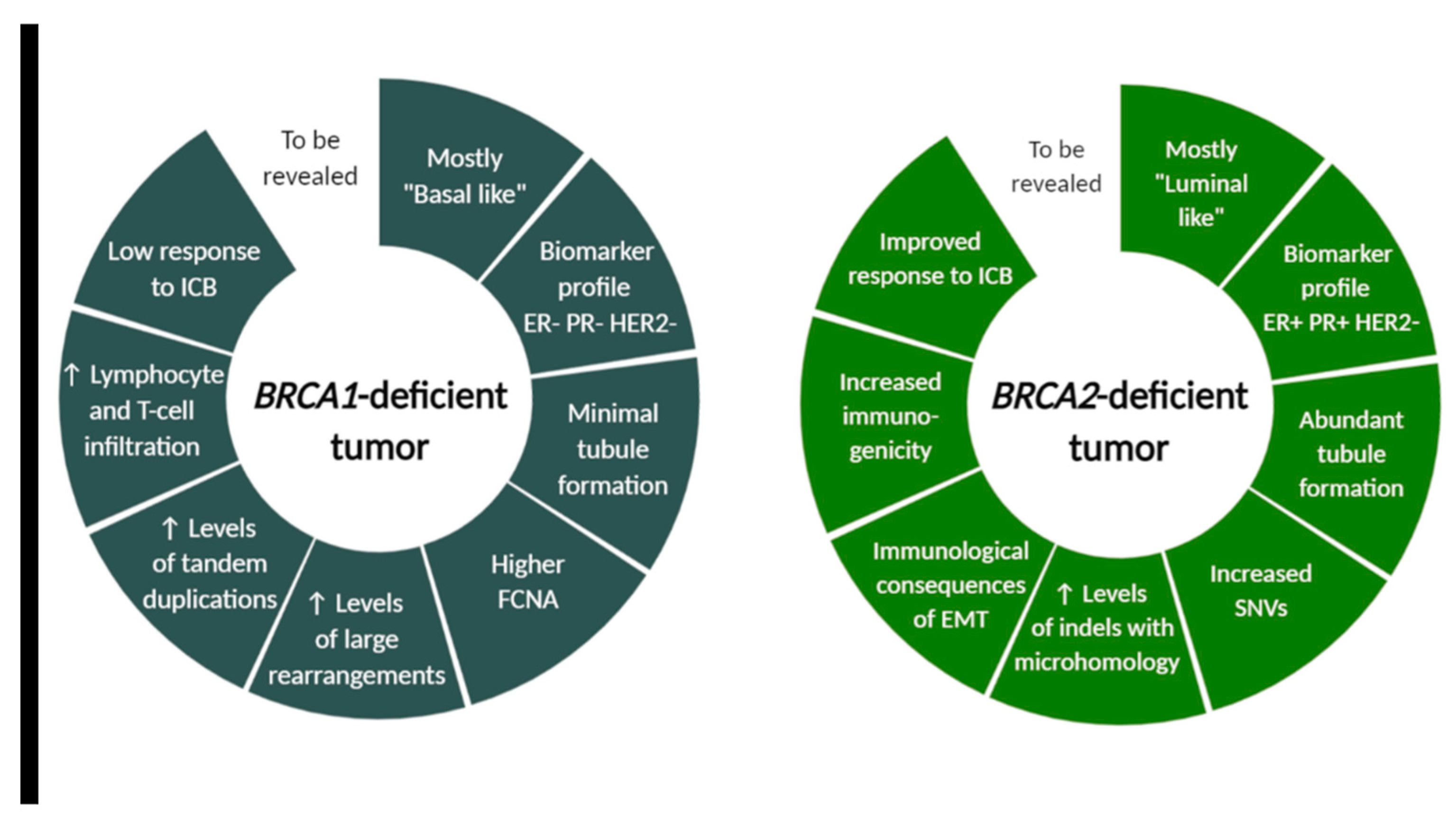
4. Other Observations from an Altered Tumor Microenvironment
5. Future Prospects and Therapeutic Strategies for BRCA-Associated Breast Cancer
6. Conclusions
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Zengel, B.; Yararbas, U.; Duran, A.; Uslu, A.; Eliyatkin, N.; Demirkiran, M.A.; Cengiz, F.; Simsek, C.; Postaci, H.; Vardar, E.; et al. Comparison of the clinicopathological features of invasive ductal, invasive lobular, and mixed (invasive ductal + invasive lobular) carcinoma of the breast. Breast Cancer 2015, 22, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Bharat, A.; Gao, F.; Margenthaler, J.A. Tumor characteristics and patient outcomes are similar between invasive lobular and mixed invasive ductal/lobular breast cancers but differ from pure invasive ductal breast cancers. Am. J. Surg. 2009, 198, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Ward, E.M.; DeSantis, C.E.; Lin, C.C.; Kramer, J.L.; Jemal, A.; Kohler, B.; Brawley, O.W.; Gansler, T. Cancer statistics: Breast cancer in situ. CA Cancer J. Clin. 2015, 65, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Chuba, P.J.; Hamre, M.R.; Yap, J.; Severson, R.K.; Lucas, D.; Shamsa, F.; Aref, A. Bilateral risk for subsequent breast cancer after lobular carcinoma-in-situ: Analysis of surveillance, epidemiology, and end results data. J. Clin. Oncol. 2005, 23, 5534–5541. [Google Scholar] [CrossRef]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Bertucci, F.; Finetti, P.; Cervera, N.; Esterni, B.; Hermitte, F.; Viens, P.; Birnbaum, D. How basal are triple-negative breast cancers? Int. J. Cancer 2008, 123, 236–240. [Google Scholar] [CrossRef]
- Zheng, G.; Yu, H.; Hemminki, A.; Forsti, A.; Sundquist, K.; Hemminki, K. Familial associations of female breast cancer with other cancers. Int. J. Cancer 2017, 141, 2253–2259. [Google Scholar] [CrossRef]
- Couto, E.; Hemminki, K. Estimates of heritable and environmental components of familial breast cancer using family history information. Br. J. Cancer 2007, 96, 1740–1742. [Google Scholar] [CrossRef]
- Claus, E.B.; Schildkraut, J.M.; Thompson, W.D.; Risch, N.J. The genetic attributable risk of breast and ovarian cancer. Cancer 1996, 77, 2318–2324. [Google Scholar] [CrossRef]
- Apostolou, P.; Fostira, F. Hereditary breast cancer: The era of new susceptibility genes. Biomed. Res. Int 2013, 2013, 747318. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, F.C.; van Overeem Hansen, T.; Sorensen, C.S. Hereditary breast and ovarian cancer: New genes in confined pathways. Nat. Rev. Cancer 2016, 16, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Kast, K.; Rhiem, K.; Wappenschmidt, B.; Hahnen, E.; Hauke, J.; Bluemcke, B.; Zarghooni, V.; Herold, N.; Ditsch, N.; Kiechle, M.; et al. Prevalence of BRCA1/2 germline mutations in 21 401 families with breast and ovarian cancer. J. Med. Genet. 2016, 53, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [PubMed]
- Lakhani, S.R.; Reis-Filho, J.S.; Fulford, L.; Penault-Llorca, F.; van der Vijver, M.; Parry, S.; Bishop, T.; Benitez, J.; Rivas, C.; Bignon, Y.J.; et al. Prediction of BRCA1 status in patients with breast cancer using estrogen receptor and basal phenotype. Clin. Cancer Res. 2005, 11, 5175–5180. [Google Scholar] [CrossRef] [PubMed]
- Mavaddat, N.; Barrowdale, D.; Andrulis, I.L.; Domchek, S.M.; Eccles, D.; Nevanlinna, H.; Ramus, S.J.; Spurdle, A.; Robson, M.; Sherman, M.; et al. Pathology of breast and ovarian cancers among BRCA1 and BRCA2 mutation carriers: Results from the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA). Cancer Epidemiol. Biomark. Prev. 2012, 21, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Stefansson, I.M.; Chappuis, P.O.; Begin, L.R.; Goffin, J.R.; Wong, N.; Trudel, M.; Akslen, L.A. Germline BRCA1 mutations and a basal epithelial phenotype in breast cancer. J. Natl. Cancer Inst. 2003, 95, 1482–1485. [Google Scholar] [CrossRef]
- Armes, J.E.; Trute, L.; White, D.; Southey, M.C.; Hammet, F.; Tesoriero, A.; Hutchins, A.M.; Dite, G.S.; McCredie, M.R.; Giles, G.G.; et al. Distinct molecular pathogeneses of early-onset breast cancers in BRCA1 and BRCA2 mutation carriers: A population-based study. Cancer Res. 1999, 59, 2011–2017. [Google Scholar]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef]
- Gardini, A.; Baillat, D.; Cesaroni, M.; Shiekhattar, R. Genome-wide analysis reveals a role for BRCA1 and PALB2 in transcriptional co-activation. EMBO J. 2014, 33, 890–905. [Google Scholar] [CrossRef]
- Fabbro, M.; Savage, K.; Hobson, K.; Deans, A.J.; Powell, S.N.; McArthur, G.A.; Khanna, K.K. BRCA1-BARD1 complexes are required for p53Ser-15 phosphorylation and a G1/S arrest following ionizing radiation-induced DNA damage. J. Biol. Chem. 2004, 279, 31251–31258. [Google Scholar] [CrossRef] [PubMed]
- Savage, K.I.; Harkin, D.P. BRCA1, a ‘complex’ protein involved in the maintenance of genomic stability. FEBS J. 2015, 282, 630–646. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef]
- Greenberg, R.A.; Sobhian, B.; Pathania, S.; Cantor, S.B.; Nakatani, Y.; Livingston, D.M. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev. 2006, 20, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Coleman, K.A.; Greenberg, R.A. The BRCA1-RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. J. Biol. Chem. 2011, 286, 13669–13680. [Google Scholar] [CrossRef]
- Wang, B.; Matsuoka, S.; Ballif, B.A.; Zhang, D.; Smogorzewska, A.; Gygi, S.P.; Elledge, S.J. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 2007, 316, 1194–1198. [Google Scholar] [CrossRef]
- Xie, J.; Peng, M.; Guillemette, S.; Quan, S.; Maniatis, S.; Wu, Y.; Venkatesh, A.; Shaffer, S.A.; Brosh, R.M., Jr.; Cantor, S.B. FANCJ/BACH1 acetylation at lysine 1249 regulates the DNA damage response. PLoS Genet. 2012, 8, e1002786. [Google Scholar] [CrossRef]
- Kim, H.; D’Andrea, A.D. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012, 26, 1393–1408. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cortez, D.; Yazdi, P.; Neff, N.; Elledge, S.J.; Qin, J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000, 14, 927–939. [Google Scholar]
- Zhang, F.; Ma, J.; Wu, J.; Ye, L.; Cai, H.; Xia, B.; Yu, X. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr. Biol. 2009, 19, 524–529. [Google Scholar] [CrossRef]
- Liu, C.; Srihari, S.; Cao, K.A.; Chenevix-Trench, G.; Simpson, P.T.; Ragan, M.A.; Khanna, K.K. A fine-scale dissection of the DNA double-strand break repair machinery and its implications for breast cancer therapy. Nucleic Acids Res. 2014, 42, 6106–6127. [Google Scholar] [CrossRef] [PubMed]
- Christou, C.M.; Kyriacou, K. BRCA1 and Its Network of Interacting Partners. Biology 2013, 2, 40–63. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.; Olschwang, S.; Keller, J.J.; Westerman, A.M.; Menko, F.H.; Boardman, L.A.; Scott, R.J.; Trimbath, J.; Giardiello, F.M.; Gruber, S.B.; et al. Relative frequency and morphology of cancers in STK11 mutation carriers. Gastroenterology 2004, 126, 1788–1794. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.H.; Mester, J.L.; Ngeow, J.; Rybicki, L.A.; Orloff, M.S.; Eng, C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin. Cancer Res. 2012, 18, 400–407. [Google Scholar] [CrossRef]
- Mai, P.L.; Best, A.F.; Peters, J.A.; DeCastro, R.M.; Khincha, P.P.; Loud, J.T.; Bremer, R.C.; Rosenberg, P.S.; Savage, S.A. Risks of first and subsequent cancers among TP53 mutation carriers in the National Cancer Institute Li-Fraumeni syndrome cohort. Cancer 2016, 122, 3673–3681. [Google Scholar] [CrossRef]
- Pharoah, P.D.; Guilford, P.; Caldas, C. Incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology 2001, 121, 1348–1353. [Google Scholar] [CrossRef]
- Shiovitz, S.; Korde, L.A. Genetics of breast cancer: A topic in evolution. Ann. Oncol. 2015, 26, 1291–1299. [Google Scholar] [CrossRef]
- Michailidou, K.; Lindstrom, S.; Dennis, J.; Beesley, J.; Hui, S.; Kar, S.; Lemacon, A.; Soucy, P.; Glubb, D.; Rostamianfar, A.; et al. Association analysis identifies 65 new breast cancer risk loci. Nature 2017, 551, 92–94. [Google Scholar] [CrossRef]
- Place, A.E.; Jin Huh, S.; Polyak, K. The microenvironment in breast cancer progression: Biology and implications for treatment. Breast Cancer Res. 2011, 13, 227. [Google Scholar] [CrossRef]
- Plava, J.; Cihova, M.; Burikova, M.; Matuskova, M.; Kucerova, L.; Miklikova, S. Recent advances in understanding tumor stroma-mediated chemoresistance in breast cancer. Mol. Cancer 2019, 18, 67. [Google Scholar] [CrossRef]
- Soysal, S.D.; Tzankov, A.; Muenst, S.E. Role of the Tumor Microenvironment in Breast Cancer. Pathobiology 2015, 82, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Halama, N.; Zoernig, I.; Berthel, A.; Kahlert, C.; Klupp, F.; Suarez-Carmona, M.; Suetterlin, T.; Brand, K.; Krauss, J.; Lasitschka, F.; et al. Tumoral Immune Cell Exploitation in Colorectal Cancer Metastases Can Be Targeted Effectively by Anti-CCR5 Therapy in Cancer Patients. Cancer Cell 2016, 29, 587–601. [Google Scholar] [CrossRef] [PubMed]
- Orlando, P.A.; Gatenby, R.A.; Brown, J.S. Tumor evolution in space: The effects of competition colonization tradeoffs on tumor invasion dynamics. Front. Oncol. 2013, 3, 45. [Google Scholar] [CrossRef] [PubMed]
- Eiro, N.; Gonzalez, L.O.; Fraile, M.; Cid, S.; Schneider, J.; Vizoso, F.J. Breast Cancer Tumor Stroma: Cellular Components, Phenotypic Heterogeneity, Intercellular Communication, Prognostic Implications and Therapeutic Opportunities. Cancers 2019, 11, 664. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.T.; Tan, Y.J.; Falasca, M.; Oon, C.E. Cancer-Associated Fibroblasts: Epigenetic Regulation and Therapeutic Intervention in Breast Cancer. Cancers 2020, 12, 2949. [Google Scholar] [CrossRef]
- Ullah, I.; Subbarao, R.B.; Rho, G.J. Human mesenchymal stem cells—Current trends and future prospective. Biosci. Rep. 2015, 35. [Google Scholar] [CrossRef]
- Kucerova, L.; Kovacovicova, M.; Polak, S.; Bohac, M.; Fedeles, J.; Palencar, D.; Matuskova, M. Interaction of human adipose tissue-derived mesenchymal stromal cells with breast cancer cells. Neoplasma 2011, 58, 361–370. [Google Scholar] [CrossRef]
- Plava, J.; Cihova, M.; Burikova, M.; Bohac, M.; Adamkov, M.; Drahosova, S.; Rusnakova, D.; Pindak, D.; Karaba, M.; Simo, J.; et al. Permanent Pro-Tumorigenic Shift in Adipose Tissue-Derived Mesenchymal Stromal Cells Induced by Breast Malignancy. Cells 2020, 9, 480. [Google Scholar] [CrossRef]
- Yu, P.F.; Huang, Y.; Xu, C.L.; Lin, L.Y.; Han, Y.Y.; Sun, W.H.; Hu, G.H.; Rabson, A.B.; Wang, Y.; Shi, Y.F. Downregulation of CXCL12 in mesenchymal stromal cells by TGFbeta promotes breast cancer metastasis. Oncogene 2017, 36, 840–849. [Google Scholar] [CrossRef]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef]
- Martin, F.T.; Dwyer, R.M.; Kelly, J.; Khan, S.; Murphy, J.M.; Curran, C.; Miller, N.; Hennessy, E.; Dockery, P.; Barry, F.P.; et al. Potential role of mesenchymal stem cells (MSCs) in the breast tumour microenvironment: Stimulation of epithelial to mesenchymal transition (EMT). Breast Cancer Res. Treat. 2010, 124, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Yang, W.M.; Chen, L.P.; Yang, D.H.; Zhou, Q.; Zhu, J.; Chen, J.J.; Huang, R.C.; Chen, Z.S.; Huang, R.P. Enhanced chemosensitization in multidrug-resistant human breast cancer cells by inhibition of IL-6 and IL-8 production. Breast Cancer Res. Treat. 2012, 135, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.R.; Lu, D.Y.; Lin, H.Y.; Yeh, W.L. Mesenchymal stem cell-induced doxorubicin resistance in triple negative breast cancer. Biomed. Res. Int 2014, 2014, 532161. [Google Scholar] [CrossRef] [PubMed]
- Yeh, W.L.; Tsai, C.F.; Chen, D.R. Peri-foci adipose-derived stem cells promote chemoresistance in breast cancer. Stem. Cell Res. Ther. 2017, 8, 177. [Google Scholar] [CrossRef] [PubMed]
- Roodhart, J.M.; Daenen, L.G.; Stigter, E.C.; Prins, H.J.; Gerrits, J.; Houthuijzen, J.M.; Gerritsen, M.G.; Schipper, H.S.; Backer, M.J.; van Amersfoort, M.; et al. Mesenchymal stem cells induce resistance to chemotherapy through the release of platinum-induced fatty acids. Cancer Cell 2011, 20, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Kucerova, L.; Skolekova, S.; Matuskova, M.; Bohac, M.; Kozovska, Z. Altered features and increased chemosensitivity of human breast cancer cells mediated by adipose tissue-derived mesenchymal stromal cells. BMC Cancer 2013, 13, 535. [Google Scholar] [CrossRef] [PubMed]
- Skolekova, S.; Matuskova, M.; Bohac, M.; Toro, L.; Durinikova, E.; Tyciakova, S.; Demkova, L.; Gursky, J.; Kucerova, L. Erratum to: Cisplatin-induced mesenchymal stromal cells-mediated mechanism contributing to decreased antitumor effect in breast cancer cells. Cell Commun. Signal. 2016, 14, 7. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef]
- Bussard, K.M.; Mutkus, L.; Stumpf, K.; Gomez-Manzano, C.; Marini, F.C. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016, 18, 84. [Google Scholar] [CrossRef]
- Hida, K.; Maishi, N.; Annan, D.A.; Hida, Y. Contribution of Tumor Endothelial Cells in Cancer Progression. Int. J. Mol. Sci. 2018, 19, 1272. [Google Scholar] [CrossRef]
- Wang, L.; Simons, D.L.; Lu, X.; Tu, T.Y.; Avalos, C.; Chang, A.Y.; Dirbas, F.M.; Yim, J.H.; Waisman, J.; Lee, P.P. Breast cancer induces systemic immune changes on cytokine signaling in peripheral blood monocytes and lymphocytes. EBioMedicine 2020, 52, 102631. [Google Scholar] [CrossRef] [PubMed]
- Saini, M.; Szczerba, B.M.; Aceto, N. Circulating Tumor Cell-Neutrophil Tango along the Metastatic Process. Cancer Res. 2019, 79, 6067–6073. [Google Scholar] [CrossRef] [PubMed]
- Miklikova, S.; Minarik, G.; Sedlackova, T.; Plava, J.; Cihova, M.; Jurisova, S.; Kalavska, K.; Karaba, M.; Benca, J.; Smolkova, B.; et al. Inflammation-Based Scores Increase the Prognostic Value of Circulating Tumor Cells in Primary Breast Cancer. Cancers 2020, 12, 1134. [Google Scholar] [CrossRef] [PubMed]
- De Giorgi, U.; Mego, M.; Scarpi, E.; Giordano, A.; Giuliano, M.; Valero, V.; Alvarez, R.H.; Ueno, N.T.; Cristofanilli, M.; Reuben, J.M. Association between circulating tumor cells and peripheral blood monocytes in metastatic breast cancer. Ther. Adv. Med. Oncol. 2019, 11, 1758835919866065. [Google Scholar] [CrossRef] [PubMed]
- McCullough, S.D.; Hu, Y.; Li, R. BRCA1 in initiation, invasion, and metastasis of breast cancer: A perspective from the tumor microenvironment. In Metastasis of Breast Cancer; Mansel, R.E., Fodstad, O., Jiang, W.G., Eds.; Springer: Dordrecht, The Netherlands, 2007; pp. 31–46. [Google Scholar] [CrossRef]
- Li, C.M.; Oren, Y.; Regev, A.; Brugge, J.S. Abstract PR06: Contribution of mutant microenvironment to hereditary cancer: Single-cell gene expression profiling of a genetically engineered mouse model of human hereditary BRCA1-related breast cancer. Cancer Res. 2018, 78, PR06. [Google Scholar] [CrossRef]
- Ghosh, S.; Lu, Y.; Katz, A.; Hu, Y.; Li, R. Tumor suppressor BRCA1 inhibits a breast cancer-associated promoter of the aromatase gene (CYP19) in human adipose stromal cells. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E246–E252. [Google Scholar] [CrossRef] [PubMed]
- Weber, F.; Shen, L.; Fukino, K.; Patocs, A.; Mutter, G.L.; Caldes, T.; Eng, C. Total-genome analysis of BRCA1/2-related invasive carcinomas of the breast identifies tumor stroma as potential landscaper for neoplastic initiation. Am. J. Hum. Genet. 2006, 78, 961–972. [Google Scholar] [CrossRef]
- Hemalatha, S.K.; Sengodan, S.K.; Nadhan, R.; Dev, J.; Sushama, R.R.; Somasundaram, V.; Thankappan, R.; Rajan, A.; Latha, N.R.; Varghese, G.R.; et al. Brcal Defective Breast Cancer Cells Induce in vitro Transformation of Cancer Associated Fibroblasts (CAFs) to Metastasis Associated Fibroblasts (MAF). Sci. Rep. 2018, 8, 13903. [Google Scholar] [CrossRef]
- Coene, E.D.; Gadelha, C.; White, N.; Malhas, A.; Thomas, B.; Shaw, M.; Vaux, D.J. A novel role for BRCA1 in regulating breast cancer cell spreading and motility. J. Cell Biol. 2011, 192, 497–512. [Google Scholar] [CrossRef]
- ElShamy, W.M.; Livingston, D.M. Identification of BRCA1-IRIS, a BRCA1 locus product. Nat. Cell Biol. 2004, 6, 954–967. [Google Scholar] [CrossRef]
- Ryan, D.; Paul, B.T.; Koziol, J.; ElShamy, W.M. The pro- and anti-tumor roles of mesenchymal stem cells toward BRCA1-IRIS-overexpressing TNBC cells. Breast Cancer Res. 2019, 21, 53. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Bai, F.; Zhang, L.H.; Scott, A.; Li, E.; Pei, X.H. Estrogen promotes estrogen receptor negative BRCA1-deficient tumor initiation and progression. Breast Cancer Res. 2018, 20, 74. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Gang, B.P.; Bassi, C.; Wakeham, A.; Baniasadi, S.P.; Hao, Z.; Li, W.Y.; Cescon, D.W.; Li, Y.T.; Molyneux, S.; et al. Estrogen controls the survival of BRCA1-deficient cells via a PI3K-NRF2-regulated pathway. Proc. Natl. Acad. Sci. USA 2014, 111, 4472–4477. [Google Scholar] [CrossRef] [PubMed]
- Saha Roy, S.; Vadlamudi, R.K. Role of estrogen receptor signaling in breast cancer metastasis. Int. J. Breast Cancer 2012, 2012, 654698. [Google Scholar] [CrossRef]
- Sacks, D.; Baxter, B.; Campbell, B.C.V.; Carpenter, J.S.; Cognard, C.; Dippel, D.; Eesa, M.; Fischer, U.; Hausegger, K.; Hirsch, J.A.; et al. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke 2018, 13, 612–632. [Google Scholar] [CrossRef]
- Micalizzi, D.S.; Ford, H.L. Epithelial-mesenchymal transition in development and cancer. Future Oncol. 2009, 5, 1129–1143. [Google Scholar] [CrossRef]
- Fedele, M.; Cerchia, L.; Chiappetta, G. The Epithelial-to-Mesenchymal Transition in Breast Cancer: Focus on Basal-Like Carcinomas. Cancers 2017, 9, 134. [Google Scholar] [CrossRef]
- Felipe Lima, J.; Nofech-Mozes, S.; Bayani, J.; Bartlett, J.M. EMT in Breast Carcinoma-A Review. J. Clin. Med. 2016, 5. [Google Scholar] [CrossRef]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef]
- Sengodan, S.K.; Sreelatha, K.H.; Nadhan, R.; Srinivas, P. Regulation of epithelial to mesenchymal transition by BRCA1 in breast cancer. Crit. Rev. Oncol. Hematol. 2018, 123, 74–82. [Google Scholar] [CrossRef]
- Bai, F.; Chan, H.L.; Scott, A.; Smith, M.D.; Fan, C.; Herschkowitz, J.I.; Perou, C.M.; Livingstone, A.S.; Robbins, D.J.; Capobianco, A.J.; et al. BRCA1 suppresses epithelial-to-mesenchymal transition and stem cell dedifferentiation during mammary and tumor development. Cancer Res. 2014, 74, 6161–6172. [Google Scholar] [CrossRef] [PubMed]
- Scimeca, M.; Antonacci, C.; Colombo, D.; Bonfiglio, R.; Buonomo, O.C.; Bonanno, E. Emerging prognostic markers related to mesenchymal characteristics of poorly differentiated breast cancers. Tumour Biol. 2016, 37, 5427–5435. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wu, J.; Zhang, C.; Sun, S.; Zhang, J.; Liu, W.; Huang, J.; Zhang, Z. BRCA mutations and survival in breast cancer: An updated systematic review and meta-analysis. Oncotarget 2016, 7, 70113–70127. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Barry, W.T.; Seah, D.S.; Tung, N.M.; Garber, J.E.; Lin, N.U. Patterns of recurrence and metastasis in BRCA1/BRCA2-associated breast cancers. Cancer 2020, 126, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.; Vaillant, F.; Wu, D.; Forrest, N.C.; Pal, B.; Hart, A.H.; Asselin-Labat, M.L.; Gyorki, D.E.; Ward, T.; Partanen, A.; et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat. Med. 2009, 15, 907–913. [Google Scholar] [CrossRef]
- Molyneux, G.; Geyer, F.C.; Magnay, F.A.; McCarthy, A.; Kendrick, H.; Natrajan, R.; Mackay, A.; Grigoriadis, A.; Tutt, A.; Ashworth, A.; et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem. Cell 2010, 7, 403–417. [Google Scholar] [CrossRef]
- Bai, F.; Smith, M.D.; Chan, H.L.; Pei, X.H. Germline mutation of Brca1 alters the fate of mammary luminal cells and causes luminal-to-basal mammary tumor transformation. Oncogene 2013, 32, 2715–2725. [Google Scholar] [CrossRef]
- Lindeman, G.J.; Visvader, J.E. Cell fate takes a slug in BRCA1-associated breast cancer. Breast Cancer Res. 2011, 13, 306. [Google Scholar] [CrossRef]
- Proia, T.A.; Keller, P.J.; Gupta, P.B.; Klebba, I.; Jones, A.D.; Sedic, M.; Gilmore, H.; Tung, N.; Naber, S.P.; Schnitt, S.; et al. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem. Cell 2011, 8, 149–163. [Google Scholar] [CrossRef]
- Mani, S.A.; Yang, J.; Brooks, M.; Schwaninger, G.; Zhou, A.; Miura, N.; Kutok, J.L.; Hartwell, K.; Richardson, A.L.; Weinberg, R.A. Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proc. Natl. Acad. Sci. USA 2007, 104, 10069–10074. [Google Scholar] [CrossRef]
- Ordonez, L.D.; Hay, T.; McEwen, R.; Polanska, U.M.; Hughes, A.; Delpuech, O.; Cadogan, E.; Powell, S.; Dry, J.; Tornillo, G.; et al. Rapid activation of epithelial-mesenchymal transition drives PARP inhibitor resistance in Brca2-mutant mammary tumours. Oncotarget 2019, 10, 2586–2606. [Google Scholar] [CrossRef] [PubMed]
- Jaspers, J.E.; Sol, W.; Kersbergen, A.; Schlicker, A.; Guyader, C.; Xu, G.; Wessels, L.; Borst, P.; Jonkers, J.; Rottenberg, S. BRCA2-deficient sarcomatoid mammary tumors exhibit multidrug resistance. Cancer Res. 2015, 75, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Bao, S.D. Roles of main pro- and anti-angiogenic factors in tumor angiogenesis. World J. Gastroenterol. 2004, 10, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Gilkes, D.M.; Semenza, G.L. Role of hypoxia-inducible factors in breast cancer metastasis. Future Oncol. 2013, 9, 1623–1636. [Google Scholar] [CrossRef]
- Salceda, S.; Caro, J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J. Biol. Chem. 1997, 272, 22642–22647. [Google Scholar] [CrossRef]
- Carmeliet, P. VEGF as a key mediator of angiogenesis in cancer. Oncology 2005, 69 (Suppl. 3), 4–10. [Google Scholar] [CrossRef]
- Liao, D.; Corle, C.; Seagroves, T.N.; Johnson, R.S. Hypoxia-inducible factor-1alpha is a key regulator of metastasis in a transgenic model of cancer initiation and progression. Cancer Res. 2007, 67, 563–572. [Google Scholar] [CrossRef]
- Masoumi Moghaddam, S.; Amini, A.; Morris, D.L.; Pourgholami, M.H. Significance of vascular endothelial growth factor in growth and peritoneal dissemination of ovarian cancer. Cancer Metastasis Rev. 2012, 31, 143–162. [Google Scholar] [CrossRef]
- Saponaro, C.; Malfettone, A.; Ranieri, G.; Danza, K.; Simone, G.; Paradiso, A.; Mangia, A. VEGF, HIF-1alpha expression and MVD as an angiogenic network in familial breast cancer. PLoS ONE 2013, 8, e53070. [Google Scholar] [CrossRef]
- Kawai, H.; Li, H.; Chun, P.; Avraham, S.; Avraham, H.K. Direct interaction between BRCA1 and the estrogen receptor regulates vascular endothelial growth factor (VEGF) transcription and secretion in breast cancer cells. Oncogene 2002, 21, 7730–7739. [Google Scholar] [CrossRef]
- Danza, K.; Pilato, B.; Lacalamita, R.; Addati, T.; Giotta, F.; Bruno, A.; Paradiso, A.; Tommasi, S. Angiogenetic axis angiopoietins/Tie2 and VEGF in familial breast cancer. Eur. J. Hum. Genet. 2013, 21, 824–830. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Furuta, S.; Wang, J.M.; Wei, S.; Jeng, Y.M.; Jiang, X.; Gu, B.; Chen, P.L.; Lee, E.Y.; Lee, W.H. Removal of BRCA1/CtIP/ZBRK1 repressor complex on ANG1 promoter leads to accelerated mammary tumor growth contributed by prominent vasculature. Cancer Cell 2006, 10, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Danza, K.; De Summa, S.; Pinto, R.; Pilato, B.; Palumbo, O.; Merla, G.; Simone, G.; Tommasi, S. MiR-578 and miR-573 as potential players in BRCA-related breast cancer angiogenesis. Oncotarget 2015, 6, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Kaakati, R.; Liu, X.; Xu, L.; Lee, A.K.; Bachelder, R.; Li, C.Y.; Hollenbeck, S.T. CRISPR/Cas9-Mediated BRCA1 Knockdown Adipose Stem Cells Promote Breast Cancer Progression. Plast. Reconstr. Surg. 2019, 143, 747–756. [Google Scholar] [CrossRef]
- Salem, A.F.; Howell, A.; Sartini, M.; Sotgia, F.; Lisanti, M.P. Downregulation of stromal BRCA1 drives breast cancer tumor growth via upregulation of HIF-1alpha, autophagy and ketone body production. Cell Cycle 2012, 11, 4167–4173. [Google Scholar] [CrossRef][Green Version]
- Russo, J.; Lynch, H.; Russo, I.H. Mammary Gland Architecture as a Determining Factor in the Susceptibility of the Human Breast to Cancer. Breast J. 2001, 7, 278–291. [Google Scholar] [CrossRef]
- Milanese, J.S.; Tibiche, C.; Zou, J.; Meng, Z.; Nantel, A.; Drouin, S.; Marcotte, R.; Wang, E. Germline variants associated with leukocyte genes predict tumor recurrence in breast cancer patients. NPJ Precis. Oncol. 2019, 3, 28. [Google Scholar] [CrossRef]
- Kotsopoulos, J. BRCA Mutations and Breast Cancer Prevention. Cancers 2018, 10, 524. [Google Scholar] [CrossRef]
- Metcalfe, K.; Eisen, A.; Senter, L.; Armel, S.; Bordeleau, L.; Meschino, W.S.; Pal, T.; Lynch, H.T.; Tung, N.M.; Kwong, A.; et al. International trends in the uptake of cancer risk reduction strategies in women with a BRCA1 or BRCA2 mutation. Br. J. Cancer 2019, 121, 15–21. [Google Scholar] [CrossRef]
- Maffini, M.V.; Calabro, J.M.; Soto, A.M.; Sonnenschein, C. Stromal regulation of neoplastic development: Age-dependent normalization of neoplastic mammary cells by mammary stroma. Am. J. Pathol. 2005, 167, 1405–1410. [Google Scholar] [CrossRef]
- Hodgson, A.; Turashvili, G. Pathology of Hereditary Breast and Ovarian Cancer. Front. Oncol. 2020, 10, 531790. [Google Scholar] [CrossRef] [PubMed]
- Min, A.; Kim, K.; Jeong, K.; Choi, S.; Kim, S.; Suh, K.J.; Lee, K.H.; Im, S.A. Homologous repair deficiency score for identifying breast cancers with defective DNA damage response. Sci. Rep. 2020, 10, 12506. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Powell, S.N. The role of the BRCA1 tumor suppressor in DNA double-strand break repair. Mol. Cancer Res. 2005, 3, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Torrisi, R.; Zuradelli, M.; Agostinetto, E.; Masci, G.; Losurdo, A.; De Sanctis, R.; Santoro, A. Platinum salts in the treatment of BRCA-associated breast cancer: A true targeted chemotherapy? Crit. Rev. Oncol. Hematol. 2019, 135, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, A.; Bertucci, A.; Bertucci, F. PARP Inhibitors in the Treatment of Early Breast Cancer: The Step Beyond? Cancers 2020, 12, 1378. [Google Scholar] [CrossRef] [PubMed]
- Litton, J.K.; Scoggins, M.E.; Hess, K.R.; Adrada, B.E.; Murthy, R.K.; Damodaran, S.; DeSnyder, S.M.; Brewster, A.M.; Barcenas, C.H.; Valero, V.; et al. Neoadjuvant Talazoparib for Patients With Operable Breast Cancer With a Germline BRCA Pathogenic Variant. J. Clin. Oncol. 2020, 38, 388–394. [Google Scholar] [CrossRef]
- Pantelidou, C.; Sonzogni, O.; De Oliveria Taveira, M.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP Inhibitor Efficacy Depends on CD8(+) T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef]
- Haque, R.; Shi, J.M.; Telford, C.; Avila, C.; Alvarado, M.; Tiller, G.E.; Dalvi, T.; Gutierrez, L.; Tyczynski, J.; Kaye, J.A. Survival Outcomes in BRCA1 or BRCA2 Mutation Carriers and the Influence of Triple-Negative Breast Cancer Subtype. Perm. J. 2018, 22, 28. [Google Scholar] [CrossRef]
- Lee, E.H.; Park, S.K.; Park, B.; Kim, S.W.; Lee, M.H.; Ahn, S.H.; Son, B.H.; Yoo, K.Y.; Kang, D. Effect of BRCA1/2 mutation on short-term and long-term breast cancer survival: A systematic review and meta-analysis. Breast Cancer Res. Treat. 2010, 122, 11–25. [Google Scholar] [CrossRef]
- Rennert, G.; Bisland-Naggan, S.; Barnett-Griness, O.; Bar-Joseph, N.; Zhang, S.; Rennert, H.S.; Narod, S.A. Clinical outcomes of breast cancer in carriers of BRCA1 and BRCA2 mutations. N. Engl. J. Med. 2007, 357, 115–123. [Google Scholar] [CrossRef]
- Nolan, E.; Savas, P.; Policheni, A.N.; Darcy, P.K.; Vaillant, F.; Mintoff, C.P.; Dushyanthen, S.; Mansour, M.; Pang, J.B.; Fox, S.B.; et al. Combined immune checkpoint blockade as a therapeutic strategy for BRCA1-mutated breast cancer. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Grandal, B.; Evrevin, C.; Laas, E.; Jardin, I.; Rozette, S.; Laot, L.; Dumas, E.; Coussy, F.; Pierga, J.Y.; Brain, E.; et al. Impact of BRCA Mutation Status on Tumor Infiltrating Lymphocytes (TILs), Response to Treatment, and Prognosis in Breast Cancer Patients Treated with Neoadjuvant Chemotherapy. Cancers 2020, 12, 3681. [Google Scholar] [CrossRef] [PubMed]
- Sonderstrup, I.M.H.; Jensen, M.B.; Ejlertsen, B.; Eriksen, J.O.; Gerdes, A.M.; Kruse, T.A.; Larsen, M.J.; Thomassen, M.; Laenkholm, A.V. Evaluation of tumor-infiltrating lymphocytes and association with prognosis in BRCA-mutated breast cancer. Acta Oncol. 2019, 58, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.X.; Leong, C.O. Association of BRCA1- and BRCA2-deficiency with mutation burden, expression of PD-L1/PD-1, immune infiltrates, and T cell-inflamed signature in breast cancer. PLoS ONE 2019, 14, e0215381. [Google Scholar] [CrossRef] [PubMed]
- Kraya, A.A.; Maxwell, K.N.; Wubbenhorst, B.; Wenz, B.M.; Pluta, J.; Rech, A.J.; Dorfman, L.M.; Lunceford, N.; Barrett, A.; Mitra, N.; et al. Genomic Signatures Predict the Immunogenicity of BRCA-Deficient Breast Cancer. Clin. Cancer Res. 2019, 25, 4363–4374. [Google Scholar] [CrossRef] [PubMed]
- Samstein, R.M.; Krishna, C.; Ma, X.; Pei, X.; Lee, K.-W.; Makarov, V.; Kuo, F.; Chung, J.; Srivastava, R.M.; Purohit, T.A.; et al. Mutations in BRCA1 and BRCA2 differentially affect the tumor microenvironment and response to checkpoint blockade immunotherapy. Nat. Cancer 2020, 1, 1188–1203. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Protein | Binding Partners | Function | Ref. |
|---|---|---|---|
| BRCA1 | MRN complex CtIP | DSB end resection | [24] |
| RAP80 MERIT40 BRCC36/45 Abraxas | stabilize/maintain DNA damage signaling and promote G2/M checkpoint arrest | [25,26] | |
| TOBP1 BACH1 (BRIP1/FANCJ) | S-phase cell cycle arrest | [24,27] | |
| Rad51 FANCD2 | repair replication forks stalled at DNA interstrand cross-links | [28] | |
| MLH1 ATM BLM MRN complex | nucleotide excision repair (NER) pathway | [29] | |
| PALB2 BRCA2 | HR-mediated DNA repair | [30] | |
| BRCA2 | BRCA1 RAD51 PALB2 | HR-mediated DNA repair | [31,32] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miklikova, S.; Trnkova, L.; Plava, J.; Bohac, M.; Kuniakova, M.; Cihova, M. The Role of BRCA1/2-Mutated Tumor Microenvironment in Breast Cancer. Cancers 2021, 13, 575. https://doi.org/10.3390/cancers13030575
Miklikova S, Trnkova L, Plava J, Bohac M, Kuniakova M, Cihova M. The Role of BRCA1/2-Mutated Tumor Microenvironment in Breast Cancer. Cancers. 2021; 13(3):575. https://doi.org/10.3390/cancers13030575
Chicago/Turabian StyleMiklikova, Svetlana, Lenka Trnkova, Jana Plava, Martin Bohac, Marcela Kuniakova, and Marina Cihova. 2021. "The Role of BRCA1/2-Mutated Tumor Microenvironment in Breast Cancer" Cancers 13, no. 3: 575. https://doi.org/10.3390/cancers13030575
APA StyleMiklikova, S., Trnkova, L., Plava, J., Bohac, M., Kuniakova, M., & Cihova, M. (2021). The Role of BRCA1/2-Mutated Tumor Microenvironment in Breast Cancer. Cancers, 13(3), 575. https://doi.org/10.3390/cancers13030575