
Shikonin Reduces Growth of Docetaxel-Resistant Prostate Cancer Cells Mainly through Necroptosis


 , ,
, ,  ,
,
Abstract
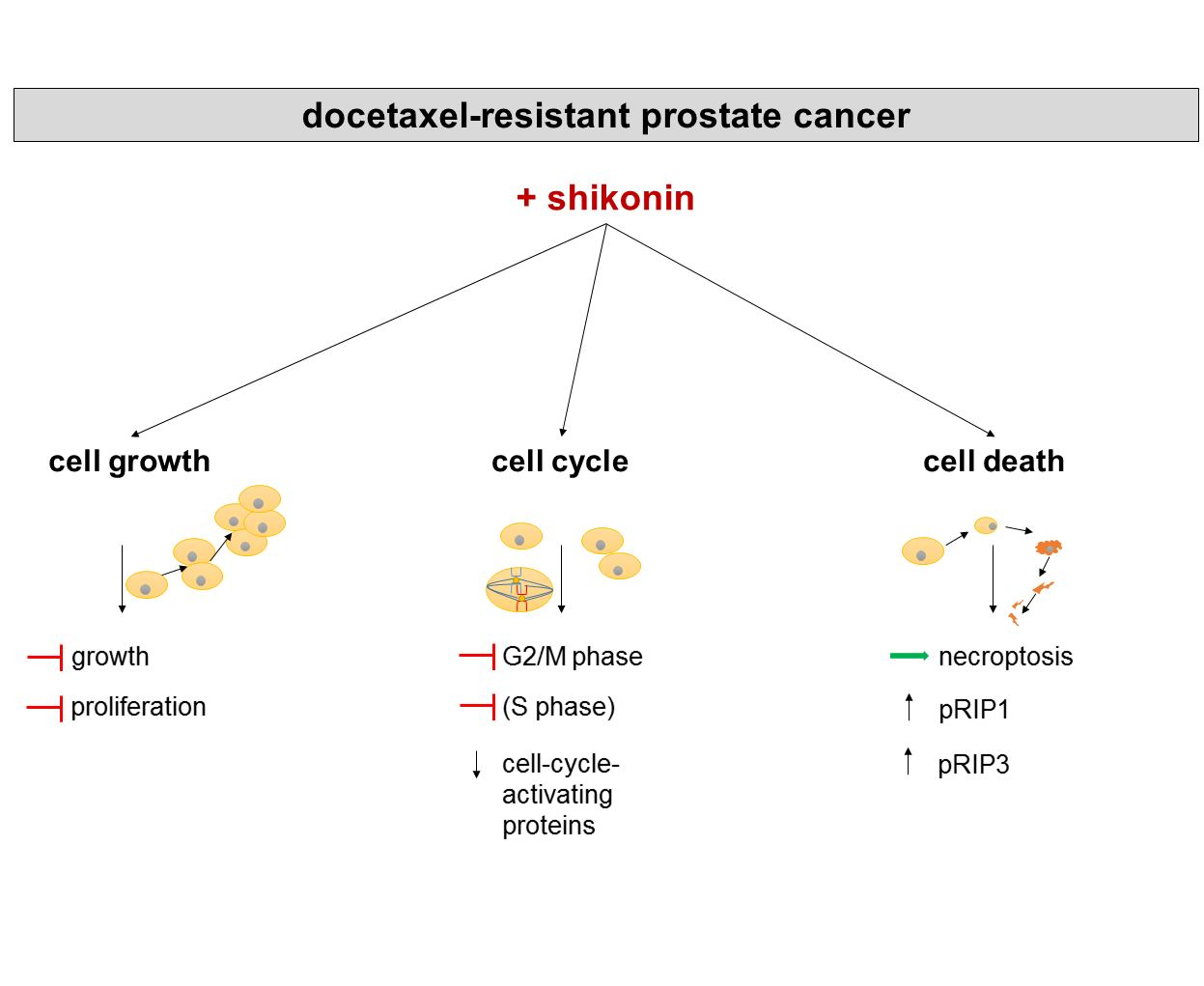
Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
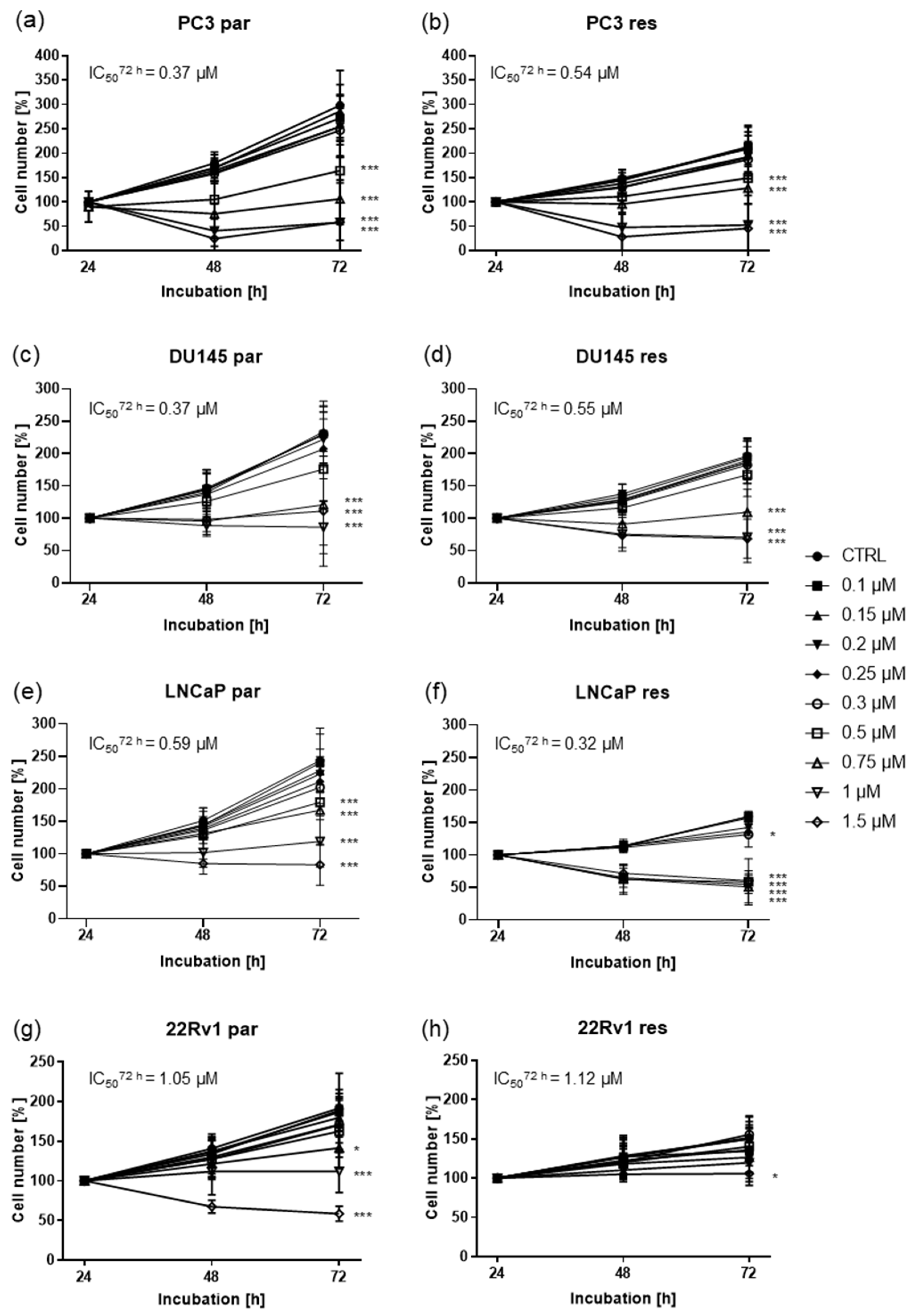
2.1. Shikonin Inhibited Cell Growth of Parental and DX-Resistant PCa Cells
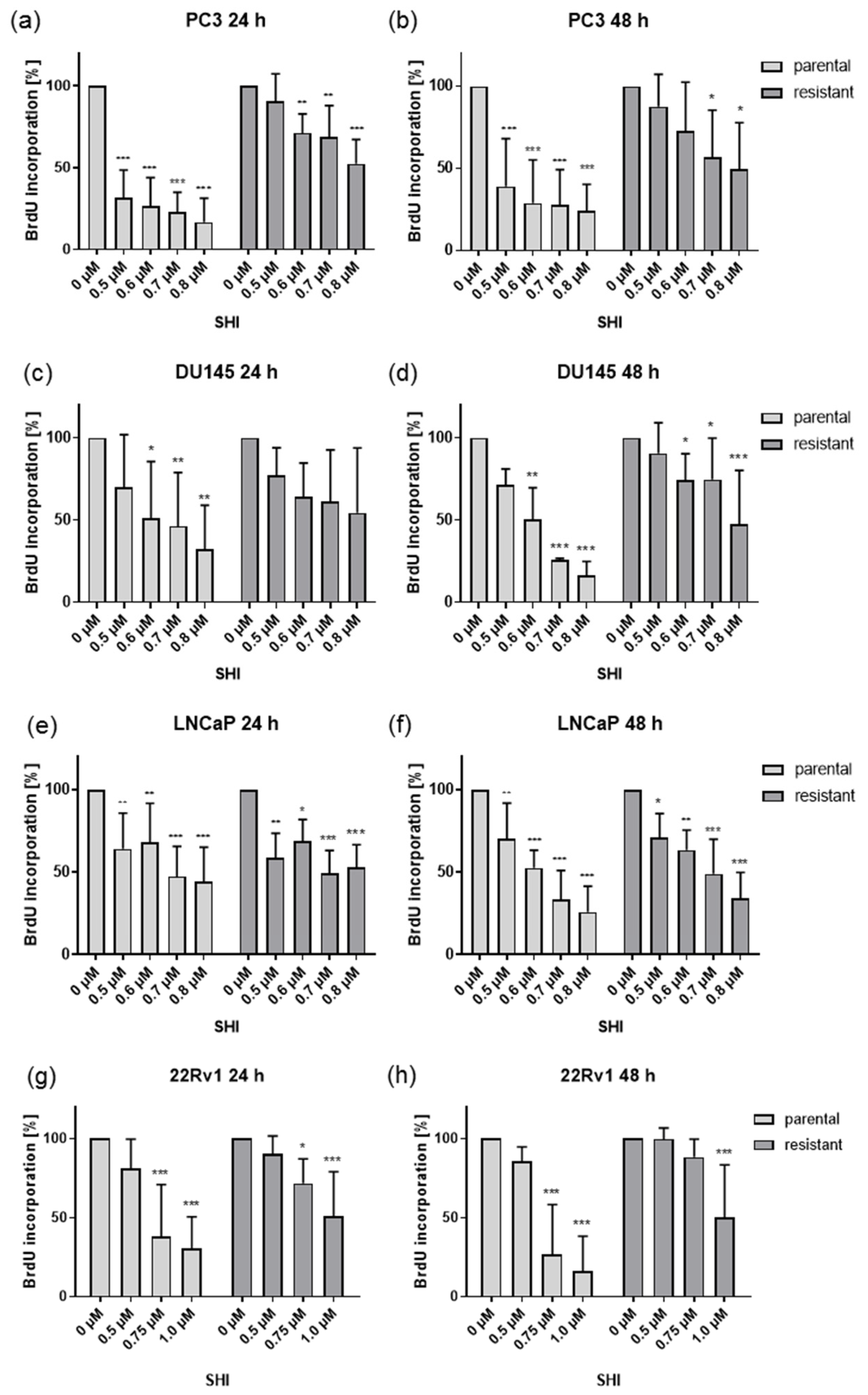
2.2. Shikonin Impaired PCa Cell Proliferation
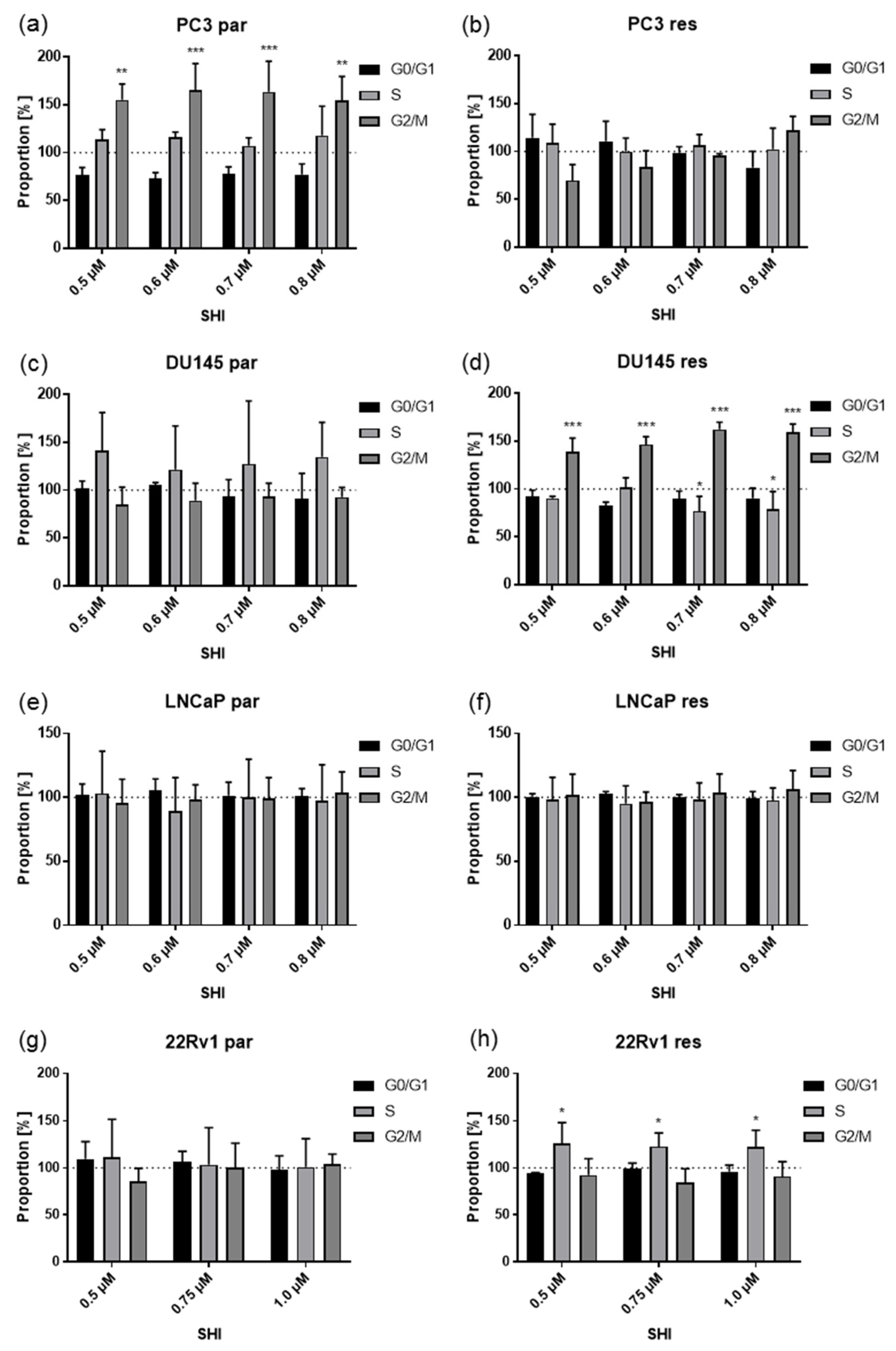
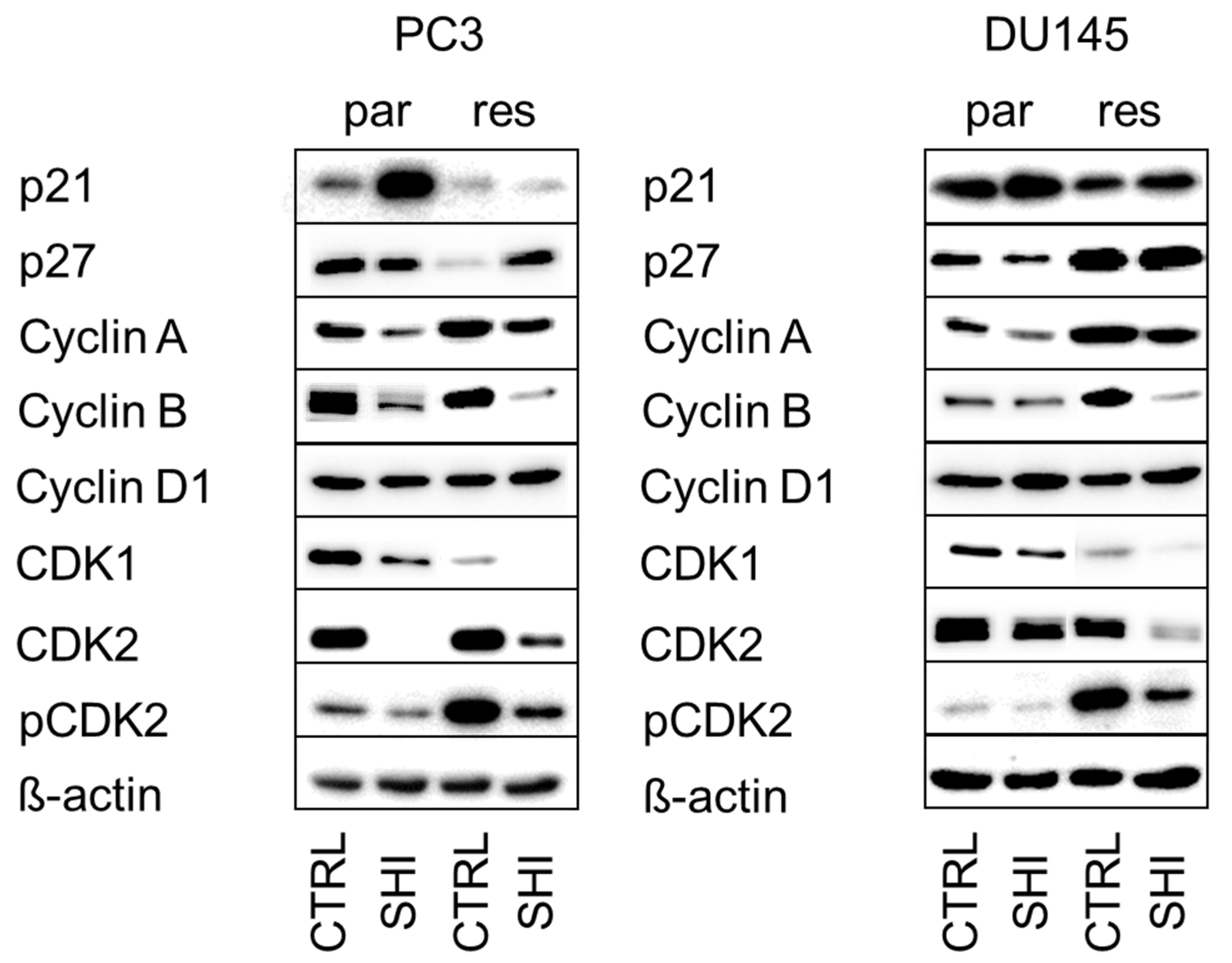
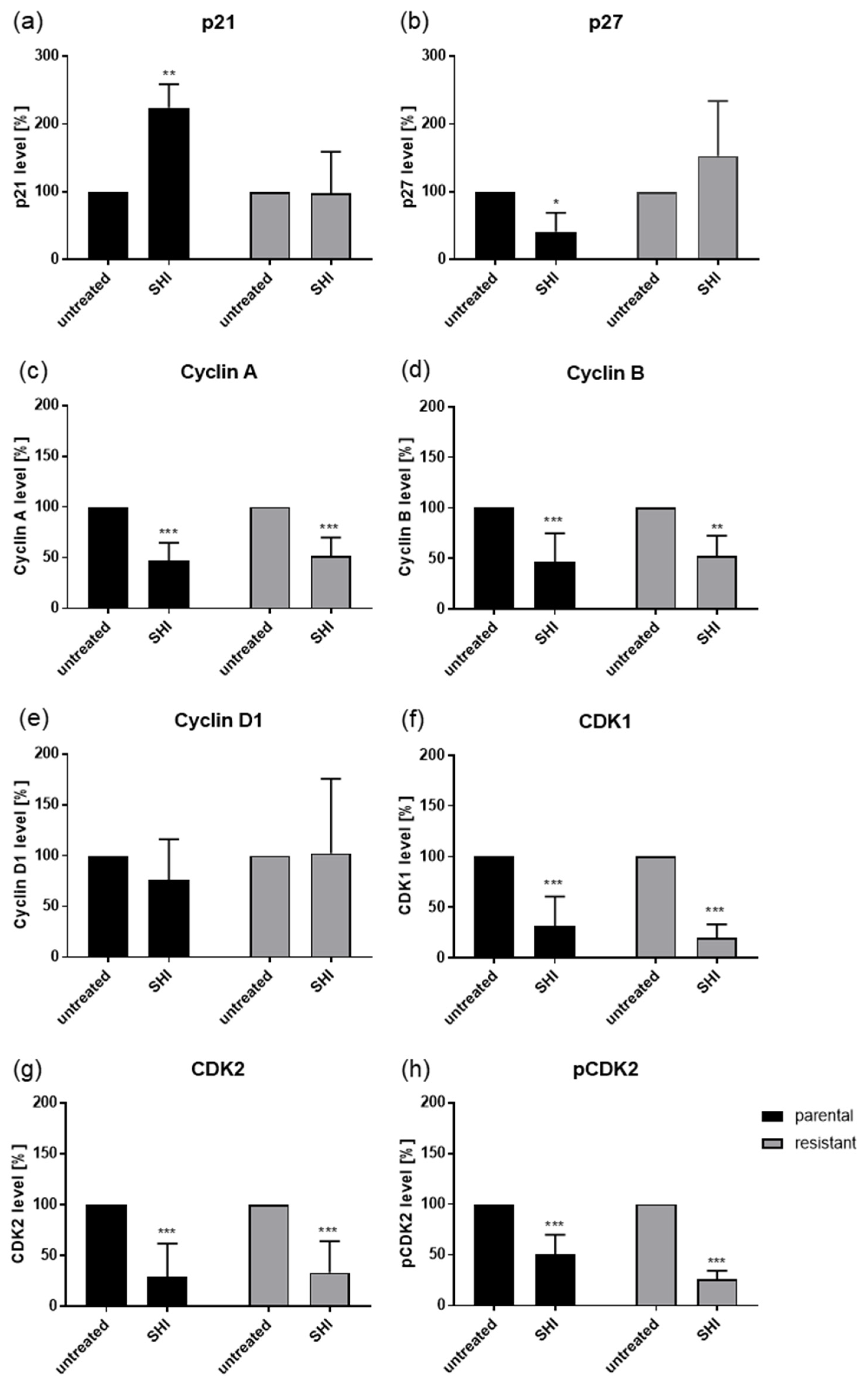
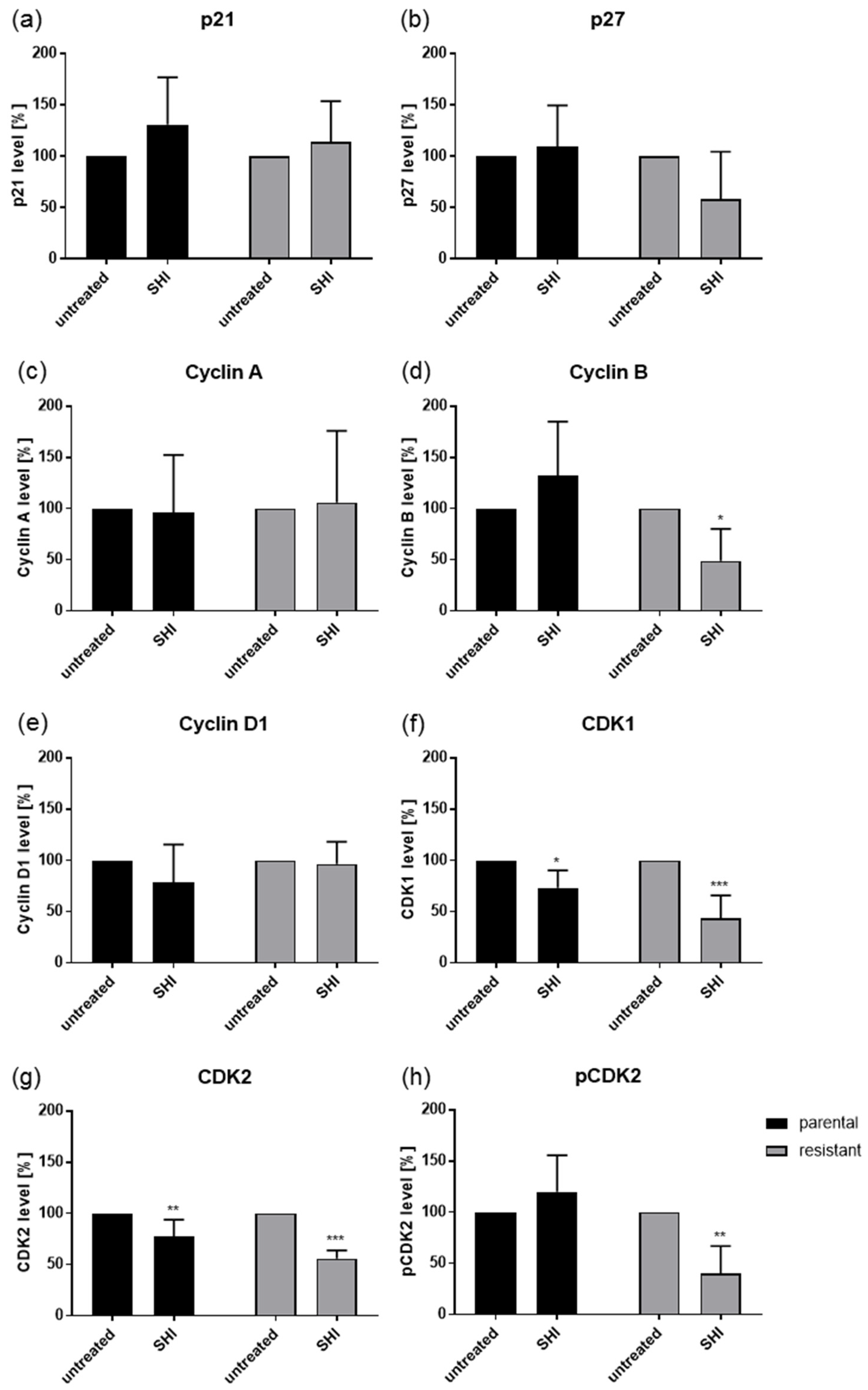
2.3. Shikonin Induced Cell Cycle Arrest and Alterations in the Expression and Activity of Cell Cycle Regulating Proteins
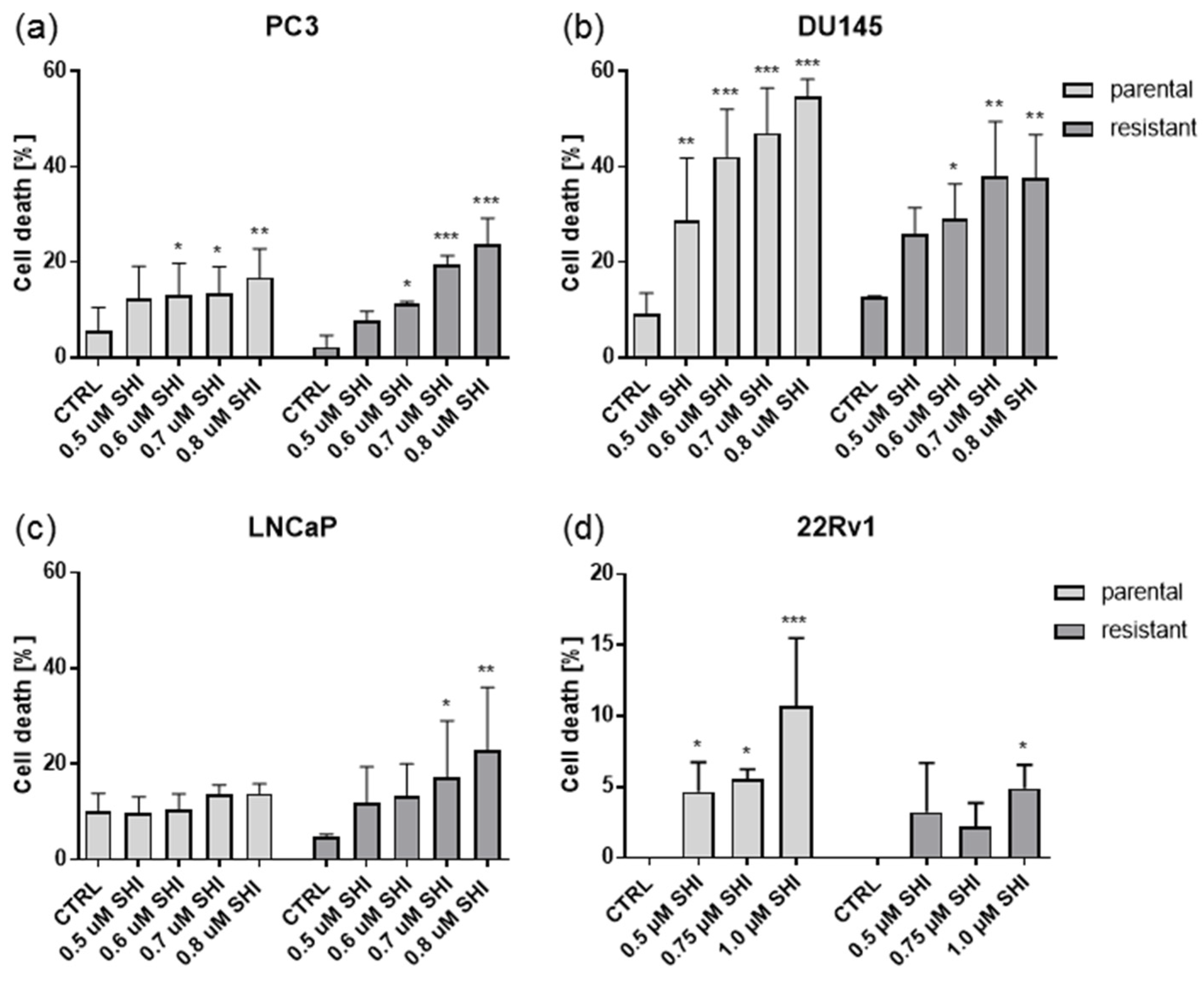
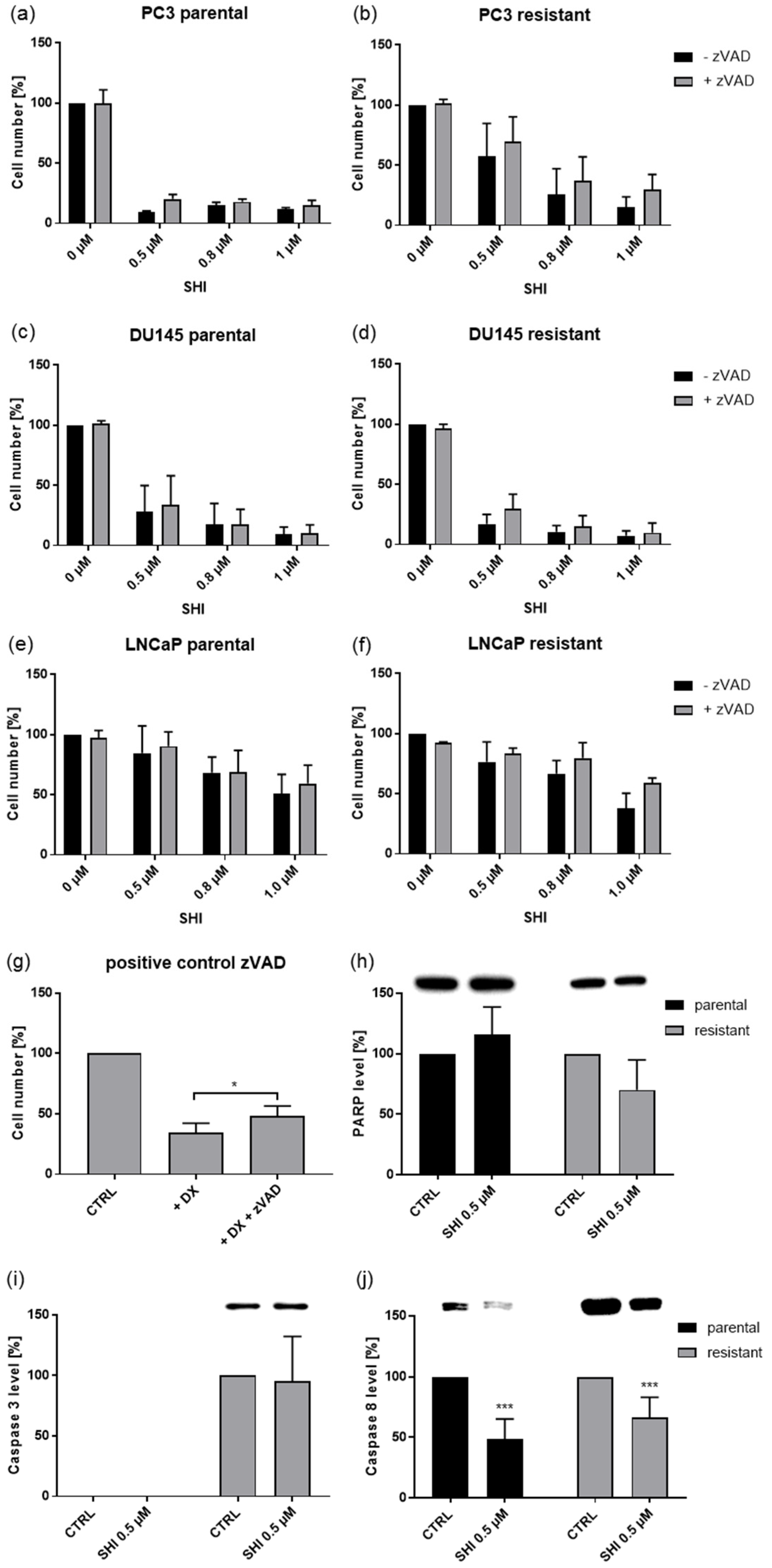
2.4. Shikonin Induced Cell Death
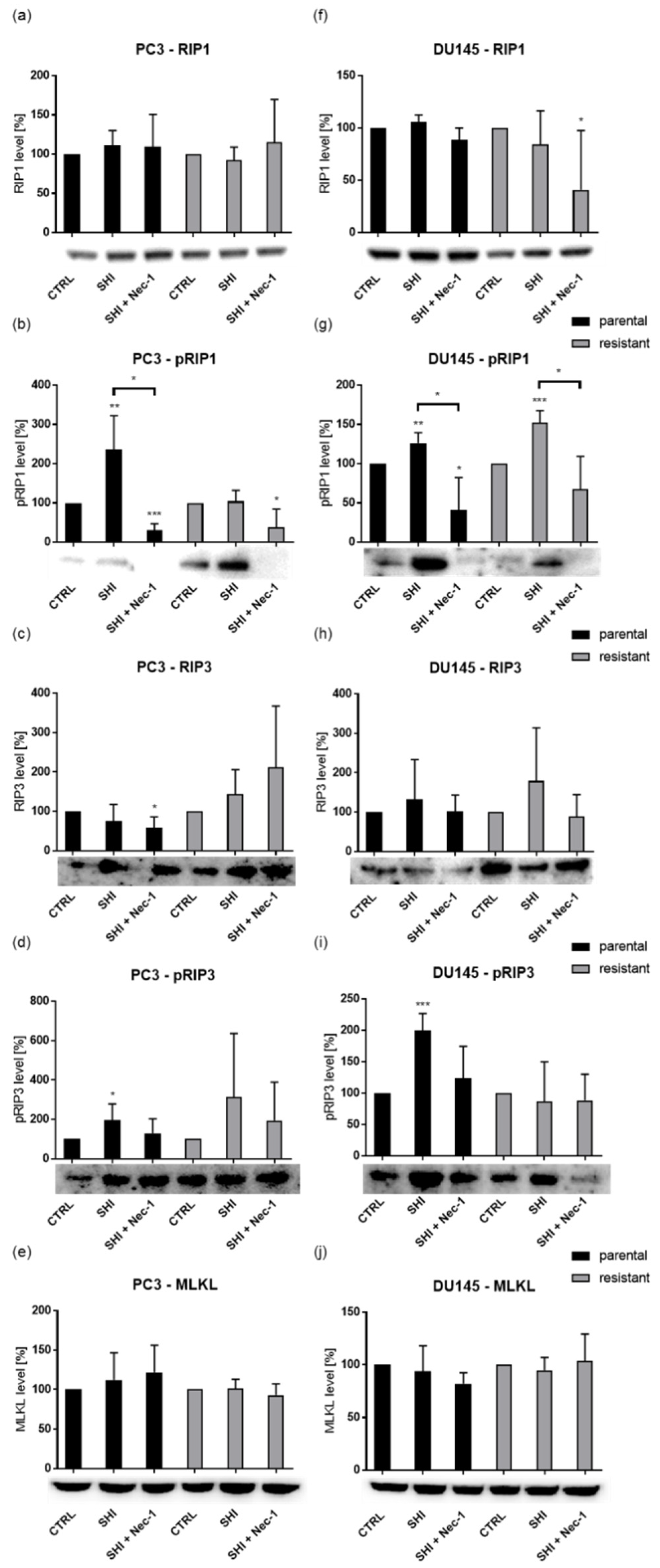
2.5. Shikonin Induced Necroptotic Effects
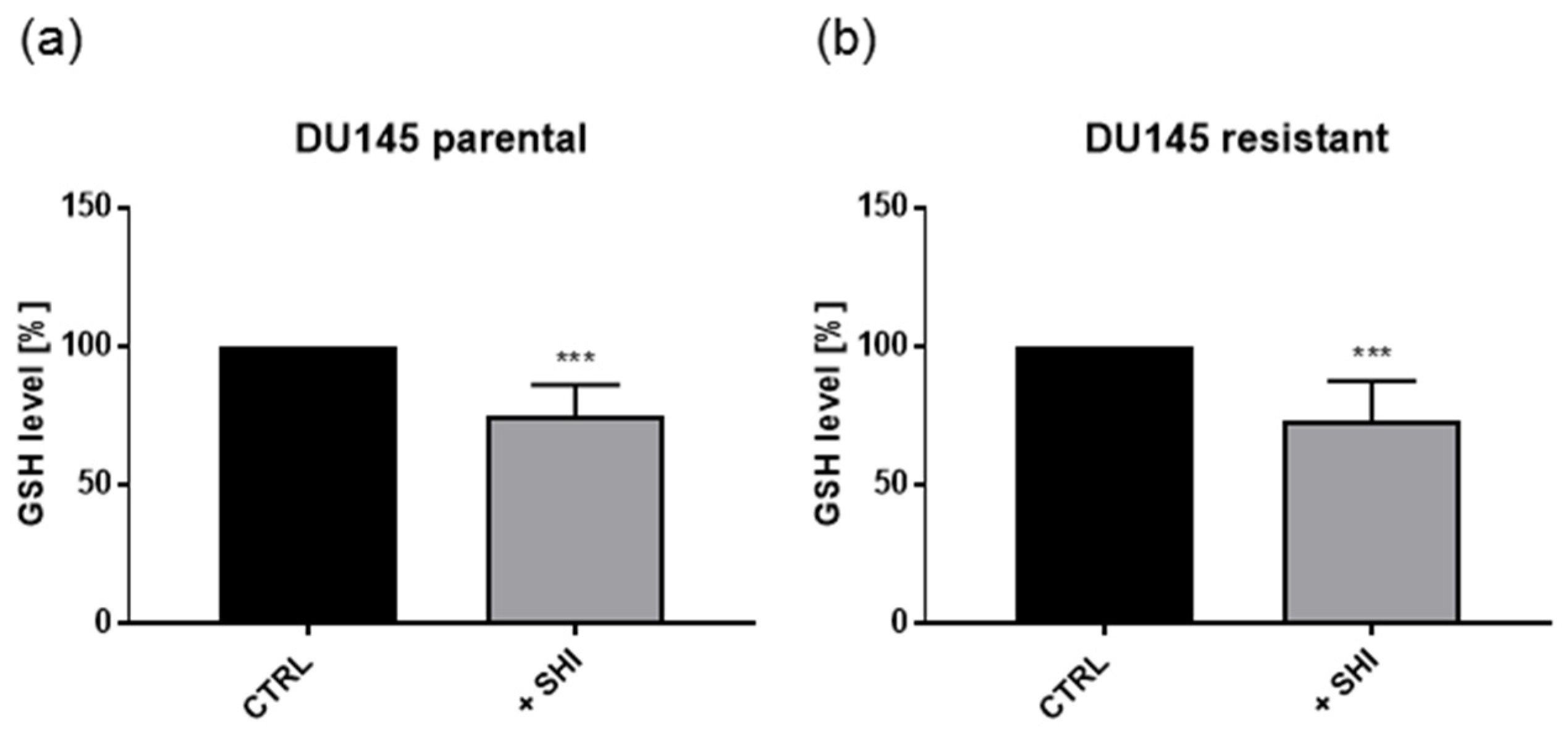
2.6. Shikonin Showed No Effects on Metabolism
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Resistance Induction and Application of Docetaxel and Shikonin
4.3. Tumor Cell Growth
4.4. Proliferation
4.5. Cell Cycle Phase Distribution
4.6. Western Blot Analysis of Cell Cycle and Cell Death Regulating Proteins
4.7. Cell Death
4.8. GSH-Assay
4.9. Evaluation of Mitochondrial Respiration and Anaerobic Glycolytic Activity
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cornford, P.; Bellmunt, J.; Bolla, M.; Briers, E.; De Santis, M.; Gross, T.; Henry, A.M.; Joniau, S.; Lam, T.B.; Mason, M.D.; et al. EAU-ESTRO-SIOG Guidelines on Prostate Cancer. Part II: Treatment of Relapsing, Metastatic, and Castration-Resistant Prostate Cancer. Eur. Urol. 2017, 71, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Tannock, I.F.; De Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Theodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus Prednisone or Mitoxantrone plus Prednisone for Advanced Prostate Cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Faivre, L.; Lesaunier, F.; Delva, R.; Gravis, G.; Rolland, F.; Priou, F.; Ferrero, J.-M.; Houede, N.; Mourey, L.; et al. Androgen deprivation therapy plus docetaxel and estramustine versus androgen deprivation therapy alone for high-risk localised prostate cancer (GETUG 12): A phase 3 randomised controlled trial. Lancet Oncol. 2015, 16, 787–794. [Google Scholar] [CrossRef]
- Rosenthal, S.A.; Hu, C.; Sartor, O.; Gomella, L.G.; Amin, M.B.; Purdy, J.; Michalski, J.M.; Garzotto, M.G.; Pervez, N.; Balogh, A.G.; et al. Effect of Chemotherapy With Docetaxel With Androgen Suppression and Radiotherapy for Localized High-Risk Prostate Cancer: The Randomized Phase III NRG Oncology RTOG 0521 Trial. J. Clin. Oncol. 2019, 37, 1159–1168. [Google Scholar] [CrossRef]
- Armstrong, A.; Garrett-Mayer, E.; De Wit, R.; Tannock, I.; Eisenberger, M. Prediction of Survival following First-Line Chemotherapy in Men with Castration-Resistant Metastatic Prostate Cancer. Clin. Cancer Res. 2009, 16, 203–211. [Google Scholar] [CrossRef]
- Thelen, P.; Gschwend, J.; Wolff, J.-M.; Miller, K. Resistenzmechanismen unter antihormoneller Therapie des fortgeschrittenen Prostatakarzinoms. Aktuel. Urol. 2016, 47, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Poonthananiwatkul, B.; Howard, R.L.; Williamson, E.M.; Lim, R.H. Cancer patients taking herbal medicines: A review of clinical purposes, associated factors, and perceptions of benefit or harm. J. Ethnopharmacol. 2015, 175, 58–66. [Google Scholar] [CrossRef]
- Saghatchian, M.; Bihan, C.; Chenailler, C.; Mazouni, C.; Dauchy, S.; Delaloge, S. Exploring frontiers: Use of complementary and alternative medicine among patients with early-stage breast cancer. Breast 2014, 23, 279–285. [Google Scholar] [CrossRef]
- Ebel, M.-D.; Rudolph, I.; Keinki, C.; Hoppe, A.; Muecke, R.; Micke, O.; Muenstedt, K.; Huebner, J. Perception of cancer patients of their disease, self-efficacy and locus of control and usage of complementary and alternative medicine. J. Cancer Res. Clin. Oncol. 2015, 141, 1449–1455. [Google Scholar] [CrossRef]
- Horneber, M.; Bueschel, G.; Dennert, G.; Less, D.; Ritter, E.; Zwahlen, M. How Many Cancer Patients Use Complementary and Alternative Medicine. Integr. Cancer Ther. 2011, 11, 187–203. [Google Scholar] [CrossRef]
- Huebner, J.; Micke, O.; Muecke, R.; Buentzel, J.; Prott, F.J.; Kleeberg, U.; Senf, B.; Muenstedt, K. User rate of comple-mentary and alternative medicine (CAM) of patients visiting a counseling facility for CAM of a German comprehensive cancer center. Anticancer Res. 2014, 34, 943–948. [Google Scholar] [PubMed]
- Christensen, C.M.; Morris, R.S.; Kapsandoy, S.C.; Archer, M.; Kuang, J.; Shane-McWhorter, L.; Bray, B.E.; Zeng-Treitler, Q. Patient needs and preferences for herb-drug-disease interaction alerts: A structured interview study. BMC Complement. Altern. Med. 2017, 17, 272. [Google Scholar] [CrossRef] [PubMed]
- Mani, J.; Juengel, E.; Arslan, I.; Bartsch, G.; Filmann, N.; Ackermann, H.; Nelson, K.; Haferkamp, A.; Engl, T.; Blaheta, R.A. Use of complementary and alternative medicine before and after organ removal due to urologic cancer. Patient Prefer. Adherence 2015, 9, 1407–1412. [Google Scholar] [CrossRef]
- Kessel, K.A.; Lettner, S.; Kessel, C.; Bier, H.; Biedermann, T.; Friess, H.; Herrschbach, P.; Gschwend, J.E.; Meyer, B.; Peschel, C.; et al. Use of Complementary and Alternative Medicine (CAM) as Part of the Oncological Treatment: Survey about Patients’ Attitude towards CAM in a University-Based Oncology Center in Germany. PLoS ONE 2016, 11, e0165801. [Google Scholar] [CrossRef] [PubMed]
- Juengel, E.; Thomas, A.; Rutz, J.; Makarevic, J.; Tsaur, I.; Nelson, K.; Haferkamp, A.; Blaheta, R.A. Amygdalin inhibits the growth of renal cell carcinoma cells in vitro. Int. J. Mol. Med. 2015, 37, 526–532. [Google Scholar] [CrossRef]
- Rutz, J.; Maxeiner, S.; Juengel, E.; Bernd, A.; Kippenberger, S.; Zöller, N.; Chun, F.K.-H.; Blaheta, R.A. Growth and Proliferation of Renal Cell Carcinoma Cells Is Blocked by Low Curcumin Concentrations Combined with Visible Light Irradiation. Int. J. Mol. Sci. 2019, 20, 1464. [Google Scholar] [CrossRef]
- Lee, H.M.; Moon, A. Amygdalin Regulates Apoptosis and Adhesion in Hs578T Triple-Negative Breast Cancer Cells. Biomol. Ther. 2016, 24, 62–66. [Google Scholar] [CrossRef]
- Wang, F.; Wang, W.; Li, J.; Zhang, J.; Wang, X.; Wang, M. Sulforaphane reverses gefitinib tolerance in human lung cancer cells via modulation of sonic hedgehog signaling. Oncol. Lett. 2017, 15, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Ma, X.; Wang, Z.; Zeng, X.; Hu, Z.; Ye, Z.; Shen, G. Curcumin induces apoptosis and protective autophagy in castration-resistant prostate cancer cells through iron chelation. Drug Des. Dev. Ther. 2017, 11, 431–439. [Google Scholar] [CrossRef]
- Singh, S.K. Combinatorial effect of curcumin with docetaxel modulates apoptotic and cell survival molecules in prostate cancer. Front. Biosci. 2017, 9, 235–245. [Google Scholar] [CrossRef]
- Markowitsch, S.D.; Schupp, P.; Lauckner, J.; Vakhrusheva, O.; Slade, K.S.; Mager, R.; Efferth, T.; Haferkamp, A.; Juengel, E. Artesunate Inhibits Growth of Sunitinib-Resistant Renal Cell Carcinoma Cells through Cell Cycle Arrest and Induction of Ferroptosis. Cancers 2020, 12, 3150. [Google Scholar] [CrossRef]
- Papageorgiou, V.P.; Assimopoulou, A.N.; Couladouros, E.A.; Hepworth, D.; Nicolaou, K.C. The Chemistry and Biology of Alkannin, Shikonin, and Related Naphthazarin Natural Products. Angew. Chem. Int. Ed. 1999, 38, 270–301. [Google Scholar] [CrossRef]
- Lu, L.; Qin, A.; Huang, H.; Zhou, P.; Zhang, C.; Liu, N.; Li, S.; Wen, G.; Zhang, C.; Dong, W.; et al. Shikonin extracted from medicinal Chinese herbs exerts anti-inflammatory effect via proteasome inhibition. Eur. J. Pharmacol. 2011, 658, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Thonsri, U.; Seubwai, W.; Waraasawapati, S.; Wongkham, S.; Boonmars, T.; Cha’On, U.; Wongkham, C. Antitumor Effect of Shikonin, a PKM2 Inhibitor, in Cholangiocarcinoma Cell Lines. Anticancer. Res. 2020, 40, 5115–5124. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Pozo, F.M.; Tian, D.; Geng, X.; Yao, X.; Zhang, Y.; Tang, J. Shikonin Inhibits Cancer Through P21 Upregulation and Apoptosis Induction. Front. Pharmacol. 2020, 11, 861. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Kim, J.M.; Shim, S.H.; Chang, H.I. Shikonin induces cell cycle arrest in human gastric cancer (AGS) by early growth response 1 (Egr1)-mediated p21 gene expression. J. Ethnopharmacol. 2014, 151, 1064–1071. [Google Scholar] [CrossRef]
- Liang, W.; Cai, A.; Chen, G.; Xi, H.; Wu, X.; Cui, J.; Zhang, K.; Zhao, X.; Yu, J.; Wei, B.; et al. Shikonin induces mitochondria-mediated apoptosis and enhances chemotherapeutic sensitivity of gastric cancer through reactive oxygen species. Sci. Rep. 2016, 6, 38267. [Google Scholar] [CrossRef]
- Chen, C.; Xiao, W.; Huang, L.; Yu, G.; Ni, J.; Yang, L.; Wan, R.; Hu, G. Shikonin induces apoptosis and necroptosis in pancreatic cancer via regulating the expression of RIP1/RIP3 and synergizes the activity of gemcitabine. Am. J. Transl. Res. 2017, 9, 5507–5517. [Google Scholar]
- Kim, H.-J.; Hwang, K.-E.; Park, D.-S.; Oh, S.-H.; Jun, H.Y.; Yoon, K.-H.; Jeong, E.-T.; Kim, H.-R.; Kim, Y.-S. Shikonin-induced necroptosis is enhanced by the inhibition of autophagy in non-small cell lung cancer cells. J. Transl. Med. 2017, 15, 1–12. [Google Scholar] [CrossRef]
- Ding, Y.; He, C.; Lu, S.; Wang, X.; Wang, C.; Wang, L.; Zhang, J.; Piao, M.; Chi, G.; Luo, Y.; et al. MLKL contributes to shikonin-induced glioma cell necroptosis via promotion of chromatinolysis. Cancer Lett. 2019, 467, 58–71. [Google Scholar] [CrossRef]
- Shahsavari, Z.; Karami-Tehrani, F.; Salami, S.; Ghasemzadeh, M. RIP1K and RIP3K provoked by shikonin induce cell cycle arrest in the triple negative breast cancer cell line, MDA-MB-468: Necroptosis as a desperate programmed suicide pathway. Tumor Biol. 2015, 37, 4479–4491. [Google Scholar] [CrossRef] [PubMed]
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A regulated inflammatory mode of cell death. J. Neuroinflamm. 2018, 15, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Fan, Z.; Luo, G.; Yang, C.; Huang, Q.; Fan, K.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. The role of necroptosis in cancer biology and therapy. Mol. Cancer 2019, 18, 1–17. [Google Scholar] [CrossRef]
- Wang, Y.; Hao, F.; Nan, Y.; Qu, L.; Na, W.; Jia, C.; Chen, X. PKM2 Inhibitor Shikonin Overcomes the Cisplatin Resistance in Bladder Cancer by Inducing Necroptosis. Int. J. Biol. Sci. 2018, 14, 1883–1891. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Jin, J.; Qian, W.; Ye, X. Shikonin Overcomes Drug Resistance and Induces Necroptosis By Regulating the Mir-92a-1-5p/Mlkl Axis in Chronic Myeloid Leukemia Cells. Blood 2019, 134, 1633. [Google Scholar] [CrossRef]
- Tang, J.-C.; Ren, Y.-G.; Zhao, J.; Long, F.; Chen, J.-Y.; Jiang, Z. Shikonin enhances sensitization of gefitinib against wild-type EGFR non-small cell lung cancer via inhibition PKM2/stat3/cyclinD1 signal pathway. Life Sci. 2018, 204, 71–77. [Google Scholar] [CrossRef]
- Zhao, X.; Zhu, Y.; Hu, J.; Jiang, L.; Li, L.; Jia, S.; Zen, K. Shikonin Inhibits Tumor Growth in Mice by Suppressing Pyruvate Kinase M2-mediated Aerobic Glycolysis. Sci. Rep. 2018, 8, 1–8. [Google Scholar] [CrossRef]
- Wiench, B.; Eichhorn, T.; Paulsen, M.; Efferth, T. Shikonin Directly Targets Mitochondria and Causes Mitochondrial Dysfunction in Cancer Cells. Evid.-Based Complement. Altern. Med. 2012, 2012, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.; Sicinski, T.O.P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef]
- Ingham, M.; Schwartz, G.K. Cell-Cycle Therapeutics Come of Age. J. Clin. Oncol. 2017, 35, 2949–2959. [Google Scholar] [CrossRef]
- Ogura, T.; Tanaka, Y.; Tamaki, H.; Harada, M. Docetaxel induces Bcl-2- and pro-apoptotic caspase-independent death of human prostate cancer DU145 cells. Int. J. Oncol. 2016, 48, 2330–2338. [Google Scholar] [CrossRef] [PubMed]
- Jeung, Y.-J.; Kim, H.-G.; Ahn, J.; Lee, H.-J.; Lee, S.-B.; Won, M.; Jung, C.-R.; Im, J.-Y.; Kim, B.-K.; Park, S.-K.; et al. Shikonin induces apoptosis of lung cancer cells via activation of FOXO3a/EGR1/SIRT1 signaling antagonized by p300. Biochim. Biophys. Acta BBA Bioenergy 2016, 1863, 2584–2593. [Google Scholar] [CrossRef] [PubMed]
- Zhai, T.; Hei, Z.; Ma, Q.; Liang, H.; Xu, Y.; Zhang, Y.; Jin, L.; Han, C.; Wang, J. Shikonin induces apoptosis and G0/G1�phase arrest of gallbladder cancer cells via the JNK signaling pathway. Oncol. Rep. 2017, 38, 3473–3480. [Google Scholar] [CrossRef][Green Version]
- Du, W.; Hao, X.; Yuan, Z.; Wang, Y.; Zhang, X.; Liu, J. Shikonin potentiates paclitaxel antitumor efficacy in esophageal cancer cells via the apoptotic pathway. Oncol. Lett. 2019, 18, 3195–3201. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Li, M.; Cui, S.; Wang, D.; Zhang, C.-Y.; Zen, K.; Li, L. Shikonin Inhibits the Proliferation of Human Breast Cancer Cells by Reducing Tumor-Derived Exosomes. Molecules 2016, 21, 777. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Z.; Li, Q.; Jiao, H.; Chong, D.; Sun, X.; Zhang, P.; Huo, Q.; Liu, H. Shikonin induces necroptosis by reactive oxygen species activation in nasopharyngeal carcinoma cell line CNE-2Z. J. Bioenerg. Biomembr. 2017, 49, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zheng, L.; Liu, J.; Zhou, Z.; Cao, X.; Lv, X.; Chen, F. Shikonin inhibits prostate cancer cells metastasis by reducing matrix metalloproteinase-2/-9 expression via AKT/mTOR and ROS/ERK1/2 pathways. Int. Immunopharmacol. 2014, 21, 447–455. [Google Scholar] [CrossRef]
- Jang, S.Y.; Jang, E.H.; Jeong, S.Y.; Kim, J.-H. Shikonin inhibits the growth of human prostate cancer cells via modulation of the androgen receptor. Int. J. Oncol. 2014, 44, 1455–1460. [Google Scholar] [CrossRef]
- Liu, Y.; Kang, X.; Niu, G.; He, S.; Zhang, T.; Bai, Y.; Li, Y.; Hao, H.; Chen, C.; Shou, Z.; et al. Shikonin induces apoptosis and prosurvival autophagy in human melanoma A375 cells via ROS-mediated ER stress and p38 pathways. Artif. Cells Nanomed. Biotechnol. 2019, 47, 626–635. [Google Scholar] [CrossRef]
- Trochon, V.; Blot, E.; Cymbalista, F.; Engelmann, C.; Tang, R.P.; Thomaïdis, A.; Vasse, M.; Soria, J.; Lu, H.; Soria, C. Apigenin inhibits endothelial-cell proliferation in G2/M phase whereas it stimulates smooth-muscle cells by inhibiting P21 and P27 expression. Int. J. Cancer 2000, 85, 691–696. [Google Scholar] [CrossRef]
- Yadav, V.; Sultana, S.; Yadav, J.; Saini, N. Gatifloxacin Induces S and G2-Phase Cell Cycle Arrest in Pancreatic Cancer Cells via p21/p27/p53. PLoS ONE 2012, 7, e47796. [Google Scholar] [CrossRef] [PubMed]
- Gavet, O.; Pines, J. Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev. Cell. 2010, 18, 533–543. [Google Scholar] [CrossRef]
- Tang, J.-C.; Zhao, J.; Long, F.; Chen, J.-Y.; Mu, B.; Jiang, Z.; Ren, Y.; Yang, J. Efficacy of Shikonin against Esophageal Cancer Cells and its possible mechanisms in vitro and in vivo. J. Cancer 2018, 9, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Assimopoulou, A.N.; Klauck, S.M.; Damianakos, H.; Chinou, I.; Kretschmer, N.; Ríos, J.-L.; Papageorgiou, V.P.; Bauer, R.; Efferth, T. Inhibition of c-MYC with involvement of ERK/JNK/MAPK and AKT pathways as a novel mechanism for shikonin and its derivatives in killing leukemia cells. Oncotarget 2015, 6, 38934–38951. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Deng, B.; Liao, Y.; Shan, L.; Yin, F.; Wang, Z.; Zeng, H.; Zuo, D.; Hua, Y.; Cai, Z. The anti-tumor effect of shikonin on osteosarcoma by inducing RIP1 and RIP3 dependent necroptosis. BMC Cancer 2013, 13, 580. [Google Scholar] [CrossRef]
- Lee, M.-J.; Kao, S.-H.; Hunag, J.-E.; Sheu, G.-T.; Yeh, C.-W.; Hseu, Y.-C.; Wang, C.-J.; Hsu, L.-S. Shikonin time-dependently induced necrosis or apoptosis in gastric cancer cells via generation of reactive oxygen species. Chem. Interact. 2014, 211, 44–53. [Google Scholar] [CrossRef]
- Diao, Y.; Ma, X.; Min, W.; Lin, S.; Kang, H.; Dai, Z.; Wang, X.; Zhao, Y. Dasatinib promotes paclitaxel-induced necroptosis in lung adenocarcinoma with phosphorylated caspase-8 by c-Src. Cancer Lett. 2016, 379, 12–23. [Google Scholar] [CrossRef]
- Mann, J.; Yang, N.; Montpetit, R.; Kirschenman, R.; Lemieux, H.; Goping, I.S. BAD sensitizes breast cancer cells to docetaxel with increased mitotic arrest and necroptosis. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Wu, X.; Wu, M.-Y.; Jiang, M.; Zhi, Q.; Bian, X.; Xu, M.-D.; Gong, F.-R.; Hou, J.; Tao, M.; Shou, L.-M.; et al. TNF-α sensitizes chemotherapy and radiotherapy against breast cancer cells. Cancer Cell Int. 2017, 17, 1–12. [Google Scholar] [CrossRef]
- Zhao, Q.; Kretschmer, N.; Bauer, R.; Efferth, T. Shikonin and its derivatives inhibit the epidermal growth factor receptor signaling and synergistically kill glioblastoma cells in combination with erlotinib. Int. J. Cancer 2015, 137, 1446–1456. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Xie, J.; Pan, Q.; Wang, B.; Hu, D.; Hu, X. Anticancer Agent Shikonin Is an Incompetent Inducer of Cancer Drug Resistance. PLoS ONE 2013, 8, e52706. [Google Scholar] [CrossRef]
- Lu, B.; Gong, X.; Wang, Z.-Q.; Ding, Y.; Wang, C.; Luo, T.-F.; Piao, M.-H.; Meng, F.-K.; Chi, G.-F.; Luo, Y.-N.; et al. Shikonin induces glioma cell necroptosis in vitro by ROS overproduction and promoting RIP1/RIP3 necrosome formation. Acta Pharmacol. Sin. 2017, 38, 1543–1553. [Google Scholar] [CrossRef]
- Rodriguez, D.; Weinlich, R.; Brown, S.L.; Guy, C.T.; Fitzgerald, P.J.; Dillon, C.P.; Oberst, A.; Quarato, G.; Low, J.; Cripps, J.G.; et al. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 2016, 23, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Lu, B.; Wang, C.; Wang, Z.; Luo, T.; Piao, M.; Meng, F.; Chi, G.; Luo, Y.; Ge, P. RIP1 and RIP3 contribute to shikonin-induced DNA double-strand breaks in glioma cells via increase of intracellular reactive oxygen species. Cancer Lett. 2017, 390, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Horoszewicz, J.S.; Leong, S.S.; Kawinski, E.; Karr, J.P.; Rosenthal, H.; Chu, T.M.; A Mirand, E.; Murphy, G.P. LNCaP model of human prostatic carcinoma. Cancer Res. 1983, 43, 1809–1818. [Google Scholar] [PubMed]
- Alimirah, F.; Chen, J.; Basrawala, Z.; Xin, H.; Choubey, D. DU-145 and PC-3 human prostate cancer cell lines express androgen receptor: Implications for the androgen receptor functions and regulation. FEBS Lett. 2006, 580, 2294–2300. [Google Scholar] [CrossRef]
- Wen, S.; Niu, Y.; Lee, S.O.; Chang, C. Androgen receptor (AR) positive vs negative roles in prostate cancer cell deaths including apoptosis, anoikis, entosis, necrosis and autophagic cell death. Cancer Treat. Rev. 2014, 40, 31–40. [Google Scholar] [CrossRef]
- Metzig, M.O.; Fuchs, D.; E Tagscherer, K.; Grone, H.; Schirmacher, P.; Roth, W. Inhibition of caspases primes colon cancer cells for 5-fluorouracil-induced TNF-α-dependent necroptosis driven by RIP1 kinase and NF-κB. Oncogene 2016, 35, 3399–3409. [Google Scholar] [CrossRef]
- Xu, Y.; Ma, H.-B.; Fang, Y.-L.; Zhang, Z.-R.; Shao, J.; Hong, M.; Huang, C.-J.; Liu, J.; Chen, R.-Q. Cisplatin-induced necroptosis in TNFα dependent and independent pathways. Cell. Signal. 2017, 31, 112–123. [Google Scholar] [CrossRef]
- Chen, J.; Xie, J.; Jiang, Z.; Wang, B.; Wang, Y.; Hu, X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene 2011, 30, 4297–4306. [Google Scholar] [CrossRef]
- Shimada, N.; Takasawa, R.; Tanuma, S.-I. Interdependence of GLO I and PKM2 in the Metabolic shift to escape apoptosis in GLO I-dependent cancer cells. Arch. Biochem. Biophys. 2018, 638, 1–7. [Google Scholar] [CrossRef]
- Shahsavari, Z.; Karami-Tehrani, F.; Salami, S. Shikonin Induced Necroptosis via Reactive Oxygen Species in the T-47D Breast Cancer Cell Line. Asian Pac. J. Cancer Prev. 2015, 16, 7261–7266. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, M.; Wass, M.N.; Cinatl, J. Drug-adapted cancer cell lines as preclinical models of acquired resistance. Cancer Drug Resist. 2019, 2, 447–456. [Google Scholar] [CrossRef]
- Michaelis, M.; Rothweiler, F.; Barth, S.; Cinatl, J.; Van Rikxoort, M.; Löschmann, N.; Voges, Y.; Breitling, R.; Von Deimling, A.; Rödel, F.; et al. Adaptation of cancer cells from different entities to the MDM2 inhibitor nutlin-3 results in the emergence of p53-mutated multi-drug-resistant cancer cells. Cell Death Dis. 2011, 2, e243. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markowitsch, S.D.; Juetter, K.M.; Schupp, P.; Hauschulte, K.; Vakhrusheva, O.; Slade, K.S.; Thomas, A.; Tsaur, I.; Cinatl, J., Jr.; Michaelis, M.; et al. Shikonin Reduces Growth of Docetaxel-Resistant Prostate Cancer Cells Mainly through Necroptosis. Cancers 2021, 13, 882. https://doi.org/10.3390/cancers13040882
Markowitsch SD, Juetter KM, Schupp P, Hauschulte K, Vakhrusheva O, Slade KS, Thomas A, Tsaur I, Cinatl J Jr., Michaelis M, et al. Shikonin Reduces Growth of Docetaxel-Resistant Prostate Cancer Cells Mainly through Necroptosis. Cancers. 2021; 13(4):882. https://doi.org/10.3390/cancers13040882
Chicago/Turabian StyleMarkowitsch, Sascha D., Kira M. Juetter, Patricia Schupp, Kristine Hauschulte, Olesya Vakhrusheva, Kimberly Sue Slade, Anita Thomas, Igor Tsaur, Jindrich Cinatl, Jr., Martin Michaelis, and et al. 2021. "Shikonin Reduces Growth of Docetaxel-Resistant Prostate Cancer Cells Mainly through Necroptosis" Cancers 13, no. 4: 882. https://doi.org/10.3390/cancers13040882
APA StyleMarkowitsch, S. D., Juetter, K. M., Schupp, P., Hauschulte, K., Vakhrusheva, O., Slade, K. S., Thomas, A., Tsaur, I., Cinatl, J., Jr., Michaelis, M., Efferth, T., Haferkamp, A., & Juengel, E. (2021). Shikonin Reduces Growth of Docetaxel-Resistant Prostate Cancer Cells Mainly through Necroptosis. Cancers, 13(4), 882. https://doi.org/10.3390/cancers13040882