
CAR-T after Stem Cell Transplantation in B-Cell Lymphoproliferative Disorders: Are They Really Autologous or Allogenic Cell Therapies?

 , ,
, , 
Abstract
Simple Summary
Abstract
1. B-Cell Lymphoproliferative Disorders and 1st Line of Treatment
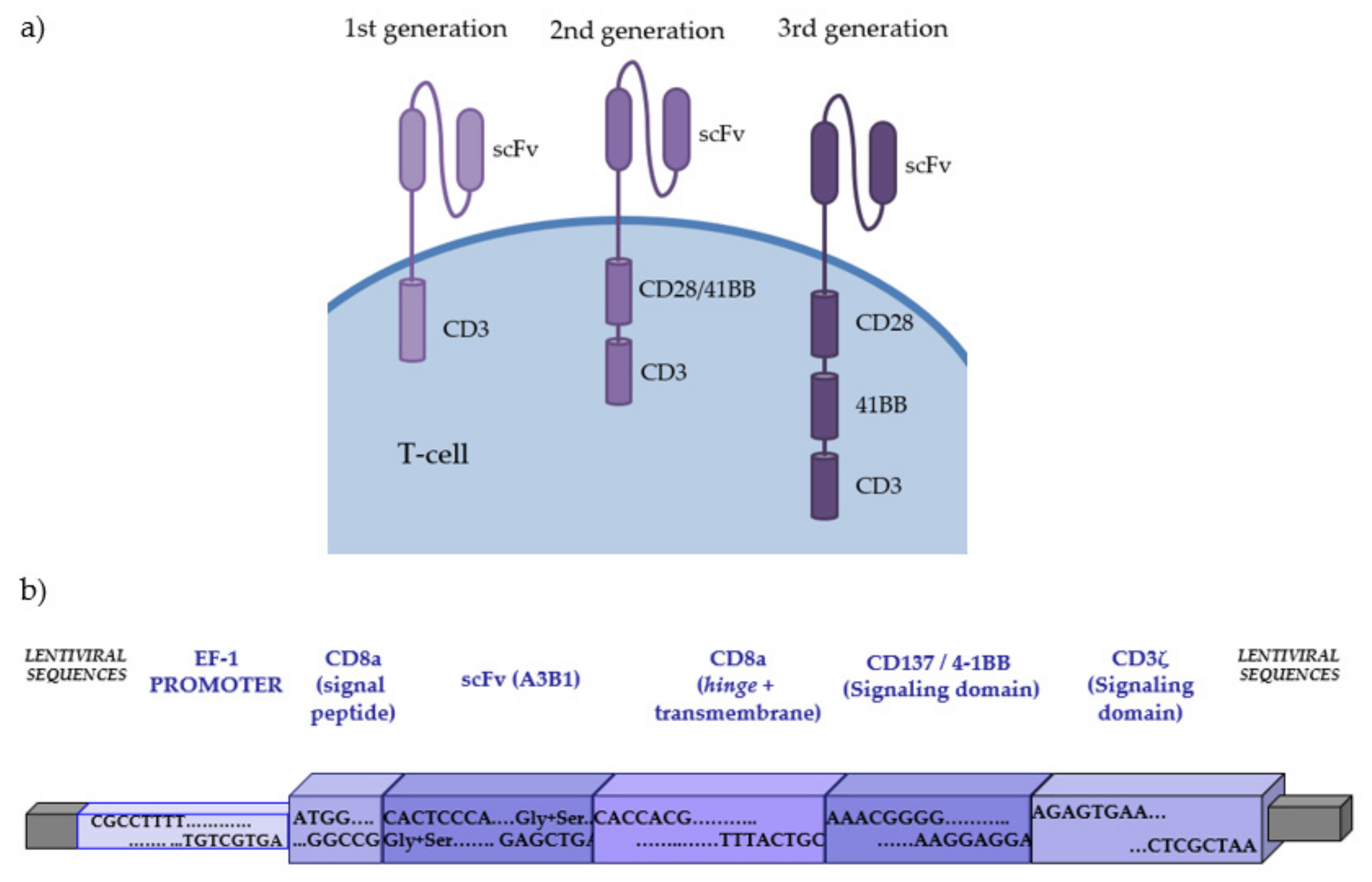
2. CAR-Redirected T-Cells (CAR-T) Offer a New and Promising Cell-Based Immunotherapy
3. Combination of HSCT and CAR-T Treatments
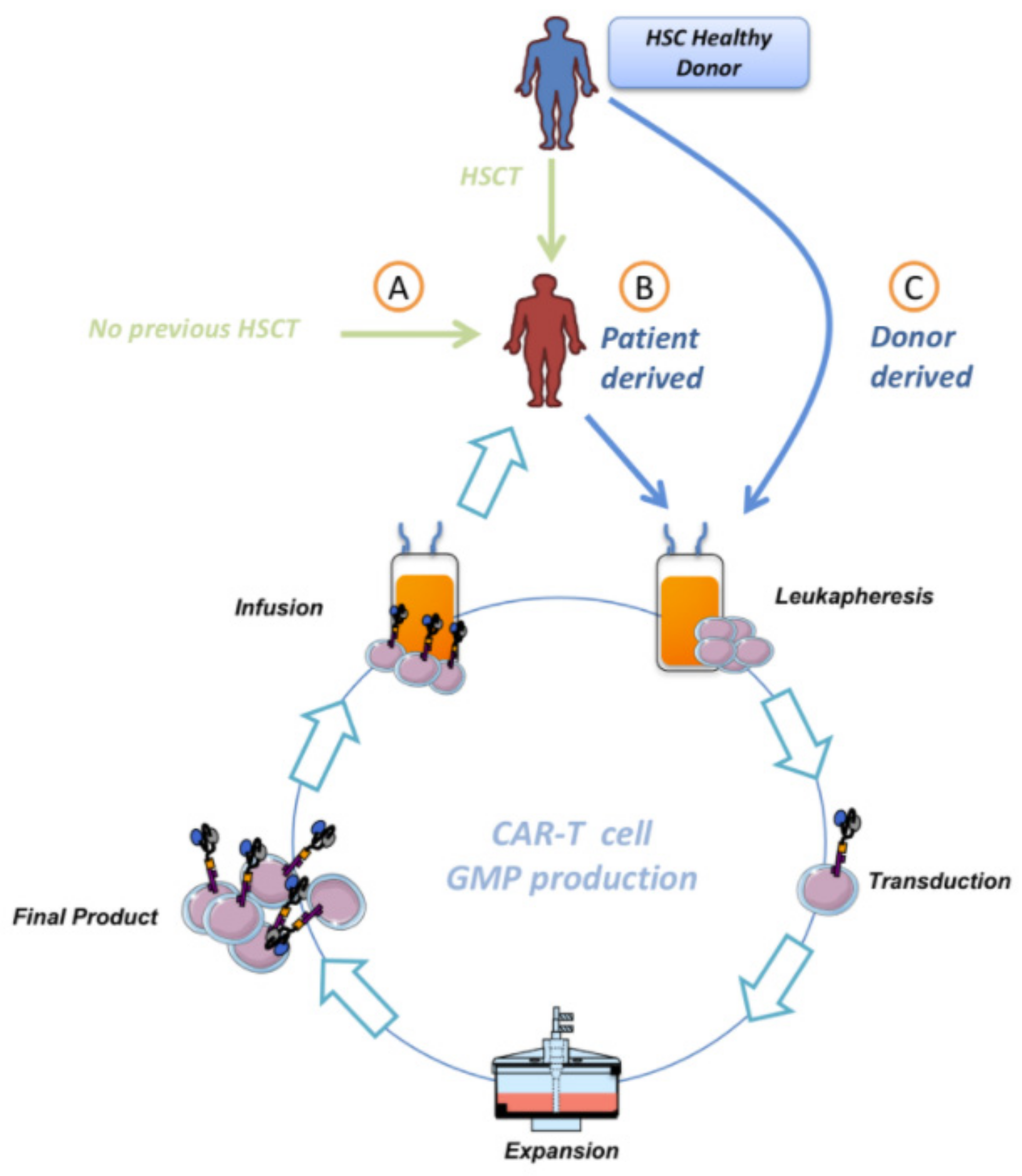
4. Chimerism before Manufacturing CAR-T
5. Autologous or Allogenic CAR-T
6. Future Perspective
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Correction Statement
Abbreviations
References
- Padala, S.A.; Kallam, A. Diffuse Large B Cell Lymphoma; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Kochenderfer, J.N.; Rosenberg, S.A. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat. Rev. Clin. Oncol. 2013, 10, 267–276. [Google Scholar] [CrossRef]
- Jacobson, C.A.; Longo, D.L. Chapter 104: Non-Hodgkin’ s Lymphoma. In Harrison’s Principles of Internal Medicine; McGraw Hill Education: New York, NY, USA, 2021; pp. 1–22. [Google Scholar]
- Lynegar, V.; Shimanovsky, A. Leukemia; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Patel, K.; Pagel, J.M. Current and future treatment strategies in chronic lymphocytic leukemia. J. Hematol. Oncol. 2021, 14, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Saadeh, S.S.; Litzow, M.R. Hematopoietic stem cell transplant in adults with acute lymphoblastic leukemia: The present state. Expert Rev. Hematol. 2018, 11, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Özgür Yurttaş, N.; Eşkazan, A.E. Novel therapeutic approaches in chronic myeloid leukemia. Leuk. Res. 2020, 91, 106337. [Google Scholar] [CrossRef]
- Takami, A. Hematopoietic stem cell transplantation for acute myeloid leukemia. Int. J. Hematol. 2018, 107, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Duarte, R.F.; Labopin, M.; Bader, P.; Basak, G.W.; Bonini, M.C.; Chabannon, C.; Corbacioglu, S.; Dreger, P.; Dufour, C.; Gennery, A.R.; et al. Indications for haematopoietic stem cell transplantation for haematological diseases, solid tumours and immune disorders: Current practice in Europe, 2019. Bone Marrow Transplant. 2019, 54, 1525–1552. [Google Scholar] [CrossRef]
- Singh, A.K.; McGuirk, J.P. Allogeneic Stem Cell Transplantation: A Historical and Scientific Overview. Cancer Res. 2016, 76, 6445–6451. [Google Scholar] [CrossRef]
- Swart, J.F.; Delemarre, E.M.; Van Wijk, F.; Boelens, J.-J.; Kuball, J.; Van Laar, J.M.; Wulffraat, N.M. Haematopoietic stem cell transplantation for autoimmune diseases. Nat. Rev. Rheumatol. 2017, 13, 244–256. [Google Scholar] [CrossRef] [PubMed]
- Passweg, J.R.; Baldomero, H.; Chabannon, C.; Basak, G.W.; de la Cámara, R.; Corbacioglu, S.; Dolstra, H.; Duarte, R.; Glass, B.; Greco, R.; et al. Hematopoietic cell transplantation and cellular therapy survey of the EBMT: Monitoring of activities and trends over 30 years. Bone Marrow Transplant. 2021, 56, 1651–1664. [Google Scholar] [CrossRef]
- Petersdorf, E.W. The major histocompatibility complex: A model for understanding graft-versus-host disease. Blood 2013, 122, 1863–1872. [Google Scholar] [CrossRef]
- Carreras, E.; Dufour, C.; Mohty, M.; Kröger, N. The EBMT Handbook; Springer International Publishing: Cham, Switzerland, 2019. [Google Scholar]
- Fürst, D.; Müller, C.; Vucinic, V.; Bunjes, D.; Herr, W.; Gramatzki, M.; Schwerdtfeger, R.; Arnold, R.; Einsele, H.; Wulf, G.; et al. High-resolution HLA matching in hematopoietic stem cell transplantation: A retrospective collaborative analysis. Blood 2013, 122, 3220–3229. [Google Scholar] [CrossRef] [PubMed]
- Blazar, B.R.; Hill, G.R.; Murphy, W.J. Dissecting the biology of allogeneic HSCT to enhance the GvT effect whilst minimizing GvHD. Nat. Rev. Clin. Oncol. 2020, 17, 475–492. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhong, J.F.; Zhang, X.; Zhang, C. Allogeneic CD19-CAR-T cell infusion after allogeneic hematopoietic stem cell transplantation in B cell malignancies. J. Hematol. Oncol. 2017, 10, 1–8. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 6230. [Google Scholar] [CrossRef]
- Rivat, C.; Santilli, G.; Gaspar, H.B.; Thrasher, A.J. Gene Therapy for Primary Immunodeficiencies. Hum. Gene Ther. 2012, 23, 668–675. [Google Scholar] [CrossRef]
- Larson, R.C.; Maus, M.V. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat. Rev. Cancer 2021, 21, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Snyder, K.M.; Suhoski, M.M.; Maus, M.V.; Kapoor, V.; June, C.H.; Mackall, C.L. 4-1BB Is Superior to CD28 Costimulation for Generating CD8+Cytotoxic Lymphocytes for Adoptive Immunotherapy. J. Immunol. 2007, 179, 4910–4918. [Google Scholar] [CrossRef]
- Ramello, M.C.; Benzaïd, I.; Kuenzi, B.M.; Lienlaf-Moreno, M.; Kandell, W.M.; Santiago, D.N.; Pabón-Saldaña, M.; Darville, L.; Fang, B.; Rix, U.; et al. An immunoproteomic approach to characterize the CAR interactome and signalosome. Sci. Signal. 2019, 12, eaap9777. [Google Scholar] [CrossRef] [PubMed]
- Benmebarek, M.-R.; Karches, C.H.; Cadilha, B.L.; Lesch, S.; Endres, S.; Kobold, S. Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells. Int. J. Mol. Sci. 2019, 20, 1283. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Lee, Y.G.; Shestova, O.; Ravikumar, P.; Hayer, K.E.; Hong, S.J.; Lu, X.M.; Pajarillo, R.; Agarwal, S.; Kuramitsu, S.; et al. Impaired Death Receptor Signaling in Leukemia Causes Antigen-Independent Resistance by Inducing CAR T-cell Dysfunction. Cancer Discov. 2020, 10, 552–567. [Google Scholar] [CrossRef] [PubMed]
- Castella, M.; Boronat, A.; Martín-Ibáñez, R.; Rodríguez, V.; Suñé, G.; Caballero, M.; Marzal, B.; Pérez-Amill, L.; Martín-Antonio, B.; Castaño, J.; et al. Development of a Novel Anti-CD19 Chimeric Antigen Receptor: A Paradigm for an Affordable CAR T Cell Production at Academic Institutions. Mol. Ther. Methods Clin. Dev. 2019, 12, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Ortíz-Maldonado, V.; Rives, S.; Castellà, M.; Alonso-Saladrigues, A.; Benítez-Ribas, D.; Caballero-Baños, M.; Baumann, T.; Cid, J.; Garcia-Rey, E.; Llanos, C.; et al. CART19-BE-01: A Multicenter Trial of ARI-0001 Cell Therapy in Patients with CD19+ Relapsed/Refractory Malignancies. Mol. Ther. 2021, 29, 636–644. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med. 2019, 25, 1341–1355. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Abramson, M.J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, D.M.A.; Wang, M.L.; Arnason, J.E.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, M.C.; et al. Pivotal Safety and Efficacy Results from Transcend NHL 001, a Multicenter Phase 1 Study of Lisocabtagene Maraleucel (liso-cel) in Relapsed/Refractory (R/R) Large B Cell Lymphomas. Blood 2019, 134, 241. [Google Scholar] [CrossRef]
- Sadelain, M.; Brentjens, R.; Rivière, I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef]
- Cappell, K.M.; Sherry, R.M.; Yang, J.C.; Goff, S.L.; Vanasse, D.A.; McIntyre, L.; Rosenberg, S.A.; Kochenderfer, J.N. Long-Term Follow-Up of Anti-CD19 Chimeric Antigen Receptor T-Cell Therapy. J. Clin. Oncol. 2020, 38, 3805–3815. [Google Scholar] [CrossRef]
- Ruella, M.; Maus, M.V. Catch me if you can: Leukemia Escape after CD19-Directed T Cell Immunotherapies. Comput. Struct. Biotechnol. J. 2016, 14, 357–362. [Google Scholar] [CrossRef]
- Dai, H.; Wu, Z.; Jia, H.; Tong, C.; Guo, Y.; Ti, D.; Liu, Y.; Zhang, W.; Wang, C.; Zhang, Y.; et al. Bispecific Chimeric Antigen Receptor Targeting Both CD19 and CD22 T Cell Therapy in Adults with Relapsed or Refractory B-Cell Acute Lymphoblastic Leukemia. SSRN Electron. J. 2020, 8, 1–10. [Google Scholar] [CrossRef]
- Tong, C.; Zhang, Y.; Liu, Y.; Ji, X.; Zhang, W.-Y.; Guo, Y.; Han, X.; Ti, D.; Dai, H.; Wang, C.; et al. Optimized tandem CD19/CD20 CAR-engineered T cells in refractory/relapsed B cell lymphoma. Blood 2020, 136, 1632–1644. [Google Scholar] [CrossRef] [PubMed]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-CAR T Cells Induce Remissions in CD19-CAR Naïve and Resistant B-ALL. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Lobato, L.G.; Ganzetti, M.; De Larrea, C.F.; Hudecek, M.; Einsele, H.; Danhof, S. CAR T-Cells in Multiple Myeloma: State of the Art and Future Directions. Front. Oncol. 2020, 10, 1243. [Google Scholar] [CrossRef] [PubMed]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): A phase 1b/2 open-label study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Berdeja, J.G.; Madduri, D.; Jakubowiak, A.J.; Agha, M.E.; Cohen, A.D.; Hari, P.; Yeh, T.-M.; Olyslager, Y.; Banerjee, A.; et al. Ciltacabtagene autoleucel, a B-cell maturation antigen (BCMA)-directed chimeric antigen receptor T-cell (CAR-T) therapy, in relapsed/refractory multiple myeloma (R/R MM): Updated results from CARTITUDE-1. J. Clin. Oncol. 2021, 39, 8005. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Frey, N.V.; Shaw, P.A.; Hexner, E.O.; Pequignot, E.; Gill, S.; Luger, S.M.; Mangan, J.K.; Loren, A.W.; Perl, A.E.; Maude, S.L.; et al. Optimizing Chimeric Antigen Receptor T-Cell Therapy for Adults with Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2020, 38, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.D.W.; Stetler-Stevenson, M.; Yuan, C.M.; Shah, N.N.; Delbrook, R.C.; Yates, R.B.; Zhang, H.; Zhang, L.; Kochenderfer, J.N.; Rosenberg, S.A.; et al. Long-Term Outcomes Following CD19 CAR T Cell Therapy for B-ALL Are Superior in Patients Receiving a Fludarabine/Cyclophosphamide Preparative Regimen and Post-CAR Hematopoietic Stem Cell Transplantation. Blood 2016, 128, 218. [Google Scholar] [CrossRef]
- Bouziana, S.; Bouzianas, D. Exploring the Dilemma of Allogeneic Hematopoietic Cell Transplantation after Chimeric Antigen Receptor T Cell Therapy: To Transplant or Not? Biol. Blood Marrow Transplant. 2020, 26, e183–e191. [Google Scholar] [CrossRef]
- Gardner, R.A.; Finney, O.; Annesley, C.; Brakke, H.; Summers, C.; Leger, K.; Bleakley, M.; Brown, C.; Mgebroff, S.; Kelly-Spratt, K.S.; et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 2017, 129, 3322–3331. [Google Scholar] [CrossRef] [PubMed]
- Curran, K.J.; Margossian, S.P.; Kernan, N.A.; Silverman, L.B.; Williams, D.A.; Shukla, N.; Kobos, R.; Forlenza, C.J.; Steinherz, P.; Prockop, S.; et al. Toxicity and response after CD19-specific CAR T-cell therapy in pediatric/young adult relapsed/refractory B-ALL. Blood 2019, 134, 2361–2368. [Google Scholar] [CrossRef]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Abstract CT102: Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014, 6, 224–225. [Google Scholar] [CrossRef]
- Clark, J.R.; Scott, S.D.; Jack, A.L.; Lee, H.; Mason, J.; Carter, G.I.; Pearce, L.; Jackson, T.; Clouston, H.; Sproul, A.; et al. Monitoring of chimerism following allogeneic haematopoietic stem cell transplantation (HSCT): Technical recommendations for the use of Short Tandem Repeat (STR) based techniques, on behalf of the United Kingdom National External Quality Assessment Service. Br. J. Haematol. 2015, 168, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Bader, P.; Niethammer, D.; Willasch, A.; Kreyenberg, H.; Klingebiel, T. How and when should we monitor chimerism after allogeneic stem cell transplantation? Bone Marrow Transplant. 2005, 35, 107–119. [Google Scholar] [CrossRef]
- Kolb, H.-J.; Simoes, B.; Schmid, C. Cellular immunotherapy after allogeneic stem cell transplantation in hematologic malignancies. Curr. Opin. Oncol. 2004, 16, 167–173. [Google Scholar] [CrossRef]
- Gabr, H.; Youssry, I.; El-Ansary, Y.; Mosallam, G.; Riad, N.M.; Hanna, M.O.F. Chimerism in pediatric hematopoietic stem cell transplantation and its correlation with the clinical outcome. Transpl. Immunol. 2017, 45, 53–58. [Google Scholar] [CrossRef]
- Delie, A.; Verlinden, A.; Beel, K.; Deeren, D.; Mazure, D.; Baron, F.; Breems, D.; De Becker, A.; Graux, C.; Lewalle, P.; et al. Use of chimerism analysis after allogeneic stem cell transplantation: Belgian guidelines and review of the current literature. Acta Clin. Belgica Int. J. Clin. Lab. Med. 2020, 1–9. [Google Scholar] [CrossRef]
- Breuer, S.; Preuner, S.; Fritsch, G.; Daxberger, H.; Koenig, M.; Poetschger, U.; Lawitschka, A.; Peters, C.; Mann, G.; Lion, T.; et al. Early recipient chimerism testing in the T- and NK-cell lineages for risk assessment of graft rejection in pediatric patients undergoing allogeneic stem cell transplantation. Leukemia 2012, 26, 509–519. [Google Scholar] [CrossRef]
- Lion, T.; Watzinger, F.; Preuner, S.; Kreyenberg, H.; Tilanus, M.; de Weger, R.; van Loon, J.; de Vries, L.; Cavé, H.; Acquaviva, C.; et al. The EuroChimerism concept for a standardized approach to chimerism analysis after allogeneic stem cell transplantation. Leukemia 2012, 26, 1821–1828. [Google Scholar] [CrossRef] [PubMed]
- Siglin, J.; Lutfi, F.; Bukhari, A.; Holtzman, N.G.; Kim, D.W.; Ali, M.M.; Rapoport, A.P.; Dahiya, S. Pseudo-Allogeneic CAR-T Therapy after Allogeneic Stem Cell Transplantation in Relapsed/Refractory B-Cell NHL. Blood 2020, 136, 22–23. [Google Scholar] [CrossRef]
- Li, Q.; Mu, J.; Yuan, J.; Yang, Z.; Wang, J.; Deng, Q. Low Level Donor Chimerism of CD19 CAR-T Cells Returned to Complete Donor Chimerism in Patients with Relapse After Allo-Hematopoietic Stem Cell Transplant. OncoTargets Ther. 2020, 13, 11471–11484. [Google Scholar] [CrossRef]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Dinofia, A.K.; Grupp, S.A. Will allogenic CAR T cells for CD19+ malignances take autologous CAR T cells ‘off the shelf’? Nat. Rev. Clin. Oncol. 2021, 18, 195–196. [Google Scholar] [CrossRef]
- Brudno, J.N.; Somerville, R.P.; Shi, V.; Rose, J.J.; Halverson, D.C.; Fowler, D.H.; Gea-Banacloche, J.C.; Pavletic, S.Z.; Hickstein, D.D.; Lu, T.L.; et al. Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor Induce Remissions of B-Cell Malignancies That Progress After Allogeneic Hematopoietic Stem-Cell Transplantation Without Causing Graft-Versus-Host Disease. J. Clin. Oncol. 2016, 34, 1112–1121. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Carpenter, R.O.; Kassim, S.H.; Rose, J.J.; Telford, W.G.; Hakim, F.T.; Halverson, D.C.; Fowler, D.H.; Hardy, N.M.; et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood 2013, 122, 4129–4139. [Google Scholar] [CrossRef]
- Cruz, C.R.Y.; Micklethwaite, K.P.; Savoldo, B.; Ramos, C.A.; Lam, S.; Ku, S.; Diouf, O.; Liu, E.; Barrett, A.J.; Ito, S.; et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: A phase 1 study. Blood 2013, 122, 2965–2973, Erratum in 2014, 123, 3364. [Google Scholar] [CrossRef]
- Graham, C.; Jozwik, A.; Pepper, A.; Benjamin, R. Allogeneic CAR-T Cells: More than Ease of Access? Cells 2018, 7, 155. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.; Wu, L. Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers 2016, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Kenderian, S.; Porter, D.L.; Gill, S. Chimeric Antigen Receptor T Cells and Hematopoietic Cell Transplantation: How Not to Put the CART Before the Horse. Biol. Blood Marrow Transplant. 2017, 23, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Huang, H. How to Combine the Two Landmark Treatment Methods—Allogeneic Hematopoietic Stem Cell Transplantation and Chimeric Antigen Receptor T Cell Therapy Together to Cure High-Risk B Cell Acute Lymphoblastic Leukemia? Front. Immunol. 2020, 11, 3300. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Patients | Product | scFv | scFv Origin | Coestimulatory Domain | CRR, % (CI 95%) | PFS/EFS, Median (CI 95%) | OS, Median (CI 95%) | Previous allo-HSCT, % | Post-allo-HSCT in CR, % | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| PEDIATRICS +/− YOUNG ADULTS | ||||||||||
| 11 kids (up to 25y) | ARI–0001 | A3B1 | Murine | 4–1BB | 79 (54–94) | 18.1 mo (14.5–ND) 82% (59–100) at 1y | NA (7.1–NA) 78% (50–100) at 1y | 55 | NA | [32] |
| 75 kids (up to 25y) | CTL019 | FM63 | Murine | 4–1BB | 81 (71–89) | 50% (35–64) at 1y | 76% (63–86) at 1y | 61 | 14 | [23] |
| 53 kids | KTE–C19 | FM63 | Murine | CD28 | 61 | 49% at 18 mo | 52% at 10 mo | 35 | 75 | [48] |
| 45 kids (up to 25y) | JCAR017 | FM63 | Murine | 4–1BB | 93 | 51% (37–70) at 1y | 69.5% (56–87) at 1y | 62 | 28 | [50] |
| 25 kids (1–23y) | 19–28z | SJ25C1 | Murine | CD28 | 75 | NA | NA | 20 | 83 | [51] |
| ADULTS | ||||||||||
| 27 adults (>18y) | ARI–0001 | A3B1 | Murine | 4–1BB | 85.2 (66–96) | 9.4 mo (3.3–20.2) 34% (12–57) at 1y | 20.2 mo (12.8–NA) 65% (40–89) at 1y | 81 | NA | [32] |
| 53 adults | 19E3/1928z | 19E3 ab | Murine | CD28 | 83 (70–92) | 50 (at 6 mo) | 12.9 mo (8.7–23.4) | 36 | 39 | [31] |
| 35 adults | CTL019 | FM63 | Murine | 4–1BB | 69 (51–83) | 5.6 mo | 19.1 mo (6.2–NA) | 37 | 38 (9 out of 24) | [47] |
| 16 adults | 19–28z | SJ25C1 | Murine | CD28 | 88 | NA | NA | 25 | 44 | [52] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartoló-Ibars, A.; Uribe-Herranz, M.; Muñoz-Sánchez, G.; Arnaldos-Pérez, C.; Ortiz-Maldonado, V.; Urbano-Ispizua, Á.; Pascal, M.; Juan, M. CAR-T after Stem Cell Transplantation in B-Cell Lymphoproliferative Disorders: Are They Really Autologous or Allogenic Cell Therapies? Cancers 2021, 13, 4664. https://doi.org/10.3390/cancers13184664
Bartoló-Ibars A, Uribe-Herranz M, Muñoz-Sánchez G, Arnaldos-Pérez C, Ortiz-Maldonado V, Urbano-Ispizua Á, Pascal M, Juan M. CAR-T after Stem Cell Transplantation in B-Cell Lymphoproliferative Disorders: Are They Really Autologous or Allogenic Cell Therapies? Cancers. 2021; 13(18):4664. https://doi.org/10.3390/cancers13184664
Chicago/Turabian StyleBartoló-Ibars, Ariadna, Mireia Uribe-Herranz, Guillermo Muñoz-Sánchez, Cristina Arnaldos-Pérez, Valentín Ortiz-Maldonado, Álvaro Urbano-Ispizua, Mariona Pascal, and Manel Juan. 2021. "CAR-T after Stem Cell Transplantation in B-Cell Lymphoproliferative Disorders: Are They Really Autologous or Allogenic Cell Therapies?" Cancers 13, no. 18: 4664. https://doi.org/10.3390/cancers13184664
APA StyleBartoló-Ibars, A., Uribe-Herranz, M., Muñoz-Sánchez, G., Arnaldos-Pérez, C., Ortiz-Maldonado, V., Urbano-Ispizua, Á., Pascal, M., & Juan, M. (2021). CAR-T after Stem Cell Transplantation in B-Cell Lymphoproliferative Disorders: Are They Really Autologous or Allogenic Cell Therapies? Cancers, 13(18), 4664. https://doi.org/10.3390/cancers13184664