
A Comparative Endocrine Trans-Differentiation Approach to Pancreatic Ductal Adenocarcinoma Cells with Different EMT Phenotypes Identifies Quasi-Mesenchymal Tumor Cells as Those with Highest Plasticity


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Pancreatic Cell Lines and Primary Culture
2.2. Quantitative Real-Time PCR
2.3. Immunoblotting
2.4. Trans-Differentiation of PDAC-Derived Cells into Pancreatic Endocrine or Insulin-Expressing Cells
2.5. Real-Time Cell Invasion Assay
2.6. Statistics
3. Results
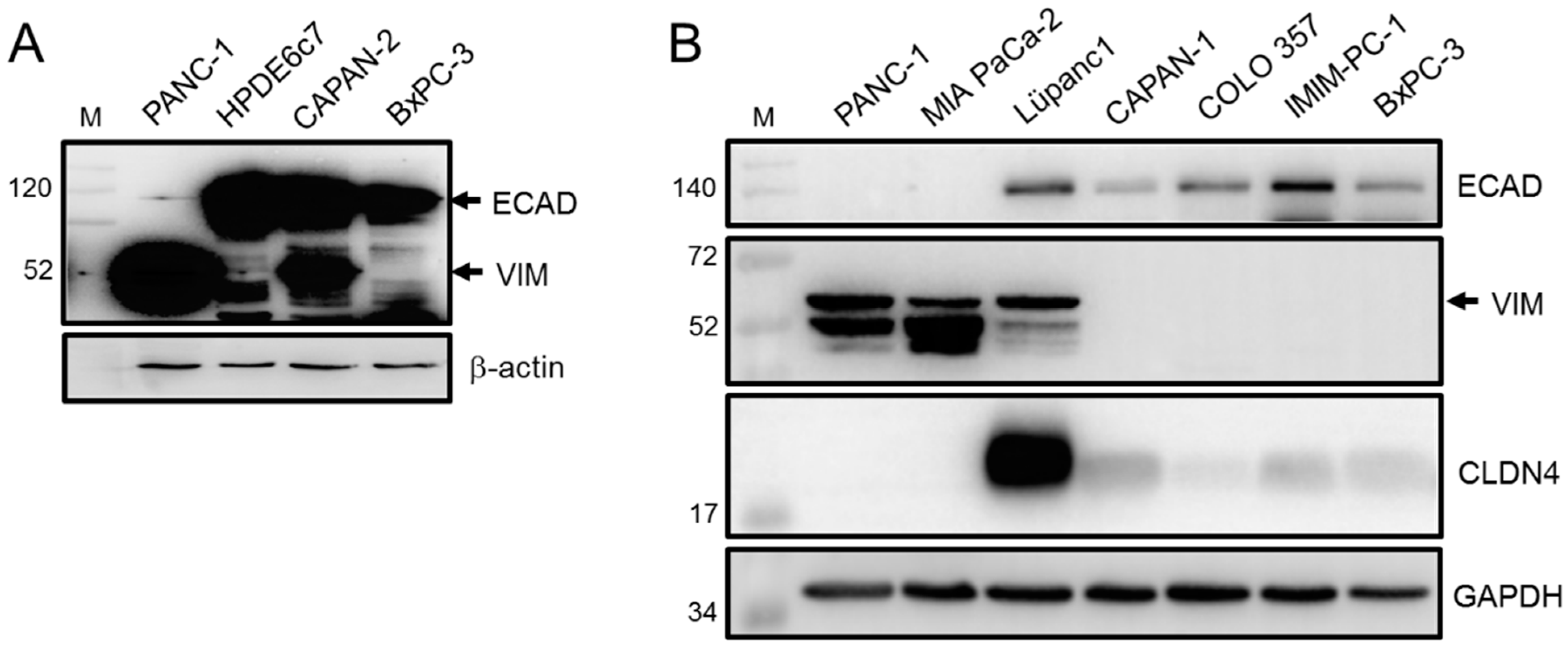
3.1. Epithelial/Mesenchymal Phenotyping of PDAC-Derived Cell Lines Based on ECAD and VIM Expression
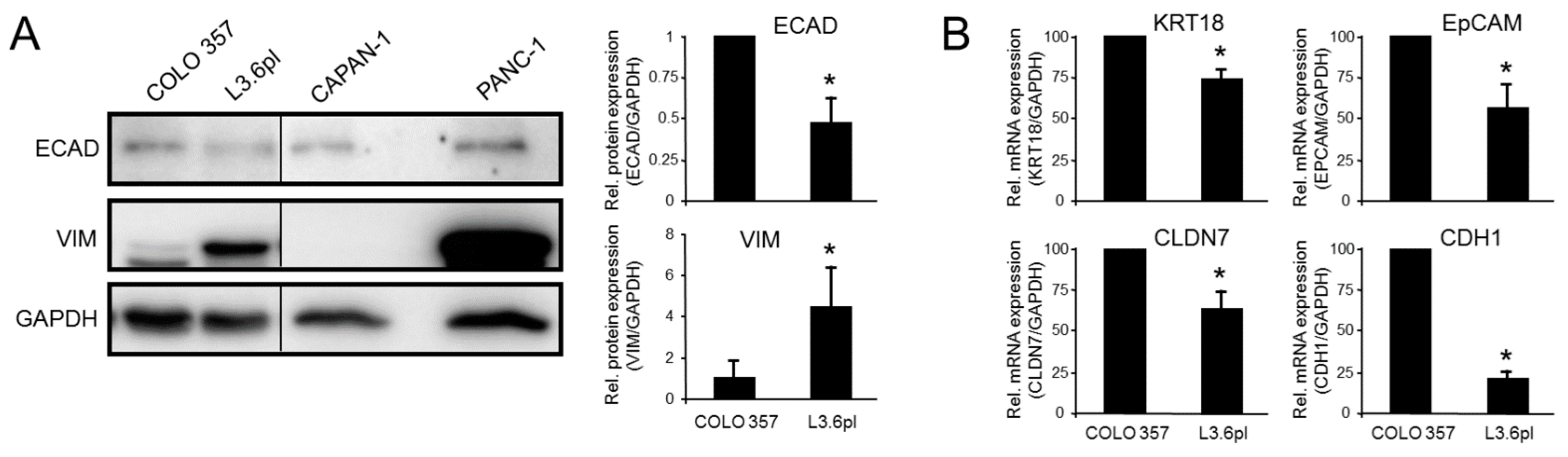
3.2. Epithelial/Mesenchymal Phenotyping of the Paired Isogenic COLO 357 and L3.6pl Cell Lines
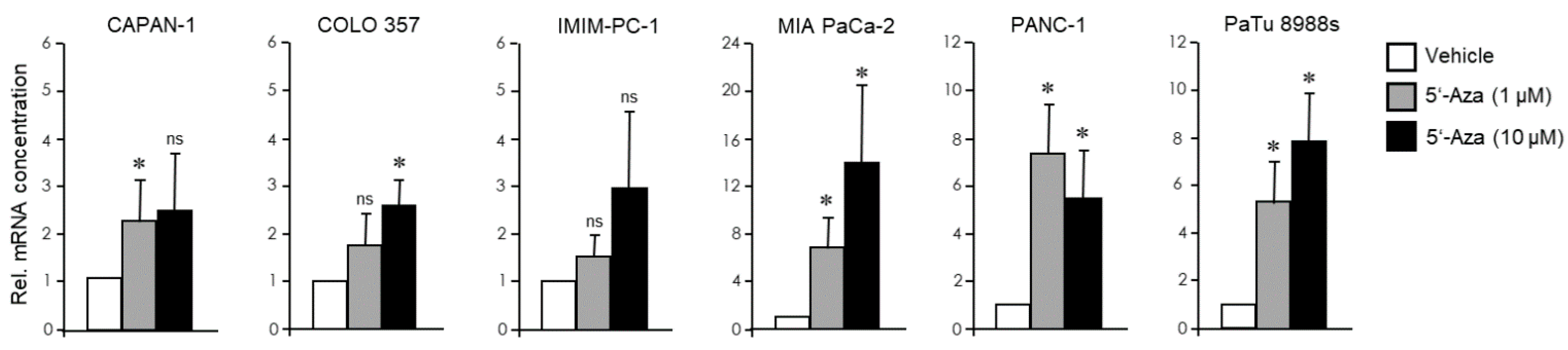
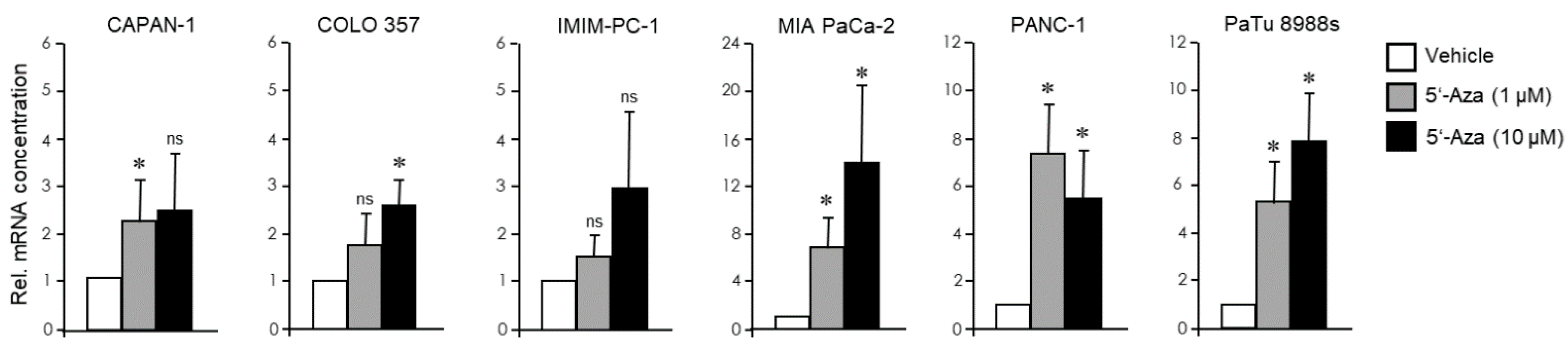
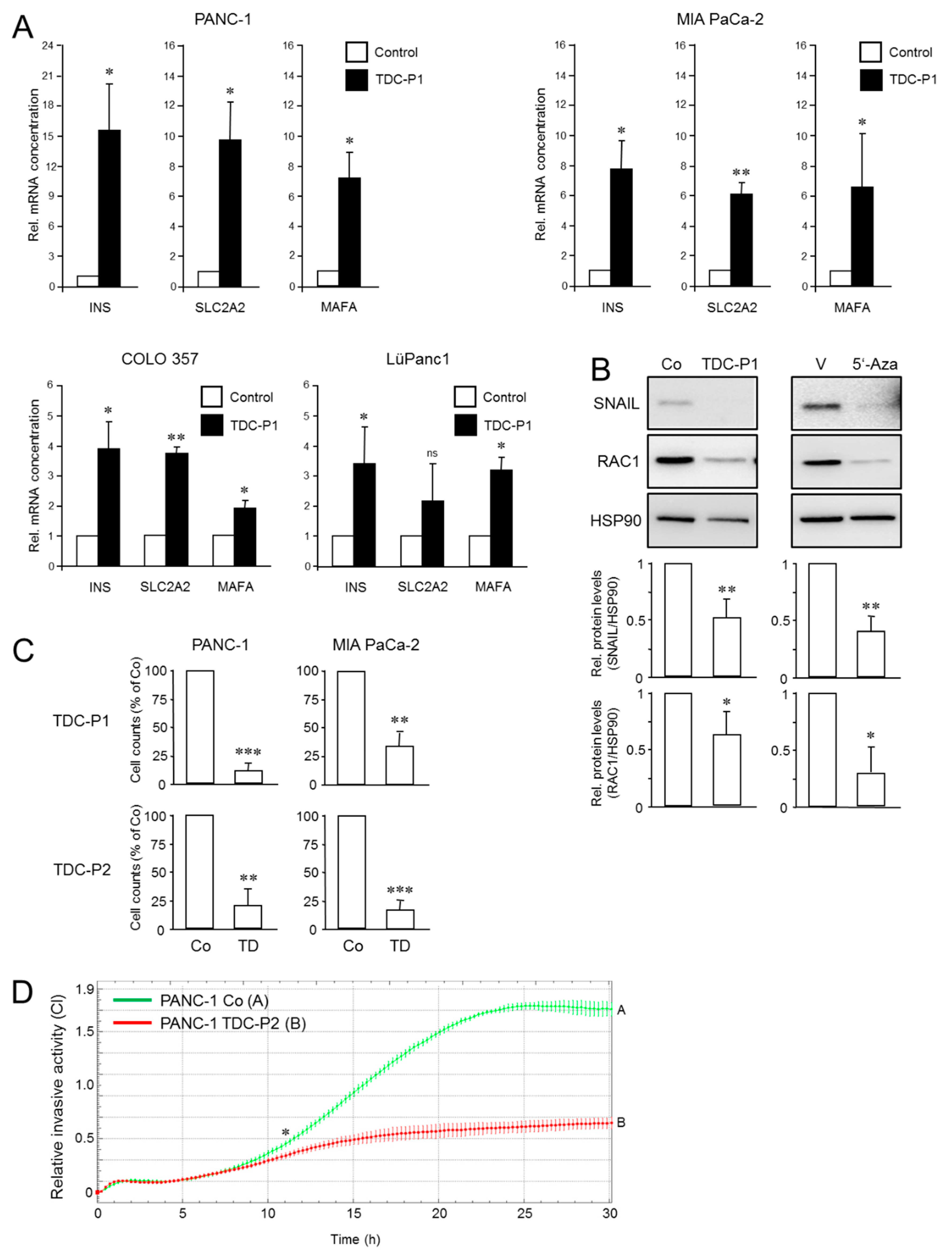
3.3. The EMT Phenotype of PDAC Cells Determines Their Trans-Differentiation Potential to Pancreatic Endocrine Precursors
3.4. The EMT Phenotype of PDAC Cells Determines Their Trans-Differentiation Potential to Insulin-Expressing Cells
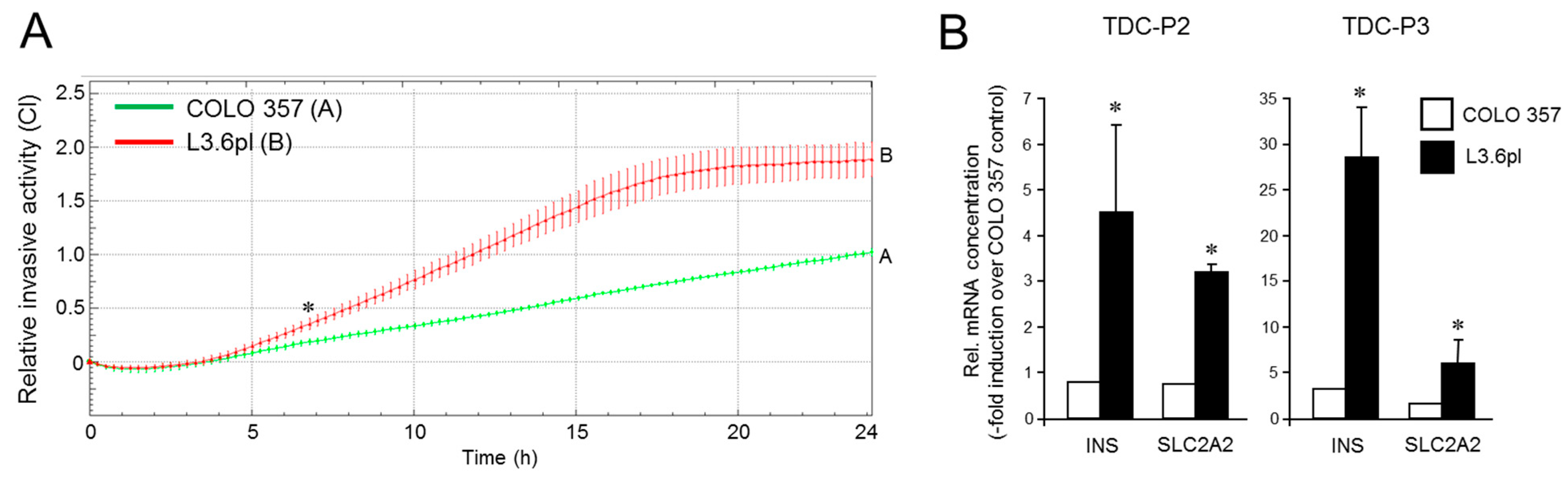
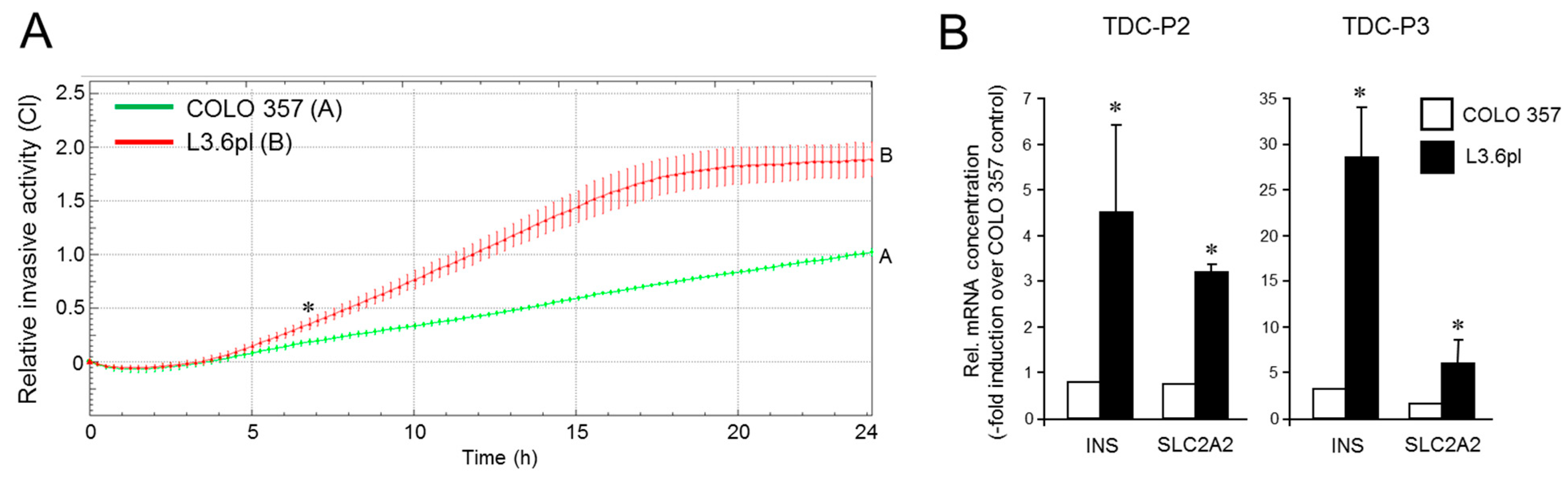
3.5. The Increased Metastatic Potential of L3.6pl Cells Compared with COLO 357 Cells Is Associated with a Higher Trans-Differentiation Potential to Insulin-Expressing Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics. CA Cancer J. Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef] [Green Version]
- Neoptolemos, J.P.; Stocken, D.D.; Friess, H.; Bassi, C.; Dunn, J.A.; Hickey, H.; Beger, H.; Fernandez-Cruz, L.; Dervenis, C.; Lacaine, F. European Study Group for Pancreatic Cancer. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N. Engl. J. Med. 2004, 350, 1200–1210. [Google Scholar] [CrossRef] [Green Version]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Muckenhuber, A.; Berger, A.K.; Schlitter, A.M.; Steiger, K.; Konukiewitz, B.; Trumpp, A.; Eils, R.; Werner, J.; Friess, H.; Esposito, I.; et al. Pancreatic Ductal Adenocarcinoma Subtyping Using the Biomarkers Hepatocyte Nuclear Factor-1A and Cytokeratin-81 Correlates with Outcome and Treatment Response. Clin. Cancer Res. 2018, 24, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Juiz, N.; Elkaoutari, A.; Bigonnet, M.; Gayet, O.; Roques, J.; Nicolle, R.; Iovanna, J.; Dusetti, N. Basal-like and classical cells coexist in pancreatic cancer revealed by single-cell analysis on biopsy-derived pancreatic cancer organoids from the classical subtype. FASEB J. 2020, 34, 12214–12228. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, K.K.; Arora, K.S.; Amzallag, A.; Williams, E.; Kulkarni, A.S.; Fernandez-Del Castillo, C.; Lillemoe, K.D.; Bardeesy, N.; Hong, T.S.; Ferrone, C.R.; et al. Quasimesenchymal phenotype predicts systemic metastasis in pancreatic ductal adenocarcinoma. Mod. Pathol. 2019, 32, 844–854. [Google Scholar] [CrossRef]
- Gupta, P.B.; Pastushenko, I.; Skibinski, A.; Blanpain, C.; Kuperwasser, C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2019, 24, 65–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellner, U.; Brabletz, T.; Keck, T. ZEB1 in Pancreatic Cancer. Cancers 2010, 2, 1617–1628. [Google Scholar] [CrossRef] [Green Version]
- Mladinich, M.; Ruan, D.; Chan, C.H. Tackling Cancer Stem Cells via Inhibition of EMT Transcription Factors. Stem Cells Int. 2016, 2016, 5285892. [Google Scholar] [CrossRef] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Wilson, M.M.; Weinberg, R.A.; Lees, J.A.; Guen, V.J. Emerging Mechanisms by which EMT Programs Control Stemness. Trends Cancer 2020, 6, 775–780. [Google Scholar] [CrossRef]
- Bhagwandin, V.J.; Bishop, J.M.; Wright, W.E.; Shay, J.W. The Metastatic Potential and Chemoresistance of Human Pancreatic Cancer Stem Cells. PLoS ONE 2016, 11, e0148807. [Google Scholar] [CrossRef]
- Rodriguez-Aznar, E.; Wiesmüller, L.; Sainz, B., Jr.; Hermann, P.C. EMT and Stemness-Key Players in Pancreatic Cancer Stem Cells. Cancers 2019, 11, 1136. [Google Scholar] [CrossRef] [Green Version]
- Dongre, A.; Rashidian, M.; Eaton, E.N.; Reinhardt, F.; Thiru, P.; Zagorulya, M.; Nepal, S.; Banaz, T.; Martner, A.; Spranger, S.; et al. Direct and Indirect Regulators of Epithelial-Mesenchymal Transition-Mediated Immunosuppression in Breast Carcinomas. Cancer Discov. 2021, 11, 1286–1305. [Google Scholar] [CrossRef] [PubMed]
- Greiner, T.U.; Kesavan, G.; Ståhlberg, A.; Semb, H. Rac1 regulates pancreatic islet morphogenesis. BMC Dev. Biol. 2009, 9, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rukstalis, J.M.; Habener, J.F. Neurogenin3: A master regulator of pancreatic islet differentiation and regeneration. Islets 2009, 1, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Rukstalis, J.M.; Habener, J.F. Snail2, a mediator of epithelial-mesenchymal transitions, expressed in progenitor cells of the developing endocrine pancreas. Gene Exp. Patterns 2007, 7, 471–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdez, I.A.; Teo, A.K.; Kulkarni, R.N. Cellular stress drives pancreatic plasticity. Sci. Transl. Med. 2015, 7, 273ps2. [Google Scholar] [CrossRef] [Green Version]
- Valdez, I.A.; Dirice, E.; Gupta, M.K.; Shirakawa, J.; Teo, A.K.K.; Kulkarni, R.N. Proinflammatory Cytokines Induce Endocrine Differentiation in Pancreatic Ductal Cells via STAT3-Dependent NGN3 Activation. Cell Rep. 2016, 15, 460–470. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Amador, J.L.; Téllez, N.; Marin, S.; Aloy-Reverté, C.; Semino, C.; Nacher, M.; Montanya, E. Epithelial to mesenchymal transition in human endocrine islet cells. PLoS ONE 2018, 13, e0191104. [Google Scholar] [CrossRef] [Green Version]
- Sintov, E.; Nathan, G.; Knoller, S.; Pasmanik-Chor, M.; Russ, H.A.; Efrat, S. Inhibition of ZEB1 expression induces redifferentiation of adult human beta cells expanded in vitro. Sci. Rep. 2015, 5, 13024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battula, V.L.; Evans, K.W.; Hollier, B.G.; Shi, Y.; Marini, F.C.; Ayyanan, A.; Wang, R.Y.; Brisken, C.; Guerra, R.; Andreeff, M.; et al. Epithelial-mesenchymal transition-derived cells exhibit multilineage differentiation potential similar to mesenchymal stem cells. Stem Cells 2010, 28, 1435–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishay-Ronen, D.; Diepenbruck, M.; Kalathur, R.K.R.; Sugiyama, N.; Tiede, S.; Ivanek, R.; Bantug, G.; Morini, M.F.; Wang, J.; Hess, C.; et al. Gain Fat—Lose Metastasis: Converting Invasive Breast Cancer Cells into Adipocytes Inhibits Cancer Metastasis. Cancer Cell 2019, 35, 17–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishay-Ronen, D.; Christofori, G. Targeting Cancer Cell Metastasis by Converting Cancer Cells into Fat. Cancer Res. 2019, 79, 5471–5475. [Google Scholar] [CrossRef]
- Hass, R.; von der Ohe, J.; Ungefroren, H. The Intimate Relationship Among EMT, MET and TME: A T(ransdifferentiation) E(nhancing) M(ix) to Be Exploited for Therapeutic Purposes. Cancers 2020, 12, 3674. [Google Scholar] [CrossRef]
- Lefebvre, B.; Belaich, S.; Longue, J.; Vandewalle, B.; Oberholzer, J.; Gmyr, V.; Pattou, F.; Kerr-Conte, J. 5′-AZA induces Ngn3 expression and endocrine differentiation in the PANC-1 human ductal cell line. Biochem. Biophys. Res. Commun. 2010, 391, 305–309. [Google Scholar] [CrossRef]
- Zhang, S.; Huang, F.; Tian, W.; Lai, J.; Qian, L.; Hong, W.; Chen, H.; Li, L.C. Andrographolide promotes pancreatic duct cells differentiation into insulin-producing cells by targeting PDX-1. Biochem. Pharmacol. 2020, 174, 113785. [Google Scholar] [CrossRef] [PubMed]
- Donadel, G.; Pastore, D.; Della-Morte, D.; Capuani, B.; Lombardo, M.F.; Pacifici, F.; Bugliani, M.; Grieco, F.A.; Marchetti, P.; Lauro, D. FGF-2b and h-PL Transform Duct and Non-Endocrine Human Pancreatic Cells into Endocrine Insulin Secreting Cells by Modulating Differentiating Genes. Int. J. Mol. Sci. 2017, 18, 2234. [Google Scholar] [CrossRef] [Green Version]
- Dubiel, E.A.; Kuehn, C.; Wang, R.; Vermette, P. In vitro morphogenesis of PANC-1 cells into islet-like aggregates using RGD-covered dextran derivative surfaces. Colloids Surf. B Biointerfaces 2012, 89, 117–125. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, H.; Saunee, N.A.; Breslin, M.B.; Lan, M.S. Insulinoma-associated antigen-1 zinc-finger transcription factor promotes pancreatic duct cell trans-differentiation. Endocrinology 2010, 151, 2030–2039. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Li, J.; Saleem, S.; Yee, S.P.; Hardikar, A.A.; Wang, R. c-Kit and stem cell factor regulate PANC-1 cell differentiation into insulin- and glucagon-producing cells. Lab. Investig. 2010, 90, 1373–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamil, L.; Benghuzzi, H.; Tucci, M. Evaluation of insulin secretion by pancreatic cells in response to increasing amounts of glucose. Biomed. Sci. Instrum. 2008, 44, 441–446. [Google Scholar] [PubMed]
- Wei, C.; Geras-Raaka, E.; Marcus-Samuels, B.; Oron, Y.; Gershengorn, M.C. Trypsin and thrombin accelerate aggregation of human endocrine pancreas precursor cells. J. Cell. Physiol. 2006, 206, 322–328. [Google Scholar] [CrossRef]
- Misiti, S.; Anastasi, E.; Sciacchitano, S.; Verga Falzacappa, C.; Panacchia, L.; Bucci, B.; Khouri, D.; D′Acquarica, I.; Brunetti, E.; Di Mario, U. 3,5,3′-Triiodo-L-thyronine enhances the differentiation of a human pancreatic duct cell line (hPANC-1) towards a beta-cell-like phenotype. J. Cell. Physiol. 2005, 204, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Hardikar, A.A.; Marcus-Samuels, B.; Geras-Raaka, E.; Raaka, B.M.; Gershengorn, M.C. Human pancreatic precursor cells secrete FGF2 to stimulate clustering into hormone-expressing islet-like cell aggregates. Proc. Natl. Acad. Sci. USA 2003, 100, 7117–7122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinn, R.; Otterbein, H.; Lehnert, H.; Ungefroren, H. RAC1B: A guardian of the epithelial phenotype and protector against epithelial-mesenchymal transition. Cells 2019, 8, 1569. [Google Scholar] [CrossRef] [Green Version]
- Witte, D.; Otterbein, H.; Förster, M.; Giehl, K.; Zeiser, R.; Lehnert, H.; Ungefroren, H. Negative regulation of TGF-β1-induced MKK6-p38 and MEK-ERK signalling and epithelial-mesenchymal transition by Rac1b. Sci. Rep. 2017, 7, 17313. [Google Scholar] [CrossRef] [Green Version]
- Luley, K.B.; Biedermann, S.B.; Künstner, A.; Busch, H.; Franzenburg, S.; Schrader, J.; Grabowski, P.; Wellner, U.F.; Keck, T.; Brabant, G.; et al. A Comprehensive Molecular Characterization of the Pancreatic Neuroendocrine Tumor Cell Lines BON-1 and QGP-1. Cancers 2020, 12, 691. [Google Scholar] [CrossRef] [Green Version]
- Benten, D.; Behrang, Y.; Unrau, L.; Weissmann, V.; Wolters-Eisfeld, G.; Burdak-Rothkamm, S.; Stahl, F.R.; Anlauf, M.; Grabowski, P.; Möbs, M.; et al. Establishment of the First Well-differentiated Human Pancreatic Neuroendocrine Tumor Model. Mol. Cancer Res. 2018, 16, 496–507. [Google Scholar] [CrossRef] [Green Version]
- Ruhnke, M.; Ungefroren, H.; Nussler, A.; Martin, F.; Brulport, M.; Schormann, W.; Hengstler, J.G.; Klapper, W.; Ulrichs, K.; Hutchinson, J.A.; et al. Differentiation of in vitro-modified human peripheral blood monocytes into hepatocyte-like and pancreatic islet-like cells. Gastroenterology 2005, 128, 1774–1786. [Google Scholar] [CrossRef]
- Rahn, S.; Zimmermann, V.; Viol, F.; Knaack, H.; Stemmer, K.; Peters, L.; Lenk, L.; Ungefroren, H.; Saur, D.; Schäfer, H.; et al. Diabetes as risk factor for pancreatic cancer: Hyperglycemia promotes epithelial-mesenchymal-transition and stem cell properties in pancreatic ductal epithelial cells. Cancer Lett. 2018, 415, 129–150. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Otterbein, H.; Wellner, U.F.; Keck, T.; Lehnert, H.; Marquardt, J.U. RAC1B Regulation of TGFB1 Reveals an Unexpected Role of Autocrine TGFβ1 in the Suppression of Cell Motility. Cancers 2020, 12, 3570. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Mellado-Gil, J.M.; Bahn, Y.J.; Pathy, S.M.; Zhang, Y.E.; Rane, S.G. Protection from beta-cell apoptosis by inhibition of TGF-beta/Smad3 signaling. Cell Death Dis. 2020, 11, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blum, B.; Roose, A.N.; Barrandon, O.; Maehr, R.; Arvanites, A.C.; Davidow, L.S.; Davis, J.C.; Peterson, Q.P.; Rubin, L.L.; Melton, D.A. Reversal of β cell de-differentiation by a small molecule inhibitor of the TGFβ pathway. eLife 2014, 3, e02809. [Google Scholar] [CrossRef] [PubMed]
- Eiden, C.; Ungefroren, H. The Ratio of RAC1B to RAC1 Expression in Breast Cancer Cell Lines as a Determinant of Epithelial/Mesenchymal Differentiation and Migratory Potential. Cells 2021, 10, 351. [Google Scholar] [CrossRef]
- Gradiz, R.; Silva, H.C.; Carvalho, L.; Botelho, M.F.; Mota-Pinto, A. MIA PaCa-2 and PANC-1—Pancreas ductal adenocarcinoma cell lines with neuroendocrine differentiation and somatostatin receptors. Sci. Rep. 2016, 6, 21648. [Google Scholar] [CrossRef] [Green Version]
- Deer, E.L.; Gonzalez-Hernandez, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and Genotype of Pancreatic Cancer Cell Lines. Pancreas 2010, 39, 425–435. [Google Scholar] [CrossRef] [Green Version]
- Suemizu, H.; Monnai, M.; Ohnishi, Y.; Ito, M.; Tamaoki, N.; Nakamura, M. Identification of a key molecular regulator of liver metastasis in human pancreatic carcinoma using a novel quantitative model of metastasis in NOD/SCID/gammacnull (NOG) mice. Int. J. Oncol. 2007, 31, 741–751. [Google Scholar] [CrossRef] [Green Version]
- Michl, P.; Barth, C.; Buchholz, M.; Lerch, M.M.; Rolke, M.; Holzmann, K.H.; Menke, A.; Fensterer, H.; Giehl, K.; Löhr, M.; et al. Claudin-4 expression decreases invasiveness and metastatic potential of pancreatic cancer. Cancer Res. 2003, 63, 6265–6271. [Google Scholar]
- Elsässer, H.P.; Lehr, U.; Agricola, B.; Kern, H.F. Establishment and characterisation of two cell lines with different grade of differentiation derived from one primary human pancreatic adenocarcinoma. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1992, 61, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.S.; Sipos, B.; Orlandini, S.; Sorio, C.; Real, F.X.; Lemoine, N.R.; Gress, T.; Bassi, C.; Klöppel, G.; Kalthoff, H.; et al. Genetic profile of 22 pancreatic carcinoma cell lines. Analysis of K-ras, p53, p16 and DPC4/Smad4. Virchows Arch. 2001, 439, 798–802. [Google Scholar] [CrossRef] [PubMed]
- Bruns, C.J.; Harbison, M.T.; Kuniyasu, H.; Eue, I.; Fidler, I.J. In vivo selection and characterization of metastatic variants from human pancreatic adenocarcinoma by using orthotopic implantation in nude mice. Neoplasia 1999, 1, 50–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vezeridis, M.P.; Tzanakakis, G.N.; Meitner, P.A.; Doremus, C.M.; Tibbetts, L.M.; Calabresi, P. In vivo selection of a highly metastatic cell line from a human pancreatic carcinoma in the nude mouse. Cancer 1992, 69, 2060–2063. [Google Scholar] [CrossRef]
- Olbrot, M.; Rud, J.; Moss, L.G.; Sharma, A. Identification of beta-cell-specific insulin gene transcription factor RIPE3b1 as mammalian MafA. Proc. Natl. Acad. Sci. USA 2002, 99, 6737–6742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Khawaga, S.; Memon, B.; Butler, A.E.; Taheri, S.; Abou-Samra, A.B.; Abdelalim, E.M. Pathways governing development of stem cell-derived pancreatic beta cells: Lessons from embryogenesis. Biol. Rev. Camb. Philos. Soc. 2018, 93, 364–389. [Google Scholar] [CrossRef]
- Reichert, M.; Rustgi, A.K. Pancreatic ductal cells in development, regeneration, and neoplasia. J. Clin. Investig. 2011, 121, 4572–4578. [Google Scholar] [CrossRef] [Green Version]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695. [Google Scholar] [CrossRef] [Green Version]
- Tao, X.; Xiang, H.; Pan, Y.; Shang, D.; Guo, J.; Gao, G.; Xiao, G.G. Pancreatitis initiated pancreatic ductal adenocarcinoma: Pathophysiology explaining clinical evidence. Pharmacol. Res. 2021, 168, 105595. [Google Scholar] [CrossRef]
- Sipos, B.; Möser, S.; Kalthoff, H.; Török, V.; Löhr, M.; Klöppel, G.A. Comprehensive characterization of pancreatic ductal carcinoma cell lines: Towards the establishment of an in vitro research platform. Virchows Arch. 2003, 442, 444–452. [Google Scholar] [CrossRef]
- Handler, J.; Cullis, J.; Avanzi, A.; Vucic, E.A.; Bar-Sagi, D. Pre-neoplastic pancreas cells enter a partially mesenchymal state following transient TGF-beta exposure. Oncogene 2018, 37, 4334–4342. [Google Scholar] [CrossRef] [PubMed]
- Kabashima, A.; Higuchi, H.; Takaishi, H.; Matsuzaki, Y.; Suzuki, S.; Izumiya, M.; Iizuka, H.; Sakai, G.; Hozawa, S.; Azuma, T.; et al. Side population of pancreatic cancer cells predominates in TGF-beta-mediated epithelial to mesenchymal transition and invasion. Int. J. Cancer 2009, 124, 2771–2779. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wu, J.; Zhang, Y.; Xue, X.; Tang, D.; Yuan, Z.; Chen, M.; Wei, J.; Zhang, J.; Miao, Y. Transforming growth factor beta-induced epithelial-mesenchymal transition increases cancer stem-like cells in the PANC-1 cell line. Oncol. Lett. 2012, 3, 229–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kali, A.; Ostapchuk, Y.O.; Belyaev, N.N. TNFalpha and TGFbeta-1 synergistically increase the cancer stem cell properties of MiaPaCa-2 cells. Oncol. Lett. 2017, 14, 4647–4658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalafalla, F.G.; Khan, M.W. Inflammation and Epithelial-Mesenchymal Transition in Pancreatic Ductal Adenocarcinoma: Fighting Against Multiple Opponents. Cancer Growth Metastasis 2017, 10, 1179064417709287. [Google Scholar] [CrossRef]
- De Cock, J.M.; Shibue, T.; Dongre, A.; Keckesova, Z.; Reinhardt, F.; Weinberg, R.A. Inflammation Triggers Zeb1-Dependent Escape from Tumor Latency. Cancer Res. 2016, 76, 6778–6784. [Google Scholar] [CrossRef] [Green Version]
- Kohler, I.; Bronsert, P.; Timme, S.; Werner, M.; Brabletz, T.; Hopt, U.T.; Schilling, O.; Bausch, D.; Keck, T.; Wellner, U.F. Detailed analysis of epithelial-mesenchymal transition and tumor budding identifies predictors of long-term survival in pancreatic ductal adenocarcinoma. J. Gastroenterol. Hepatol. 2015, 30 (Suppl. S1), 78–84. [Google Scholar] [CrossRef]
- Oshimori, N.; Oristian, D.; Fuchs, E. TGF-β promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 2015, 160, 963–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinz, S.; LaBarge, M.A. Hijacking EMT: Better Fat Than Dead. Cancer Cell 2019, 35, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Takehara, M.; Sato, Y.; Kimura, T.; Noda, K.; Miyamoto, H.; Fujino, Y.; Miyoshi, J.; Nakamura, F.; Wada, H.; Bando, Y.; et al. Cancer-associated adipocytes promote pancreatic cancer progression through SAA1 expression. Cancer Sci. 2020, 111, 2883–2894. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidtlein, P.M.; Volz, C.; Braun, R.; Thürling, I.; Lapshyna, O.; Wellner, U.F.; Konukiewitz, B.; Lehnert, H.; Marquardt, J.-U.; Ungefroren, H. A Comparative Endocrine Trans-Differentiation Approach to Pancreatic Ductal Adenocarcinoma Cells with Different EMT Phenotypes Identifies Quasi-Mesenchymal Tumor Cells as Those with Highest Plasticity. Cancers 2021, 13, 4663. https://doi.org/10.3390/cancers13184663
Schmidtlein PM, Volz C, Braun R, Thürling I, Lapshyna O, Wellner UF, Konukiewitz B, Lehnert H, Marquardt J-U, Ungefroren H. A Comparative Endocrine Trans-Differentiation Approach to Pancreatic Ductal Adenocarcinoma Cells with Different EMT Phenotypes Identifies Quasi-Mesenchymal Tumor Cells as Those with Highest Plasticity. Cancers. 2021; 13(18):4663. https://doi.org/10.3390/cancers13184663
Chicago/Turabian StyleSchmidtlein, Paula M., Clara Volz, Rüdiger Braun, Isabel Thürling, Olha Lapshyna, Ulrich F. Wellner, Björn Konukiewitz, Hendrik Lehnert, Jens-Uwe Marquardt, and Hendrik Ungefroren. 2021. "A Comparative Endocrine Trans-Differentiation Approach to Pancreatic Ductal Adenocarcinoma Cells with Different EMT Phenotypes Identifies Quasi-Mesenchymal Tumor Cells as Those with Highest Plasticity" Cancers 13, no. 18: 4663. https://doi.org/10.3390/cancers13184663
APA StyleSchmidtlein, P. M., Volz, C., Braun, R., Thürling, I., Lapshyna, O., Wellner, U. F., Konukiewitz, B., Lehnert, H., Marquardt, J.-U., & Ungefroren, H. (2021). A Comparative Endocrine Trans-Differentiation Approach to Pancreatic Ductal Adenocarcinoma Cells with Different EMT Phenotypes Identifies Quasi-Mesenchymal Tumor Cells as Those with Highest Plasticity. Cancers, 13(18), 4663. https://doi.org/10.3390/cancers13184663