
Histologic Transformation in EGFR-Mutant Lung Adenocarcinomas: Mechanisms and Therapeutic Implications

Abstract
Simple Summary
Abstract
1. Introduction
2. Targeting EGFR in Lung Cancer
3. Mechanisms of Resistance to EGFR TKIs
3.1. EGFR Target-Dependent Mechanisms of Resistance
3.2. EGFR Target-Independent Mechanisms of Resistance
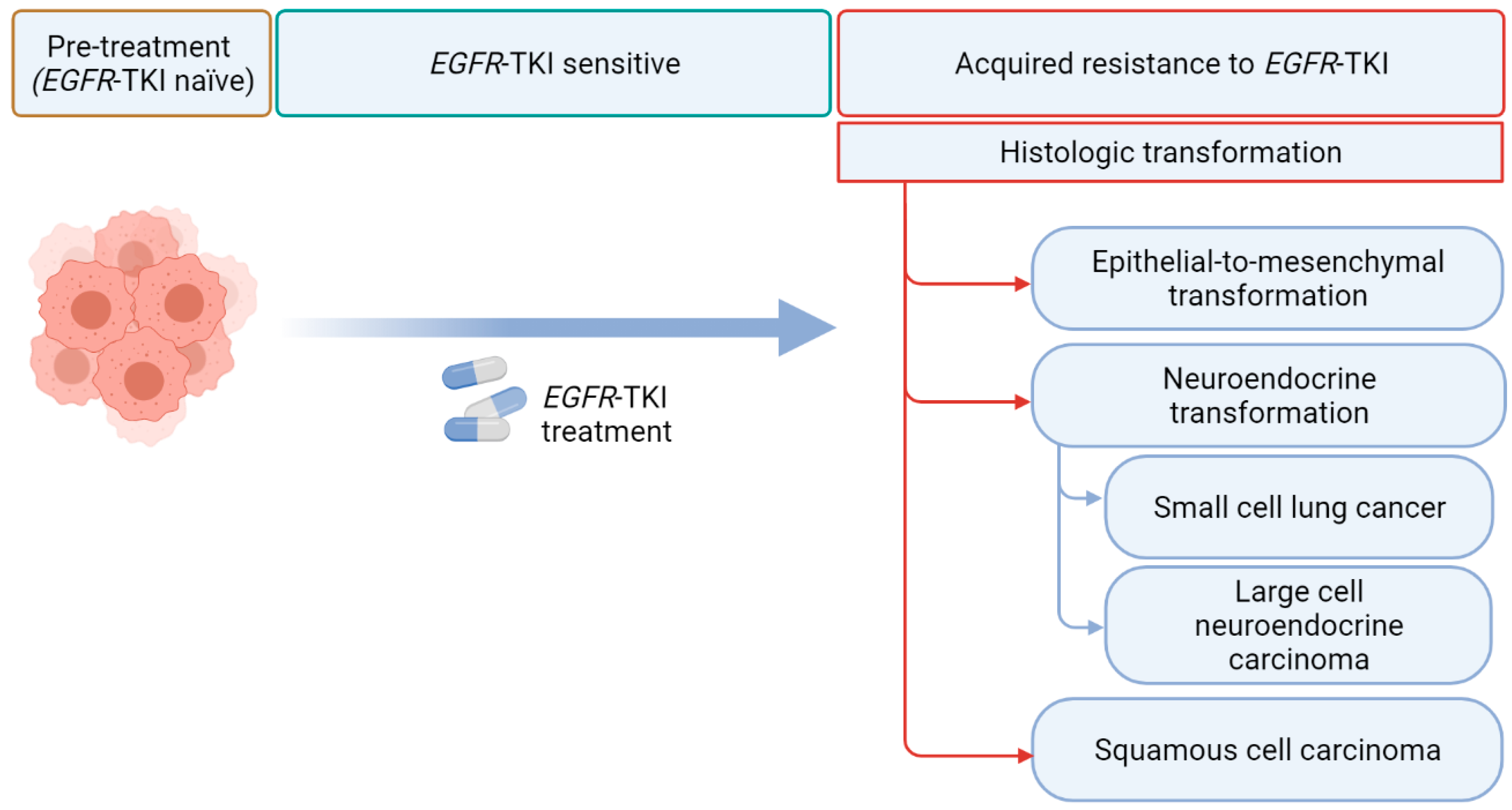
3.3. Histologic and Phenotypic Transformations
4. Histologic Transformations in EGFR TKIs Treated NSCLCs
4.1. Epithelial-to-Mesenchymal Transformation
Novel Therapies
4.2. Neuroendocrine Transformation
4.2.1. SCLC
Novel Therapies
4.2.2. LCNEC
4.3. SCC Transformation
Novel Therapies
5. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, Y.-L.; Yuan, J.-Q.; Wang, K.-F.; Fu, X.-H.; Han, X.-R.; Threapleton, D.; Yang, Z.-Y.; Mao, C.; Tang, J.-L. The prevalence of EGFR mutation in patients with non-small cell lung cancer: A systematic review and meta-analysis. Oncotarget 2016, 7, 78985–78993. [Google Scholar] [CrossRef] [PubMed]
- Mass, R.D. The HER receptor family: A rich target for therapeutic development. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 932–940. [Google Scholar] [CrossRef]
- Lee, C.K.; Davies, L.; Wu, Y.-L.; Mitsudomi, T.; Inoue, A.; Rosell, R.; Zhou, C.; Nakagawa, K.; Thongprasert, S.; Fukuoka, M.; et al. Gefitinib or erlotinib vs chemotherapy for EGFR Mutation-positive lung cancer: Individual patient data meta-analysis of overall survival. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Tsuboi, M.; He, J.; John, T.; Grohe, C.; Majem, M.; Goldman, J.W.; Laktionov, K.; Kim, S.-W.; Kato, T.; et al. Osimertinib in resected EGFR-mutated non–small-cell lung cancer. N. Engl. J. Med. 2020, 383, 1711–1723. [Google Scholar] [CrossRef]
- Howlader, N.; Forjaz, G.; Mooradian, M.J.; Meza, R.; Kong, C.Y.; Cronin, K.A.; Mariotto, A.B.; Lowy, D.R.; Feuer, E.J. The effect of advances in lung-cancer treatment on population mortality. N. Engl. J. Med. 2020, 387, 640–649. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [PubMed]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef]
- Santoni-Rugiu, E.; Melchior, L.C.; Urbanska, E.M.; Jakobsen, J.N.; de Stricker, K.; Grauslund, M.; Sørensen, J.B. Intrinsic resistance to EGFR-tyrosine kinase inhibitors in EGFR-mutant non-small cell lung cancer: Differences and similarities with acquired resistance. Cancers 2019, 11, 923. [Google Scholar] [CrossRef]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef]
- Ferrer, L.; Giaj Levra, M.; Brevet, M.; Antoine, M.; Mazieres, J.; Rossi, G.; Chiari, R.; Westeel, V.; Poudenx, M.; Letreut, J.; et al. A Brief report of transformation from NSCLC to SCLC: Molecular and therapeutic characteristics. J. Thorac. Oncol. 2019, 14, 130–134. [Google Scholar] [CrossRef]
- Marcoux, N.; Gettinger, S.N.; O’Kane, G.; Arbour, K.C.; Neal, J.W.; Husain, H.; Evans, T.L.; Brahmer, J.R.; Muzikansky, A.; Bonomi, P.D.; et al. EGFR-mutant adenocarcinomas that transform to small-cell lung cancer and other neuroendocrine carcinomas: Clinical outcomes. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 37, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Moriya, R.; Hokari, S.; Shibata, S.; Koizumi, T.; Tetsuka, T.; Ito, K.; Hashidate, H.; Tsukada, H. Histological transformation to large cell neuroendocrine carcinoma from lung adenocarcinoma harboring an EGFR mutation: An autopsy case report. Intern. Med. 2017, 56, 2013–2017. [Google Scholar] [CrossRef][Green Version]
- Izumi, H.; Yamasaki, A.; Ueda, Y.; Sumikawa, T.; Maeta, H.; Nakamoto, S.; Shimizu, E. Squamous cell carcinoma transformation from EGFR-mutated lung adenocarcinoma: A case report and literature review. Clin. Lung Cancer 2018, 19, e63–e66. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.Y.; Grandis, J.R.; Shin, D.M. Targeting epidermal growth factor receptor: Novel therapeutics in the management of cancer. Expert Rev. Anticancer Ther. 2003, 3, 367–380. [Google Scholar] [CrossRef]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Khaddour, K.; Jonna, S.; Deneka, A.; Patel, J.D.; Abazeed, M.E.; Golemis, E.; Borghaei, H.; Boumber, Y. Targeting the epidermal growth factor receptor in EGFR-mutated lung cancer: Current and emerging therapies. Cancers 2021, 13, 3164. [Google Scholar] [CrossRef]
- Schoenfeld, A.J.; Chan, J.M.; Kubota, D.; Sato, H.; Rizvi, H.; Daneshbod, Y.; Chang, J.C.; Paik, P.K.; Offin, M.; Arcila, M.E.; et al. Tumor analyses reveal squamous transformation and off-target alterations as early resistance mechanisms to first-line osimertinib in EGFR-mutant lung cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 2654–2663. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Cheng, Y.; Zhou, C.; Ohe, Y.; Imamura, F.; Cho, B.C.; Lin, M.-C.; Majem, M.; Shah, R.; Rukazenkov, Y.; et al. Mechanisms of acquired resistance to first-line osimertinib: Preliminary data from the phase III FLAURA study. Ann. Oncol. 2018, 29, viii740. [Google Scholar] [CrossRef]
- Yun, C.-H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.-K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.-M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Passaro, A.; Jänne, P.A.; Mok, T.; Peters, S. Overcoming therapy resistance in EGFR -mutant lung cancer. Nat. Cancer 2021, 2, 377–391. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Marck, V.L.V.; Bracke, M.E. Epithelial-Mesenchymal Transitions in Human Cancer; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Jakobsen, K.R.; Demuth, C.; Sorensen, B.S.; Nielsen, A.L. The role of epithelial to mesenchymal transition in resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Transl. Lung Cancer Res. 2016, 5, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Prudkin, L.; Liu, D.D.; Ozburn, N.C.; Sun, M.; Behrens, C.; Tang, X.; Brown, K.C.; Bekele, B.N.; Moran, C.; Wistuba, I.I. Epithelial-to-mesenchymal transition in the development and progression of adenocarcinoma and squamous cell carcinoma of the lung. Mod. Pathol. Off. J. U. S. Can. Acad. Pathol. Inc 2009, 22, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.; Buck, E.; Petti, F.; Griffin, G.; Brown, E.; Ramnarine, N.; Iwata, K.K.; Gibson, N.; Haley, J.D. Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res. 2005, 65, 9455–9462. [Google Scholar] [CrossRef]
- Song, K.-A.; Niederst, M.J.; Lochmann, T.L.; Hata, A.N.; Kitai, H.; Ham, J.; Floros, K.V.; Hicks, M.A.; Hu, H.; Mulvey, H.E.; et al. Epithelial-to-mesenchymal transition antagonizes response to targeted therapies in lung cancer by suppressing BIM. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 197–208. [Google Scholar] [CrossRef]
- Shaurova, T.; Zhang, L.; Goodrich, D.W.; Hershberger, P.A. Understanding lineage plasticity as a path to targeted therapy failure in EGFR-mutant non-small cell lung cancer. Front. Genet. 2020, 11, 281. [Google Scholar] [CrossRef]
- Richardson, F.; Young, G.D.; Sennello, R.; Wolf, J.; Argast, G.M.; Mercado, P.; Davies, A.; Epstein, D.M.; Wacker, B. The evaluation of E-Cadherin and vimentin as biomarkers of clinical outcomes among patients with non-small cell lung cancer treated with erlotinib as second- or third-line therapy. Anticancer Res. 2012, 32, 537–552. [Google Scholar]
- Lee, A.-F.; Chen, M.-C.; Chen, C.-J.; Yang, C.-J.; Huang, M.-S.; Liu, Y.-P. Reverse epithelial-mesenchymal transition contributes to the regain of drug sensitivity in tyrosine kinase inhibitor-resistant non-small cell lung cancer cells. PLoS ONE 2017, 12, e0180383. [Google Scholar] [CrossRef]
- Xie, M.; Zhang, L.; He, C.; Xu, F.; Liu, J.; Hu, Z.; Zhao, L.; Tian, Y. Activation of Notch-1 enhances epithelial-mesenchymal transition in gefitinib-acquired resistant lung cancer cells. J. Cell. Biochem. 2012, 113, 1501–1513. [Google Scholar] [CrossRef]
- Yochum, Z.A.; Cades, J.; Wang, H.; Chatterjee, S.; Simons, B.W.; O’Brien, J.P.; Khetarpal, S.K.; Lemtiri-Chlieh, G.; Myers, K.V.; Huang, E.H.-B.; et al. Targeting the EMT transcription factor TWIST1 overcomes resistance to EGFR inhibitors in EGFR-mutant non-small-cell lung cancer. Oncogene 2019, 38, 656–670. [Google Scholar] [CrossRef]
- Izumchenko, E.; Chang, X.; Michailidi, C.; Kagohara, L.; Ravi, R.; Paz, K.; Brait, M.; Hoque, M.O.; Ling, S.; Bedi, A.; et al. The TGFβ-miR200-MIG6 pathway orchestrates the EMT-associated kinase switch that induces resistance to EGFR inhibitors. Cancer Res. 2014, 74, 3995–4005. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Shien, K.; Tomida, S.; Okayasu, K.; Suzawa, K.; Hashida, S.; Torigoe, H.; Watanabe, M.; Yamamoto, H.; Soh, J.; et al. Targeting the miR-200c/LIN28B axis in acquired EGFR-TKI resistance non-small cell lung cancer cells harboring EMT features. Sci. Rep. 2017, 7, 40847. [Google Scholar] [CrossRef] [PubMed]
- Legras, A.; Pécuchet, N.; Imbeaud, S.; Pallier, K.; Didelot, A.; Roussel, H.; Gibault, L.; Fabre, E.; Le Pimpec-Barthes, F.; Laurent-Puig, P.; et al. Epithelial-to-mesenchymal transition and microRNAs in lung cancer. Cancers 2017, 9, 101. [Google Scholar] [CrossRef]
- Park, K.-S.; Raffeld, M.; Moon, Y.W.; Xi, L.; Bianco, C.; Pham, T.; Lee, L.C.; Mitsudomi, T.; Yatabe, Y.; Okamoto, I.; et al. CRIPTO1 expression in EGFR-mutant NSCLC elicits intrinsic EGFR-inhibitor resistance. J. Clin. Invest. 2014, 124, 3003–3015. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Zeng, S.; Wang, Z.; Wu, M.; Ma, Y.; Ye, X.; Zhang, B.; Liu, H. Cancer-associated fibroblasts promote epithelial-mesenchymal transition and EGFR-TKI resistance of non-small cell lung cancers via HGF/IGF-1/ANXA2 signaling. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Nurwidya, F.; Takahashi, F.; Kobayashi, I.; Murakami, A.; Kato, M.; Minakata, K.; Nara, T.; Hashimoto, M.; Yagishita, S.; Baskoro, H.; et al. Treatment with insulin-like growth factor 1 receptor inhibitor reverses hypoxia-induced epithelial-mesenchymal transition in non-small cell lung cancer. Biochem. Biophys. Res. Commun. 2014, 455, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef]
- Weng, C.-H.; Chen, L.-Y.; Lin, Y.-C.; Shih, J.-Y.; Lin, Y.-C.; Tseng, R.-Y.; Chiu, A.-C.; Yeh, Y.-H.; Liu, C.; Lin, Y.-T.; et al. Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per se is a common mechanism for acquired resistance to EGFR TKI. Oncogene 2019, 38, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.; Diao, L.; Cuentas, E.R.P.; Denning, W.L.; Chen, L.; Fan, Y.; Byers, L.A.; Wang, J.; Papadimitrakopoulou, V.; Behrens, C.; et al. Epithelial-mesenchymal transition is associated with a distinct tumor microenvironment including elevation of inflammatory signals and multiple immune checkpoints in lung adenocarcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 3630–3642. [Google Scholar] [CrossRef]
- Hsieh, M.-S.; Lin, M.-W.; Lee, Y.-H. Lung adenocarcinoma with sarcomatoid transformation after tyrosine kinase inhibitor treatment and chemotherapy. Lung Cancer 2019, 137, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Roca, E.; Gurizzan, C.; Amoroso, V.; Vermi, W.; Ferrari, V.; Berruti, A. Outcome of patients with lung adenocarcinoma with transformation to small-cell lung cancer following tyrosine kinase inhibitors treatment: A systematic review and pooled analysis. Cancer Treat. Rev. 2017, 59, 117–122. [Google Scholar] [CrossRef]
- Meuwissen, R.; Linn, S.C.; Linnoila, R.I.; Zevenhoven, J.; Mooi, W.J.; Berns, A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 2003, 4, 181–189. [Google Scholar] [CrossRef]
- Byers, L.A.; Wang, J.; Nilsson, M.B.; Fujimoto, J.; Saintigny, P.; Yordy, J.; Giri, U.; Peyton, M.; Fan, Y.H.; Diao, L.; et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov. 2012, 2, 798–811. [Google Scholar] [CrossRef]
- Peifer, M.; Fernández-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef]
- Meder, L.; König, K.; Ozretić, L.; Schultheis, A.M.; Ueckeroth, F.; Ade, C.P.; Albus, K.; Boehm, D.; Rommerscheidt-Fuss, U.; Florin, A.; et al. NOTCH, ASCL1, p53 and RB alterations define an alternative pathway driving neuroendocrine and small cell lung carcinomas. Int. J. Cancer 2016, 138, 927–938. [Google Scholar] [CrossRef]
- Niederst, M.J.; Sequist, L.V.; Poirier, J.T.; Mermel, C.H.; Lockerman, E.L.; Garcia, A.R.; Katayama, R.; Costa, C.; Ross, K.N.; Moran, T.; et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat. Commun. 2015, 6, 6377. [Google Scholar] [CrossRef]
- Lee, J.-K.; Lee, J.; Kim, S.; Kim, S.; Youk, J.; Park, S.; An, Y.; Keam, B.; Kim, D.-W.; Heo, D.S.; et al. Clonal history and genetic predictors of transformation into small-cell carcinomas from lung adenocarcinomas. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 3065–3074. [Google Scholar] [CrossRef] [PubMed]
- Oser, M.G.; Niederst, M.J.; Sequist, L.V.; Engelman, J.A. Transformation from non-small-cell lung cancer to small-cell lung cancer: Molecular drivers and cells of origin. Lancet Oncol. 2015, 16, e165–e172. [Google Scholar] [CrossRef]
- Offin, M.; Chan, J.M.; Tenet, M.; Rizvi, H.A.; Shen, R.; Riely, G.J.; Rekhtman, N.; Daneshbod, Y.; Quintanal-Villalonga, A.; Penson, A.; et al. Concurrent RB1 and TP53 Alterations define a subset of EGFR-mutant lung cancers at risk for histologic transformation and inferior clinical outcomes. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2019, 14, 1784–1793. [Google Scholar] [CrossRef]
- Gong, Z.; Lu, R.; Xie, S.; Jiang, M.; Liu, K.; Xiao, R.; Shen, J.; Wang, Y.; Guo, L. Overexpression of pro-gastrin releasing peptide promotes the cell proliferation and progression in small cell lung cancer. Biochem. Biophys. Res. Commun. 2016, 479, 312–318. [Google Scholar] [CrossRef]
- Liu, Y. Small cell lung cancer transformation from EGFR-mutated lung adenocarcinoma: A case report and literatures review. Cancer Biol. Ther. 2018, 19, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, X.-Y.; Tang, Y.; Xu, Y.; Guo, W.-H.; Li, Y.-C.; Liu, X.-K.; Huang, C.-Y.; Wang, Y.-S.; Wei, Y.-Q. Rapid increase of serum neuron specific enolase level and tachyphylaxis of EGFR-tyrosine kinase inhibitor indicate small cell lung cancer transformation from EGFR positive lung adenocarcinoma? Lung Cancer Amst. Neth. 2013, 81, 302–305. [Google Scholar] [CrossRef]
- Kato, Y.; Tanaka, Y.; Hino, M.; Gemma, A. ProGRP as early predictive marker of non–small-cell lung cancer to small-cell lung cancer transformation after EGFR-TKI treatment. Respir. Med. Case Rep. 2019, 27, 100837. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; Chung, S.; Brough, R.; Vail, P.; Franco, J.; Lord, C.J.; Knudsen, E.S. Targeting the vulnerability of RB tumor suppressor loss in triple-negative breast cancer. Cell Rep. 2018, 22, 1185–1199. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Du, J.; Parsons, S.H.; Merzoug, F.F.; Webster, Y.; Iversen, P.W.; Chio, L.-C.; Van Horn, R.D.; Lin, X.; Blosser, W.; et al. Aurora a kinase inhibition is synthetic lethal with loss of the RB1 tumor suppressor gene. Cancer Discov. 2019, 9, 248–263. [Google Scholar] [CrossRef]
- Shah, K.N.; Bhatt, R.; Rotow, J.; Rohrberg, J.; Olivas, V.; Wang, V.E.; Hemmati, G.; Martins, M.M.; Maynard, A.; Kuhn, J.; et al. Aurora kinase A drives the evolution of resistance to third-generation EGFR inhibitors in lung cancer. Nat. Med. 2019, 25, 111–118. [Google Scholar] [CrossRef]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbé, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef]
- Lantuejoul, S.; Fernandez-Cuesta, L.; Damiola, F.; Girard, N.; McLeer, A. New molecular classification of large cell neuroendocrine carcinoma and small cell lung carcinoma with potential therapeutic impacts. Transl. Lung Cancer Res. 2020, 9, 2233–2244. [Google Scholar] [CrossRef]
- George, J.; Walter, V.; Peifer, M.; Alexandrov, L.B.; Seidel, D.; Leenders, F.; Maas, L.; Müller, C.; Dahmen, I.; Delhomme, T.M.; et al. Integrative genomic profiling of large-cell neuroendocrine carcinomas reveals distinct subtypes of high-grade neuroendocrine lung tumors. Nat. Commun. 2018, 9, 1048. [Google Scholar] [CrossRef]
- Belluomini, L.; Caliò, A.; Giovannetti, R.; Motton, M.; Mazzarotto, R.; Micheletto, C.; Infante, M.V.; Scarpa, A.; Milella, M.; Pilotto, S. Molecular predictors of EGFR-mutant NSCLC transformation into LCNEC after frontline osimertinib: Digging under the surface. ESMO Open 2021, 6, 100028. [Google Scholar] [CrossRef] [PubMed]
- Baglivo, S.; Ludovini, V.; Sidoni, A.; Metro, G.; Ricciuti, B.; Siggillino, A.; Rebonato, A.; Messina, S.; Crinò, L.; Chiari, R. Large cell neuroendocrine carcinoma transformation and EGFR-T790M mutation as coexisting mechanisms of acquired resistance to EGFR-TKIs in lung cancer. Mayo Clin. Proc. 2017, 92, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Shim, J.H.; Lee, B.; Cho, I.; Park, W.-Y.; Kim, Y.; Lee, S.-H.; Choi, Y.L.; Han, J.; Ahn, J.S.; et al. Paired genomic analysis of squamous cell carcinoma transformed from EGFR-mutated lung adenocarcinoma. Lung Cancer Amst. Neth. 2019, 134, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Matsui, A.; Shimoyama, Y.; Omote, N.; Morise, M.; Hase, T.; Tanaka, I.; Suzuki, K.; Hasegawa, Y. An EGFR-mutated lung adenocarcinoma undergoing squamous cell carcinoma transformation exhibited a durable response to afatinib. Intern. Med. 2018, 57, 3429–3432. [Google Scholar] [CrossRef]
- Jin, R.; Peng, L.; Shou, J.; Wang, J.; Jin, Y.; Liang, F.; Zhao, J.; Wu, M.; Li, Q.; Zhang, B.; et al. EGFR-mutated squamous cell lung cancer and its association with outcomes. Front. Oncol. 2021, 11, 2262. [Google Scholar] [CrossRef]
- Ruffini, E.; Rena, O.; Oliaro, A.; Filosso, P.L.; Bongiovanni, M.; Arslanian, A.; Papalia, E.; Maggi, G. Lung tumors with mixed histologic pattern. Clinico-pathologic characteristics and prognostic significance. Eur. J. Cardio-Thorac. Surg. Off. J. Eur. Assoc. Cardio-Thorac. Surg. 2002, 22, 701–707. [Google Scholar] [CrossRef]
- Han, X.; Li, F.; Fang, Z.; Gao, Y.; Li, F.; Fang, R.; Yao, S.; Sun, Y.; Li, L.; Zhang, W.; et al. Transdifferentiation of lung adenocarcinoma in mice with Lkb1 deficiency to squamous cell carcinoma. Nat. Commun. 2014, 5, 3261. [Google Scholar] [CrossRef]
- Koivunen, J.P.; Kim, J.; Lee, J.; Rogers, A.M.; Park, J.O.; Zhao, X.; Naoki, K.; Okamoto, I.; Nakagawa, K.; Yeap, B.Y.; et al. Mutations in the LKB1 tumour suppressor are frequently detected in tumours from Caucasian but not Asian lung cancer patients. Br. J. Cancer 2008, 99, 245–252. [Google Scholar] [CrossRef]
- Gao, B.; Sun, Y.; Zhang, J.; Ren, Y.; Fang, R.; Han, X.; Shen, L.; Liu, X.-Y.; Pao, W.; Chen, H.; et al. Spectrum of LKB1, EGFR, and KRAS mutations in chinese lung adenocarcinomas. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2010, 5, 1130–1135. [Google Scholar] [CrossRef] [PubMed]
- Cescon, D.W.; Bratman, S.V.; Chan, S.M.; Siu, L.L. Circulating tumor DNA and liquid biopsy in oncology. Nat. Cancer 2020, 1, 276–290. [Google Scholar] [CrossRef]
- Remon, J.; Menis, J.; Hasan, B.; Peric, A.; De Maio, E.; Novello, S.; Reck, M.; Berghmans, T.; Wasag, B.; Besse, B.; et al. The APPLE trial: Feasibility and activity of AZD9291 (osimertinib) treatment on positive PLASMA T790M in EGFR-mutant NSCLC patients. EORTC 1613. Clin. Lung Cancer 2017, 18, 583–588. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Histologic Type | Novel Pathways and Potential Therapeutics |
|---|---|
| Small cell lung cancer |
|
| Squamous cell cancer |
|
| Epithelial-to-mesenchymal transformation |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pathak, R.; Villaflor, V.M. Histologic Transformation in EGFR-Mutant Lung Adenocarcinomas: Mechanisms and Therapeutic Implications. Cancers 2021, 13, 4641. https://doi.org/10.3390/cancers13184641
Pathak R, Villaflor VM. Histologic Transformation in EGFR-Mutant Lung Adenocarcinomas: Mechanisms and Therapeutic Implications. Cancers. 2021; 13(18):4641. https://doi.org/10.3390/cancers13184641
Chicago/Turabian StylePathak, Ranjan, and Victoria M. Villaflor. 2021. "Histologic Transformation in EGFR-Mutant Lung Adenocarcinomas: Mechanisms and Therapeutic Implications" Cancers 13, no. 18: 4641. https://doi.org/10.3390/cancers13184641
APA StylePathak, R., & Villaflor, V. M. (2021). Histologic Transformation in EGFR-Mutant Lung Adenocarcinomas: Mechanisms and Therapeutic Implications. Cancers, 13(18), 4641. https://doi.org/10.3390/cancers13184641