
Intermittent and Periodic Fasting, Hormones, and Cancer Prevention


Abstract
Simple Summary
Abstract
1. Introduction
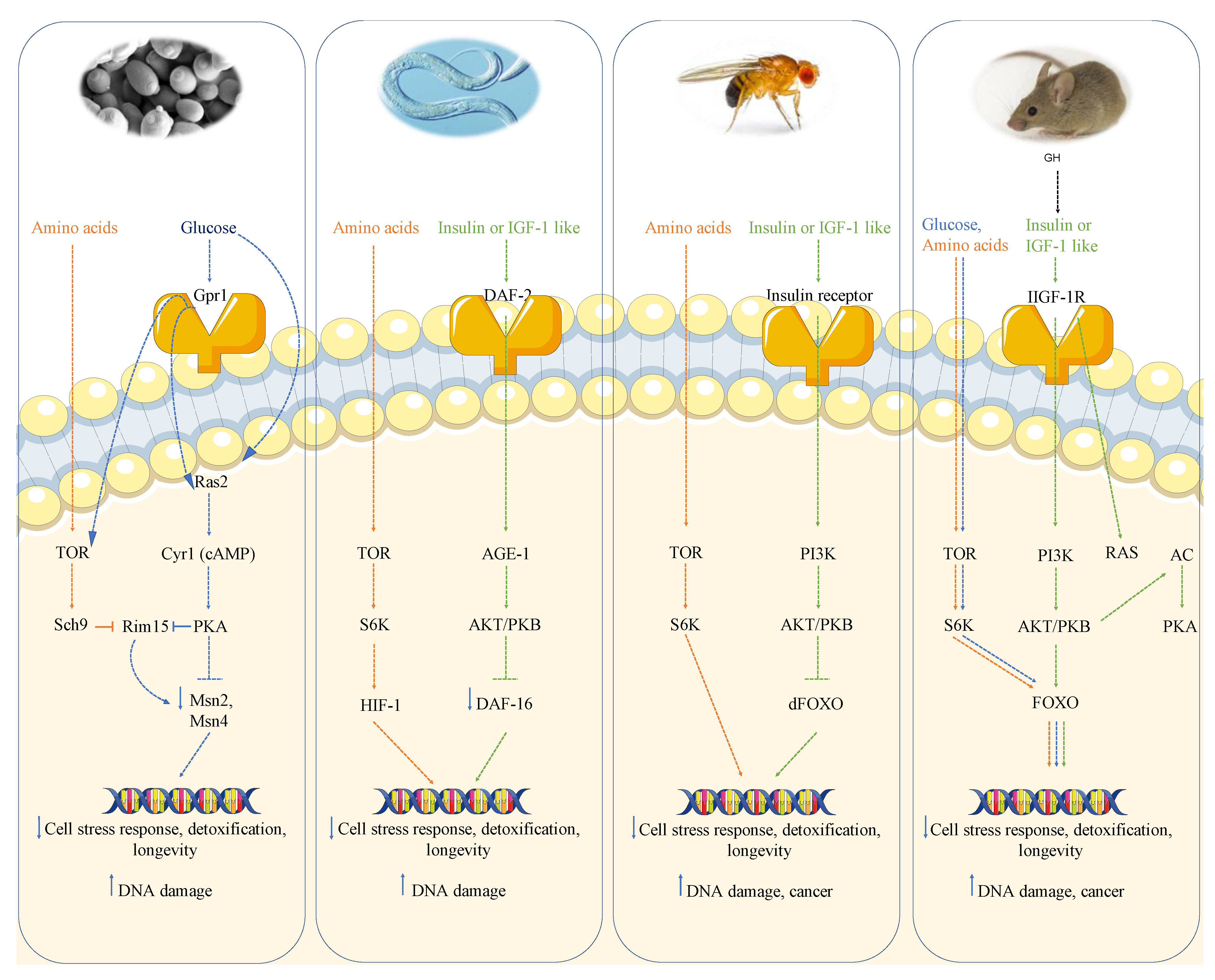
1.1. Growth Genes, Longevity, and Cancer: From Yeast to Humans
1.1.1. Growth Genes Aging and DNA Damage in Yeast
1.1.2. Growth Genes Aging and DNA Damage in Worms and Flies
1.1.3. Growth Genes, Aging and Cancer in Mice
1.1.4. Growth Genes and Cancer in Humans
1.2. Calorie Restriction (CR) and Cancer
1.3. Fasting and Fasting Mimicking Diets
1.4. Fasting Mimicking Diet, Hormones and Cancer Prevention
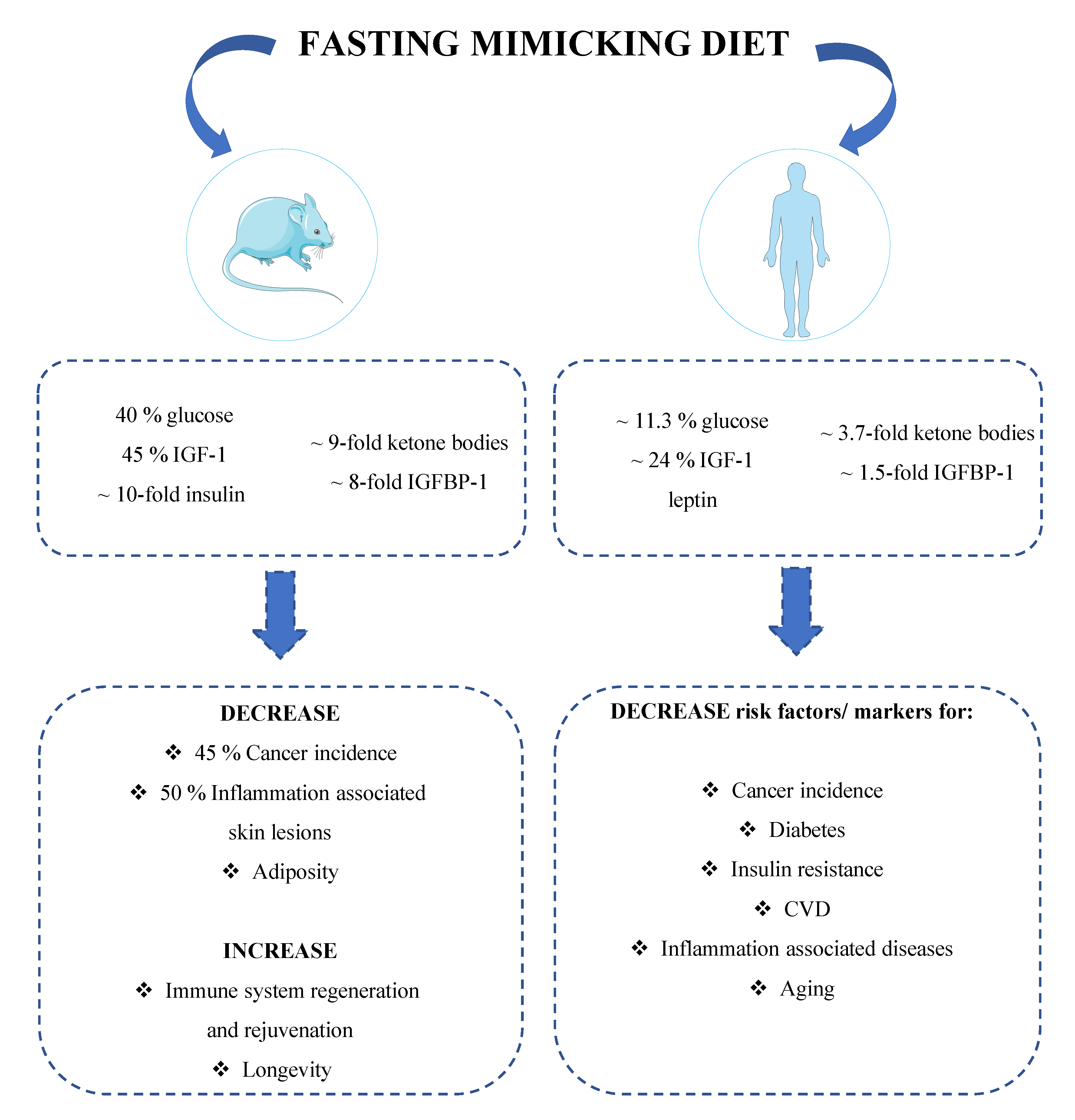
1.4.1. Mice Studies
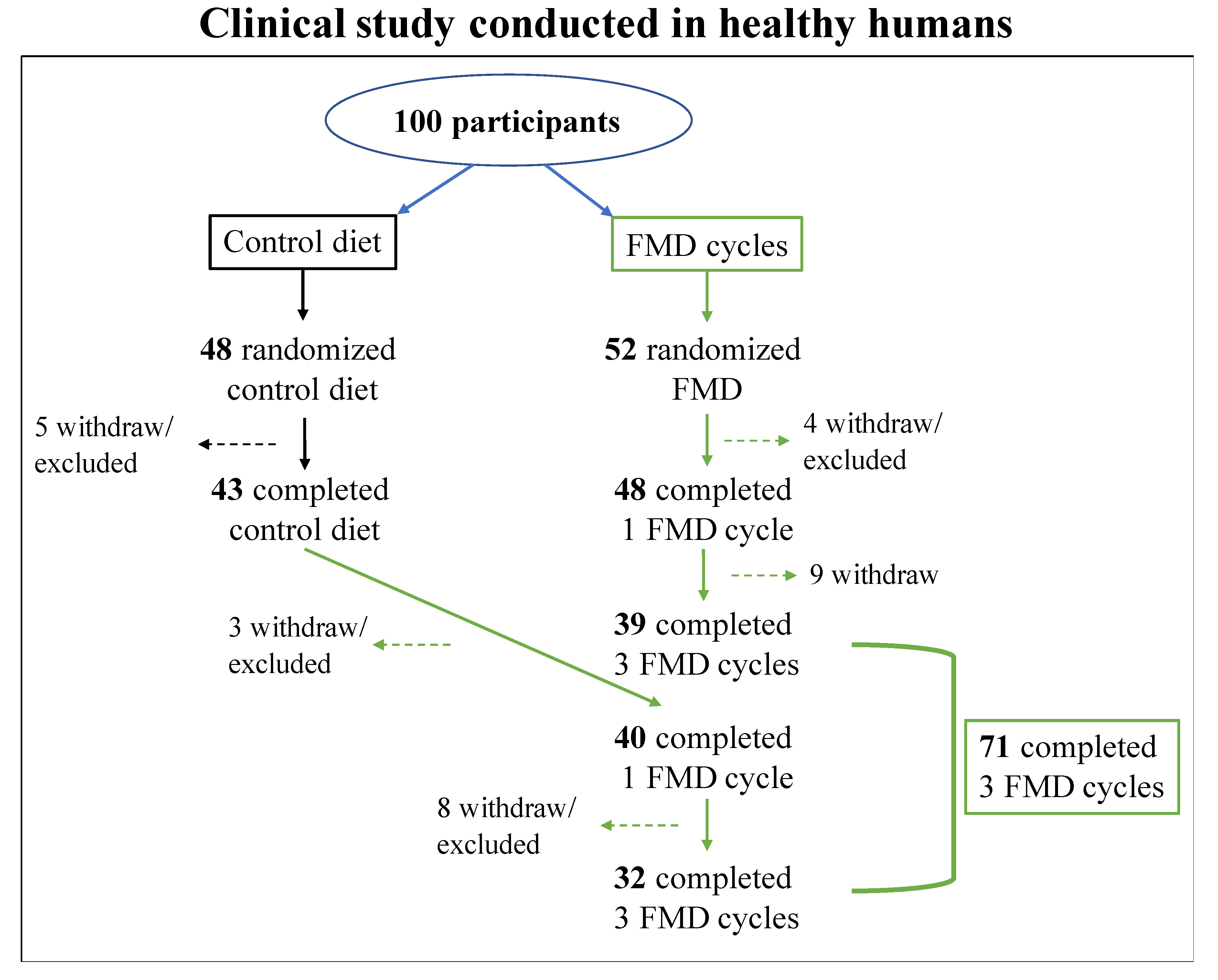
1.4.2. Human Studies

1.5. Alternative Interventions to Reduce Age-Related Diseases Risk Factors

2. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sugimura, T. Nutrition and dietary carcinogens. Carcinogenesis 2000, 21, 387–395. [Google Scholar] [CrossRef]
- Kushi, L.H.; Byers, T.; Doyle, C.; Bandera, E.V.; McCullough, M.; McTiernan, A.; Gansler, T.; Andrews, K.S.; Thun, M.J.; American Cancer Society, N.; et al. American Cancer Society Guidelines on Nutrition and Physical Activity for cancer prevention: Reducing the risk of cancer with healthy food choices and physical activity. CA Cancer J. Clin. 2006, 56, 254–281. [Google Scholar] [CrossRef]
- Kaaks, R. Nutrition, hormones, and breast cancer: Is insulin the missing link? Cancer Causes Control 1996, 7, 605–625. [Google Scholar] [CrossRef] [PubMed]
- Gunter, M.J.; Hoover, D.R.; Yu, H.; Wassertheil-Smoller, S.; Rohan, T.E.; Manson, J.E.; Li, J.; Ho, G.Y.; Xue, X.; Anderson, G.L.; et al. Insulin, insulin-like growth factor-I, and risk of breast cancer in postmenopausal women. J. Natl. Cancer Inst. 2009, 101, 48–60. [Google Scholar] [CrossRef]
- Guevara-Aguirre, J.; Rosenbloom, A.L.; Balasubramanian, P.; Teran, E.; Guevara-Aguirre, M.; Guevara, C.; Procel, P.; Alfaras, I.; De Cabo, R.; Di Biase, S.; et al. GH Receptor Deficiency in Ecuadorian Adults Is Associated With Obesity and Enhanced Insulin Sensitivity. J. Clin. Endocrinol. Metab. 2015, 100, 2589–2596. [Google Scholar] [CrossRef] [PubMed]
- Guevara-Aguirre, J.; Balasubramanian, P.; Guevara-Aguirre, M.; Wei, M.; Madia, F.; Cheng, C.W.; Hwang, D.; Martin-Montalvo, A.; Saavedra, J.; Ingles, S.; et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci. Transl. Med. 2011, 3, 70ra13. [Google Scholar] [CrossRef]
- Giovannucci, E. Insulin and colon cancer. Cancer Causes Control 1995, 6, 164–179. [Google Scholar] [CrossRef] [PubMed]
- Bartke, A.; Sun, L.Y.; Longo, V. Somatotropic signaling: Trade-offs between growth, reproductive development, and longevity. Physiol. Rev. 2013, 93, 571–598. [Google Scholar] [CrossRef]
- Pollak, M.N. Insulin-like growth factors and neoplasia. Novartis Found. Symp. 2004, 262, 84–98. [Google Scholar]
- Levine, M.E.; Suarez, J.A.; Brandhorst, S.; Balasubramanian, P.; Cheng, C.W.; Madia, F.; Fontana, L.; Mirisola, M.G.; Guevara-Aguirre, J.; Wan, J.; et al. Low protein intake is associated with a major reduction in IGF-1, cancer, and overall mortality in the 65 and younger but not older population. Cell Metab. 2014, 19, 407–417. [Google Scholar] [CrossRef]
- Anisimov, V.N.; Bartke, A. The key role of growth hormone-insulin-IGF-1 signaling in aging and cancer. Crit. Rev. Oncol. Hematol. 2013, 87, 201–223. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Kaaks, R.; Lukanova, A. Effects of weight control and physical activity in cancer prevention: Role of endogenous hormone metabolism. Ann. N. Y. Acad. Sci. 2002, 963, 268–281. [Google Scholar] [CrossRef]
- Kershaw, E.E.; Flier, J.S. Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef]
- Berrino, F.; Bellati, C.; Secreto, G.; Camerini, E.; Pala, V.; Panico, S.; Allegro, G.; Kaaks, R. Reducing bioavailable sex hormones through a comprehensive change in diet: The diet and androgens (DIANA) randomized trial. Cancer Epidemiol. Biomark. Prev. 2001, 10, 25–33. [Google Scholar]
- Pugeat, M.; Crave, J.C.; Elmidani, M.; Nicolas, M.H.; Garoscio-Cholet, M.; Lejeune, H.; Dechaud, H.; Tourniaire, J. Pathophysiology of sex hormone binding globulin (SHBG): Relation to insulin. J. Steroid Biochem. Mol. Biol. 1991, 40, 841–849. [Google Scholar] [CrossRef]
- Guevara-Aguirre, J.; Teran, E.; Lescano, D.; Guevara, A.; Guevara, C.; Longo, V.; Gavilanes, A.W.D. Growth hormone receptor deficiency in humans associates to obesity, increased body fat percentage, a healthy brain and a coordinated insulin sensitivity. Growth Horm. IGF Res. 2020, 51, 58–64. [Google Scholar] [CrossRef]
- Weiderpass, E.; Partanen, T.; Kaaks, R.; Vainio, H.; Porta, M.; Kauppinen, T.; Ojajarvi, A.; Boffetta, P.; Malats, N. Occurrence, trends and environment etiology of pancreatic cancer. Scand. J. Work Environ. Health 1998, 24, 165–174. [Google Scholar] [CrossRef]
- Longo, V.D.; Finch, C.E. Evolutionary medicine: From dwarf model systems to healthy centenarians? Science 2003, 299, 1342–1346. [Google Scholar] [CrossRef]
- Fabrizio, P.; Pozza, F.; Pletcher, S.D.; Gendron, C.M.; Longo, V.D. Regulation of longevity and stress resistance by Sch9 in yeast. Science 2001, 292, 288–290. [Google Scholar] [CrossRef]
- Kenyon, C. The plasticity of aging: Insights from long-lived mutants. Cell 2005, 120, 449–460. [Google Scholar] [CrossRef]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending healthy life span—from yeast to humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Foury, F. Human genetic diseases: A cross-talk between man and yeast. Gene 1997, 195, 1–10. [Google Scholar] [CrossRef]
- Mortimer, R.K.; Johnston, J.R. Life span of individual yeast cells. Nature 1959, 183, 1751–1752. [Google Scholar] [CrossRef]
- Longo, V.D.; Shadel, G.S.; Kaeberlein, M.; Kennedy, B. Replicative and chronological aging in Saccharomyces cerevisiae. Cell Metab. 2012, 16, 18–31. [Google Scholar] [CrossRef]
- Longo, V.D.; Ellerby, L.M.; Bredesen, D.E.; Valentine, J.S.; Gralla, E.B. Human Bcl-2 reverses survival defects in yeast lacking superoxide dismutase and delays death of wild-type yeast. J. Cell Biol. 1997, 137, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Mirisola, M.G.; Longo, V.D. Acetic acid and acidification accelerate chronological and replicative aging in yeast. Cell Cycle 2012, 11, 3532–3533. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wei, M.; Mirisola, M.G.; Longo, V.D. Assessing chronological aging in Saccharomyces cerevisiae. Methods Mol. Biol. 2013, 965, 463–472. [Google Scholar] [CrossRef]
- Longo, V.D. Mutations in signal transduction proteins increase stress resistance and longevity in yeast, nematodes, fruit flies, and mammalian neuronal cells. Neurobiol. Aging 1999, 20, 479–486. [Google Scholar] [CrossRef]
- Wei, M.; Fabrizio, P.; Hu, J.; Ge, H.; Cheng, C.; Li, L.; Longo, V.D. Life span extension by calorie restriction depends on Rim15 and transcription factors downstream of Ras/PKA, Tor, and Sch9. PLoS Genet. 2008, 4, e13. [Google Scholar] [CrossRef]
- Toda, T.; Cameron, S.; Sass, P.; Wigler, M. SCH9, a gene of Saccharomyces cerevisiae that encodes a protein distinct from, but functionally and structurally related to, cAMP-dependent protein kinase catalytic subunits. Genes Dev. 1988, 2, 517–527. [Google Scholar] [CrossRef]
- McCormick, M.A.; Delaney, J.R.; Tsuchiya, M.; Tsuchiyama, S.; Shemorry, A.; Sim, S.; Chou, A.C.; Ahmed, U.; Carr, D.; Murakami, C.J.; et al. A Comprehensive Analysis of Replicative Lifespan in 4698 Single-Gene Deletion Strains Uncovers Conserved Mechanisms of Aging. Cell Metab. 2015, 22, 895–906. [Google Scholar] [CrossRef]
- Fabrizio, P.; Hoon, S.; Shamalnasab, M.; Galbani, A.; Wei, M.; Giaever, G.; Nislow, C.; Longo, V.D. Genome-wide screen in Saccharomyces cerevisiae identifies vacuolar protein sorting, autophagy, biosynthetic, and tRNA methylation genes involved in life span regulation. PLoS Genet. 2010, 6, e1001024. [Google Scholar] [CrossRef] [PubMed]
- Mirisola, M.G.; Taormina, G.; Fabrizio, P.; Wei, M.; Hu, J.; Longo, V.D. Serine- and threonine/valine-dependent activation of PDK and Tor orthologs converge on Sch9 to promote aging. PLoS Genet. 2014, 10, e1004113. [Google Scholar] [CrossRef]
- Thevelein, J.M.; de Winde, J.H. Novel sensing mechanisms and targets for the cAMP-protein kinase A pathway in the yeast Saccharomyces cerevisiae. Mol. Microbiol. 1999, 33, 904–918. [Google Scholar] [CrossRef]
- Hlavata, L.; Aguilaniu, H.; Pichova, A.; Nystrom, T. The oncogenic RAS2(val19) mutation locks respiration, independently of PKA, in a mode prone to generate ROS. EMBO J. 2003, 22, 3337–3345. [Google Scholar] [CrossRef]
- Johnson, T.E. Increased life-span of age-1 mutants in Caenorhabditis elegans and lower Gompertz rate of aging. Science 1990, 249, 908–912. [Google Scholar] [CrossRef]
- Kenyon, C.; Chang, J.; Gensch, E.; Rudner, A.; Tabtiang, R. A C. elegans mutant that lives twice as long as wild type. Nature 1993, 366, 461–464. [Google Scholar] [CrossRef]
- Hansen, M.; Chandra, A.; Mitic, L.L.; Onken, B.; Driscoll, M.; Kenyon, C. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet. 2008, 4, e24. [Google Scholar] [CrossRef]
- Johnson, T.E. Caenorhabditis elegans 2007: The premier model for the study of aging. Exp. Gerontol. 2008, 43, 1–4. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mehta, R.; Steinkraus, K.A.; Sutphin, G.L.; Ramos, F.J.; Shamieh, L.S.; Huh, A.; Davis, C.; Chandler-Brown, D.; Kaeberlein, M. Proteasomal regulation of the hypoxic response modulates aging in C. elegans. Science 2009, 324, 1196–1198. [Google Scholar] [CrossRef]
- Pinkston, J.M.; Garigan, D.; Hansen, M.; Kenyon, C. Mutations that increase the life span of C. elegans inhibit tumor growth. Science 2006, 313, 971–975. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Fabrizio, P. Regulation of longevity and stress resistance: A molecular strategy conserved from yeast to humans? Cell Mol. Life Sci. 2002, 59, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Honda, Y.; Honda, S. The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. FASEB J. 1999, 13, 1385–1393. [Google Scholar] [CrossRef]
- LaFever, L.; Drummond-Barbosa, D. Direct control of germline stem cell division and cyst growth by neural insulin in Drosophila. Science 2005, 309, 1071–1073. [Google Scholar] [CrossRef]
- Clancy, D.J.; Gems, D.; Harshman, L.G.; Oldham, S.; Stocker, H.; Hafen, E.; Leevers, S.J.; Partridge, L. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science 2001, 292, 104–106. [Google Scholar] [CrossRef] [PubMed]
- Tatar, M.; Kopelman, A.; Epstein, D.; Tu, M.P.; Yin, C.M.; Garofalo, R.S. A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science 2001, 292, 107–110. [Google Scholar] [CrossRef]
- Igaki, T.; Pagliarini, R.A.; Xu, T. Loss of cell polarity drives tumor growth and invasion through JNK activation in Drosophila. Curr. Biol. 2006, 16, 1139–1146. [Google Scholar] [CrossRef]
- Bjedov, I.; Toivonen, J.M.; Kerr, F.; Slack, C.; Jacobson, J.; Foley, A.; Partridge, L. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 2010, 11, 35–46. [Google Scholar] [CrossRef]
- Orr, W.C.; Sohal, R.S. Extension of life-span by overexpression of superoxide dismutase and catalase in Drosophila melanogaster. Science 1994, 263, 1128–1130. [Google Scholar] [CrossRef]
- Lin, Y.J.; Seroude, L.; Benzer, S. Extended life-span and stress resistance in the Drosophila mutant methuselah. Science 1998, 282, 943–946. [Google Scholar] [CrossRef] [PubMed]
- Selman, C.; Tullet, J.M.; Wieser, D.; Irvine, E.; Lingard, S.J.; Choudhury, A.I.; Claret, M.; Al-Qassab, H.; Carmignac, D.; Ramadani, F.; et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 2009, 326, 140–144. [Google Scholar] [CrossRef]
- Enns, L.C.; Morton, J.F.; Treuting, P.R.; Emond, M.J.; Wolf, N.S.; Dai, D.F.; McKnight, G.S.; Rabinovitch, P.S.; Ladiges, W.C. Disruption of protein kinase A in mice enhances healthy aging. PLoS ONE 2009, 4, e5963. [Google Scholar] [CrossRef]
- Brown-Borg, H.M.; Borg, K.E.; Meliska, C.J.; Bartke, A. Dwarf mice and the ageing process. Nature 1996, 384, 33. [Google Scholar] [CrossRef]
- Coschigano, K.T.; Clemmons, D.; Bellush, L.L.; Kopchick, J.J. Assessment of growth parameters and life span of GHR/BP gene-disrupted mice. Endocrinology 2000, 141, 2608–2613. [Google Scholar] [CrossRef] [PubMed]
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Geloen, A.; Even, P.C.; Cervera, P.; Le Bouc, Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003, 421, 182–187. [Google Scholar] [CrossRef]
- Miquet, J.G.; Gonzalez, L.; Matos, M.N.; Hansen, C.E.; Louis, A.; Bartke, A.; Turyn, D.; Sotelo, A.I. Transgenic mice overexpressing GH exhibit hepatic upregulation of GH-signaling mediators involved in cell proliferation. J. Endocrinol. 2008, 198, 317–330. [Google Scholar] [CrossRef]
- Carboni, J.M.; Lee, A.V.; Hadsell, D.L.; Rowley, B.R.; Lee, F.Y.; Bol, D.K.; Camuso, A.E.; Gottardis, M.; Greer, A.F.; Ho, C.P.; et al. Tumor development by transgenic expression of a constitutively active insulin-like growth factor I receptor. Cancer Res. 2005, 65, 3781–3787. [Google Scholar] [CrossRef]
- Ikeno, Y.; Hubbard, G.B.; Lee, S.; Cortez, L.A.; Lew, C.M.; Webb, C.R.; Berryman, D.E.; List, E.O.; Kopchick, J.J.; Bartke, A. Reduced incidence and delayed occurrence of fatal neoplastic diseases in growth hormone receptor/binding protein knockout mice. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 522–529. [Google Scholar] [CrossRef]
- Pollak, M.; Blouin, M.J.; Zhang, J.C.; Kopchick, J.J. Reduced mammary gland carcinogenesis in transgenic mice expressing a growth hormone antagonist. Br. J. Cancer 2001, 85, 428–430. [Google Scholar] [CrossRef]
- Divisova, J.; Kuiatse, I.; Lazard, Z.; Weiss, H.; Vreeland, F.; Hadsell, D.L.; Schiff, R.; Osborne, C.K.; Lee, A.V. The growth hormone receptor antagonist pegvisomant blocks both mammary gland development and MCF-7 breast cancer xenograft growth. Breast Cancer Res. Treat. 2006, 98, 315–327. [Google Scholar] [CrossRef]
- Longo, V.D.; Lieber, M.R.; Vijg, J. Turning anti-ageing genes against cancer. Nat. Rev. Mol. Cell Biol. 2008, 9, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Mattson, M.P. Fasting: Molecular mechanisms and clinical applications. Cell Metab. 2014, 19, 181–192. [Google Scholar] [CrossRef]
- Rodriguez-Viciana, P.; Tetsu, O.; Oda, K.; Okada, J.; Rauen, K.; McCormick, F. Cancer targets in the Ras pathway. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 461–467. [Google Scholar] [CrossRef]
- Yoeli-Lerner, M.; Toker, A. Akt/PKB signaling in cancer: A function in cell motility and invasion. Cell Cycle 2006, 5, 603–605. [Google Scholar] [CrossRef]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef]
- Consortium, A.P.G. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef]
- Renehan, A.G.; Zwahlen, M.; Minder, C.; O’Dwyer, S.T.; Shalet, S.M.; Egger, M. Insulin-like growth factor (IGF)-I, IGF binding protein-3, and cancer risk: Systematic review and meta-regression analysis. Lancet 2004, 363, 1346–1353. [Google Scholar] [CrossRef]
- Suh, Y.; Atzmon, G.; Cho, M.O.; Hwang, D.; Liu, B.; Leahy, D.J.; Barzilai, N.; Cohen, P. Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proc. Natl. Acad. Sci. USA 2008, 105, 3438–3442. [Google Scholar] [CrossRef] [PubMed]
- Dominici, F.P.; Arostegui Diaz, G.; Bartke, A.; Kopchick, J.J.; Turyn, D. Compensatory alterations of insulin signal transduction in liver of growth hormone receptor knockout mice. J. Endocrinol. 2000, 166, 579–590. [Google Scholar] [CrossRef]
- Dominici, F.P.; Hauck, S.; Argentino, D.P.; Bartke, A.; Turyn, D. Increased insulin sensitivity and upregulation of insulin receptor, insulin receptor substrate (IRS)-1 and IRS-2 in liver of Ames dwarf mice. J. Endocrinol. 2002, 173, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Bartke, A. Insulin resistance and cognitive aging in long-lived and short-lived mice. J. Gerontol. A Biol. Sci. Med. Sci. 2005, 60, 133–134. [Google Scholar] [CrossRef]
- Masternak, M.M.; Panici, J.A.; Bonkowski, M.S.; Hughes, L.F.; Bartke, A. Insulin sensitivity as a key mediator of growth hormone actions on longevity. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 516–521. [Google Scholar] [CrossRef]
- Steuerman, R.; Shevah, O.; Laron, Z. Congenital IGF1 deficiency tends to confer protection against post-natal development of malignancies. Eur. J. Endocrinol. 2011, 164, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Hursting, S.D.; Lavigne, J.A.; Berrigan, D.; Perkins, S.N.; Barrett, J.C. Calorie restriction, aging, and cancer prevention: Mechanisms of action and applicability to humans. Annu. Rev. Med. 2003, 54, 131–152. [Google Scholar] [CrossRef] [PubMed]
- Barrows, C.H., Jr.; Kokkonen, G. The effect of various dietary restricted regimes on biochemical variables in the mouse. Growth 1978, 42, 71–85. [Google Scholar]
- Weindruch, R.; Walford, R.L.; Fligiel, S.; Guthrie, D. The retardation of aging in mice by dietary restriction: Longevity, cancer, immunity and lifetime energy intake. J. Nutr. 1986, 116, 641–654. [Google Scholar] [CrossRef]
- Lane, M.A.; Mattison, J.; Ingram, D.K.; Roth, G.S. Caloric restriction and aging in primates: Relevance to humans and possible CR mimetics. Microsc. Res. Tech. 2002, 59, 335–338. [Google Scholar] [CrossRef]
- Anderson, R.M.; Shanmuganayagam, D.; Weindruch, R. Caloric restriction and aging: Studies in mice and monkeys. Toxicol. Pathol. 2009, 37, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Grandison, R.C.; Piper, M.D.; Partridge, L. Amino-acid imbalance explains extension of lifespan by dietary restriction in Drosophila. Nature 2009, 462, 1061–1064. [Google Scholar] [CrossRef]
- Colman, R.J.; Beasley, T.M.; Kemnitz, J.W.; Johnson, S.C.; Weindruch, R.; Anderson, R.M. Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat. Commun. 2014, 5, 3557. [Google Scholar] [CrossRef]
- McCay, C.M.; Crowell, M.F.; Maynard, L.A. The effect of retarded growth upon the length of life span and upon the ultimate body size. 1935. Nutrition 1989, 5, 155–171. [Google Scholar]
- Patel, N.V.; Finch, C.E. The glucocorticoid paradox of caloric restriction in slowing brain aging. Neurobiol. Aging 2002, 23, 707–717. [Google Scholar] [CrossRef]
- Hursting, S.D.; Switzer, B.R.; French, J.E.; Kari, F.W. The growth hormone: Insulin-like growth factor 1 axis is a mediator of diet restriction-induced inhibition of mononuclear cell leukemia in Fischer rats. Cancer Res. 1993, 53, 2750–2757. [Google Scholar] [PubMed]
- Tomas, F.M.; Chandler, C.S.; Coyle, P.; Bourgeois, C.S.; Burgoyne, J.L.; Rofe, A.M. Effects of insulin and insulin-like growth factors on protein and energy metabolism in tumour-bearing rats. Biochem. J. 1994, 301 Pt 3, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Dunn, S.E.; Kari, F.W.; French, J.; Leininger, J.R.; Travlos, G.; Wilson, R.; Barrett, J.C. Dietary restriction reduces insulin-like growth factor I levels, which modulates apoptosis, cell proliferation, and tumor progression in p53-deficient mice. Cancer Res. 1997, 57, 4667–4672. [Google Scholar]
- Hursting, S.D.; Slaga, T.J.; Fischer, S.M.; DiGiovanni, J.; Phang, J.M. Mechanism-based cancer prevention approaches: Targets, examples, and the use of transgenic mice. J. Natl. Cancer Inst. 1999, 91, 215–225. [Google Scholar] [CrossRef]
- Pearson, K.J.; Lewis, K.N.; Price, N.L.; Chang, J.W.; Perez, E.; Cascajo, M.V.; Tamashiro, K.L.; Poosala, S.; Csiszar, A.; Ungvari, Z.; et al. Nrf2 mediates cancer protection but not prolongevity induced by caloric restriction. Proc. Natl. Acad. Sci. USA 2008, 105, 2325–2330. [Google Scholar] [CrossRef]
- Jiang, W.; Zhu, Z.; Thompson, H.J. Dietary energy restriction modulates the activity of AMP-activated protein kinase, Akt, and mammalian target of rapamycin in mammary carcinomas, mammary gland, and liver. Cancer Res. 2008, 68, 5492–5499. [Google Scholar] [CrossRef]
- Moore, T.; Beltran, L.; Carbajal, S.; Strom, S.; Traag, J.; Hursting, S.D.; DiGiovanni, J. Dietary energy balance modulates signaling through the Akt/mammalian target of rapamycin pathways in multiple epithelial tissues. Cancer Prev. Res. 2008, 1, 65–76. [Google Scholar] [CrossRef]
- Albanes, D. Total calories, body weight, and tumor incidence in mice. Cancer Res. 1987, 47, 1987–1992. [Google Scholar]
- Klurfeld, D.M.; Welch, C.B.; Lloyd, L.M.; Kritchevsky, D. Inhibition of DMBA-induced mammary tumorigenesis by caloric restriction in rats fed high-fat diets. Int. J. Cancer 1989, 43, 922–925. [Google Scholar] [CrossRef]
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.K.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.B.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W.; et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009, 325, 201–204. [Google Scholar] [CrossRef]
- Solon-Biet, S.M.; McMahon, A.C.; Ballard, J.W.; Ruohonen, K.; Wu, L.E.; Cogger, V.C.; Warren, A.; Huang, X.; Pichaud, N.; Melvin, R.G.; et al. The ratio of macronutrients, not caloric intake, dictates cardiometabolic health, aging, and longevity in ad libitum-fed mice. Cell Metab. 2014, 19, 418–430. [Google Scholar] [CrossRef] [PubMed]
- Strom, A.; Jensen, R.A. Mortality from circulatory diseases in Norway 1940–1945. Lancet 1951, 1, 126–129. [Google Scholar] [CrossRef]
- Hindhede, M. The Effect of Food Restriction During War on Mortality in Copenhagen. JAMA 1920, 74, 381–382. [Google Scholar] [CrossRef][Green Version]
- Fontana, L.; Klein, S. Aging, adiposity, and calorie restriction. JAMA 2007, 297, 986–994. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, N.J.; Clifton, P.M.; Noakes, M.; Fenech, M. Weight loss in obese men is associated with increased telomere length and decreased abasic sites in rectal mucosa. Rejuvenation Res. 2009, 12, 169–176. [Google Scholar] [CrossRef]
- Bartke, A. Mutations prolong life in flies; implications for aging in mammals. Trends Endocrinol. Metab. 2001, 12, 233–234. [Google Scholar] [CrossRef]
- Heilbronn, L.K.; de Jonge, L.; Frisard, M.I.; DeLany, J.P.; Larson-Meyer, D.E.; Rood, J.; Nguyen, T.; Martin, C.K.; Volaufova, J.; Most, M.M.; et al. Effect of 6-month calorie restriction on biomarkers of longevity, metabolic adaptation, and oxidative stress in overweight individuals: A randomized controlled trial. JAMA 2006, 295, 1539–1548. [Google Scholar] [CrossRef]
- Dirks, A.J.; Leeuwenburgh, C. Caloric restriction in humans: Potential pitfalls and health concerns. Mech. Ageing Dev. 2006, 127, 1–7. [Google Scholar] [CrossRef]
- Holloszy, J.O.; Fontana, L. Caloric restriction in humans. Exp. Gerontol. 2007, 42, 709–712. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Partridge, L. Promoting health and longevity through diet: From model organisms to humans. Cell 2015, 161, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Roberts, S.B.; Bhapkar, M.V.; Villareal, D.T.; Fontana, L.; Martin, C.K.; Racette, S.B.; Fuss, P.J.; Kraus, W.E.; Wong, W.W.; et al. Body-composition changes in the Comprehensive Assessment of Long-term Effects of Reducing Intake of Energy (CALERIE)-2 study: A 2-y randomized controlled trial of calorie restriction in nonobese humans. Am. J. Clin. Nutr. 2017, 105, 913–927. [Google Scholar] [CrossRef]
- Trepanowski, J.F.; Canale, R.E.; Marshall, K.E.; Kabir, M.M.; Bloomer, R.J. Impact of caloric and dietary restriction regimens on markers of health and longevity in humans and animals: A summary of available findings. Nutr. J. 2011, 10, 107. [Google Scholar] [CrossRef]
- Bruce-Keller, A.J.; Umberger, G.; McFall, R.; Mattson, M.P. Food restriction reduces brain damage and improves behavioral outcome following excitotoxic and metabolic insults. Ann. Neurol. 1999, 45, 8–15. [Google Scholar] [CrossRef]
- Johnson, J.B.; Summer, W.; Cutler, R.G.; Martin, B.; Hyun, D.H.; Dixit, V.D.; Pearson, M.; Nassar, M.; Telljohann, R.; Maudsley, S.; et al. Alternate day calorie restriction improves clinical findings and reduces markers of oxidative stress and inflammation in overweight adults with moderate asthma. Free Radic. Biol. Med. 2007, 42, 665–674. [Google Scholar] [CrossRef]
- Harvie, M.N.; Pegington, M.; Mattson, M.P.; Frystyk, J.; Dillon, B.; Evans, G.; Cuzick, J.; Jebb, S.A.; Martin, B.; Cutler, R.G.; et al. The effects of intermittent or continuous energy restriction on weight loss and metabolic disease risk markers: A randomized trial in young overweight women. Int. J. Obes. 2011, 35, 714–727. [Google Scholar] [CrossRef]
- Regmi, P.; Chaudhary, R.; Page, A.J.; Hutchison, A.T.; Vincent, A.D.; Liu, B.; Heilbronn, L. Early or delayed time-restricted feeding prevents metabolic impact of obesity in mice. J. Endocrinol. 2021, 248, 75–86. [Google Scholar] [CrossRef]
- Chaix, A.; Lin, T.; Le, H.D.; Chang, M.W.; Panda, S. Time-Restricted Feeding Prevents Obesity and Metabolic Syndrome in Mice Lacking a Circadian Clock. Cell Metab. 2019, 29, 303–319.e304. [Google Scholar] [CrossRef]
- Raffaghello, L.; Lee, C.; Safdie, F.M.; Wei, M.; Madia, F.; Bianchi, G.; Longo, V.D. Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proc. Natl. Acad. Sci. USA 2008, 105, 8215–8220. [Google Scholar] [CrossRef]
- Brandhorst, S.; Choi, I.Y.; Wei, M.; Cheng, C.W.; Sedrakyan, S.; Navarrete, G.; Dubeau, L.; Yap, L.P.; Park, R.; Vinciguerra, M.; et al. A Periodic Diet that Mimics Fasting Promotes Multi-System Regeneration, Enhanced Cognitive Performance, and Healthspan. Cell Metab. 2015, 22, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Panda, S. Fasting, Circadian Rhythms, and Time-Restricted Feeding in Healthy Lifespan. Cell Metab. 2016, 23, 1048–1059. [Google Scholar] [CrossRef] [PubMed]
- Fond, G.; Macgregor, A.; Leboyer, M.; Michalsen, A. Fasting in mood disorders: Neurobiology and effectiveness. A review of the literature. Psychiatry Res. 2013, 209, 253–258. [Google Scholar] [CrossRef]
- Maniaci, G.; La Cascia, C.; Giammanco, A.; Ferraro, L.; Chianetta, R.; Di Peri, R.; Sardella, Z.; Citarrella, R.; Mannella, Y.; Larcan, S.; et al. Efficacy of a fasting-mimicking diet in functional therapy for depression: A randomised controlled pilot trial. J. Clin. Psychol. 2020, 76, 1807–1817. [Google Scholar] [CrossRef]
- Gonidakis, S.; Finkel, S.E.; Longo, V.D. Genome-wide screen identifies Escherichia coli TCA-cycle-related mutants with extended chronological lifespan dependent on acetate metabolism and the hypoxia-inducible transcription factor ArcA. Aging Cell 2010, 9, 868–881. [Google Scholar] [CrossRef]
- Lee, G.D.; Wilson, M.A.; Zhu, M.; Wolkow, C.A.; de Cabo, R.; Ingram, D.K.; Zou, S. Dietary deprivation extends lifespan in Caenorhabditis elegans. Aging Cell 2006, 5, 515–524. [Google Scholar] [CrossRef]
- Greer, E.L.; Dowlatshahi, D.; Banko, M.R.; Villen, J.; Hoang, K.; Blanchard, D.; Gygi, S.P.; Brunet, A. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr. Biol. 2007, 17, 1646–1656. [Google Scholar] [CrossRef]
- Honjoh, S.; Yamamoto, T.; Uno, M.; Nishida, E. Signalling through RHEB-1 mediates intermittent fasting-induced longevity in C. elegans. Nature 2009, 457, 726–730. [Google Scholar] [CrossRef]
- Mattson, M.P.; Longo, V.D.; Harvie, M. Impact of intermittent fasting on health and disease processes. Ageing Res. Rev. 2017, 39, 46–58. [Google Scholar] [CrossRef]
- Singh, R.; Manchanda, S.; Kaur, T.; Kumar, S.; Lakhanpal, D.; Lakhman, S.S.; Kaur, G. Middle age onset short-term intermittent fasting dietary restriction prevents brain function impairments in male Wistar rats. Biogerontology 2015, 16, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Wan, R.; Camandola, S.; Mattson, M.P. Intermittent fasting and dietary supplementation with 2-deoxy-D-glucose improve functional and metabolic cardiovascular risk factors in rats. FASEB J. 2003, 17, 1133–1134. [Google Scholar] [CrossRef] [PubMed]
- Castello, L.; Froio, T.; Maina, M.; Cavallini, G.; Biasi, F.; Leonarduzzi, G.; Donati, A.; Bergamini, E.; Poli, G.; Chiarpotto, E. Alternate-day fasting protects the rat heart against age-induced inflammation and fibrosis by inhibiting oxidative damage and NF-kB activation. Free Radic. Biol. Med. 2010, 48, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, F.M.; da Cunha, F.M.; Caldeira da Silva, C.C.; Chausse, B.; Romano, R.L.; Garcia, C.C.; Colepicolo, P.; Medeiros, M.H.; Kowaltowski, A.J. Long-term intermittent feeding, but not caloric restriction, leads to redox imbalance, insulin receptor nitration, and glucose intolerance. Free Radic. Biol. Med. 2011, 51, 1454–1460. [Google Scholar] [CrossRef] [PubMed]
- Varady, K.A.; Bhutani, S.; Church, E.C.; Klempel, M.C. Short-term modified alternate-day fasting: A novel dietary strategy for weight loss and cardioprotection in obese adults. Am. J. Clin. Nutr. 2009, 90, 1138–1143. [Google Scholar] [CrossRef]
- Varady, K.A.; Hudak, C.S.; Hellerstein, M.K. Modified alternate-day fasting and cardioprotection: Relation to adipose tissue dynamics and dietary fat intake. Metabolism 2009, 58, 803–811. [Google Scholar] [CrossRef]
- Dorighello, G.G.; Rovani, J.C.; Luhman, C.J.; Paim, B.A.; Raposo, H.F.; Vercesi, A.E.; Oliveira, H.C. Food restriction by intermittent fasting induces diabetes and obesity and aggravates spontaneous atherosclerosis development in hypercholesterolaemic mice. Br. J. Nutr. 2014, 111, 979–986. [Google Scholar] [CrossRef]
- Goodrick, C.L.; Ingram, D.K.; Reynolds, M.A.; Freeman, J.R.; Cider, N. Effects of intermittent feeding upon body weight and lifespan in inbred mice: Interaction of genotype and age. Mech. Ageing Dev. 1990, 55, 69–87. [Google Scholar] [CrossRef]
- Cheng, C.W.; Adams, G.B.; Perin, L.; Wei, M.; Zhou, X.; Lam, B.S.; Da Sacco, S.; Mirisola, M.; Quinn, D.I.; Dorff, T.B.; et al. Prolonged fasting reduces IGF-1/PKA to promote hematopoietic-stem-cell-based regeneration and reverse immunosuppression. Cell Stem Cell 2014, 14, 810–823. [Google Scholar] [CrossRef]
- Lee, C.; Safdie, F.M.; Raffaghello, L.; Wei, M.; Madia, F.; Parrella, E.; Hwang, D.; Cohen, P.; Bianchi, G.; Longo, V.D. Reduced levels of IGF-I mediate differential protection of normal and cancer cells in response to fasting and improve chemotherapeutic index. Cancer Res. 2010, 70, 1564–1572. [Google Scholar] [CrossRef]
- Cotterill, A.M.; Holly, J.M.; Wass, J.A. The regulation of insulin-like growth factor binding protein (IGFBP)-1 during prolonged fasting. Clin. Endocrinol. 1993, 39, 357–362. [Google Scholar] [CrossRef]
- Thissen, J.P.; Ketelslegers, J.M.; Underwood, L.E. Nutritional regulation of the insulin-like growth factors. Endocr. Rev. 1994, 15, 80–101. [Google Scholar] [CrossRef]
- Giovannucci, E.; Pollak, M.; Platz, E.A.; Willett, W.C.; Stampfer, M.J.; Majeed, N.; Colditz, G.A.; Speizer, F.E.; Hankinson, S.E. Insulin-like growth factor I (IGF-I), IGF-binding protein-3 and the risk of colorectal adenoma and cancer in the Nurses’ Health Study. Growth Horm. IGF Res. 2000, 10 (Suppl. A), S30–S31. [Google Scholar] [CrossRef]
- Lee, C.; Longo, V.D. Fasting vs. dietary restriction in cellular protection and cancer treatment: From model organisms to patients. Oncogene 2011, 30, 3305–3316. [Google Scholar] [CrossRef]
- Murphy, N.; Carreras-Torres, R.; Song, M.; Chan, A.T.; Martin, R.M.; Papadimitriou, N.; Dimou, N.; Tsilidis, K.K.; Banbury, B.; Bradbury, K.E.; et al. Circulating Levels of Insulin-like Growth Factor 1 and Insulin-like Growth Factor Binding Protein 3 Associate With Risk of Colorectal Cancer Based on Serologic and Mendelian Randomization Analyses. Gastroenterology 2020, 158, 1300–1312.e20. [Google Scholar] [CrossRef]
- Safdie, F.M.; Dorff, T.; Quinn, D.; Fontana, L.; Wei, M.; Lee, C.; Cohen, P.; Longo, V.D. Fasting and cancer treatment in humans: A case series report. Aging 2009, 1, 988–1007. [Google Scholar] [CrossRef] [PubMed]
- de Groot, S.; Vreeswijk, M.P.; Welters, M.J.; Gravesteijn, G.; Boei, J.J.; Jochems, A.; Houtsma, D.; Putter, H.; van der Hoeven, J.J.; Nortier, J.W.; et al. The effects of short-term fasting on tolerance to (neo) adjuvant chemotherapy in HER2-negative breast cancer patients: A randomized pilot study. BMC Cancer 2015, 15, 652. [Google Scholar] [CrossRef]
- Bauersfeld, S.P.; Kessler, C.S.; Wischnewsky, M.; Jaensch, A.; Steckhan, N.; Stange, R.; Kunz, B.; Bruckner, B.; Sehouli, J.; Michalsen, A. The effects of short-term fasting on quality of life and tolerance to chemotherapy in patients with breast and ovarian cancer: A randomized cross-over pilot study. BMC Cancer 2018, 18, 476. [Google Scholar] [CrossRef]
- Dorff, T.B.; Groshen, S.; Garcia, A.; Shah, M.; Tsao-Wei, D.; Pham, H.; Cheng, C.W.; Brandhorst, S.; Cohen, P.; Wei, M.; et al. Safety and feasibility of fasting in combination with platinum-based chemotherapy. BMC Cancer 2016, 16, 360. [Google Scholar] [CrossRef]
- Wei, M.; Brandhorst, S.; Shelehchi, M.; Mirzaei, H.; Cheng, C.W.; Budniak, J.; Groshen, S.; Mack, W.J.; Guen, E.; Di Biase, S.; et al. Fasting-mimicking diet and markers/risk factors for aging, diabetes, cancer, and cardiovascular disease. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Caffa, I.; Spagnolo, V.; Vernieri, C.; Valdemarin, F.; Becherini, P.; Wei, M.; Brandhorst, S.; Zucal, C.; Driehuis, E.; Ferrando, L.; et al. Author Correction: Fasting-mimicking diet and hormone therapy induce breast cancer regression. Nature 2020, 588, E33. [Google Scholar] [CrossRef]
- de Groot, S.; Lugtenberg, R.T.; Cohen, D.; Welters, M.J.P.; Ehsan, I.; Vreeswijk, M.P.G.; Smit, V.; de Graaf, H.; Heijns, J.B.; Portielje, J.E.A.; et al. Fasting mimicking diet as an adjunct to neoadjuvant chemotherapy for breast cancer in the multicentre randomized phase 2 DIRECT trial. Nat. Commun. 2020, 11, 3083. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Raffaghello, L.; Brandhorst, S.; Safdie, F.M.; Bianchi, G.; Martin-Montalvo, A.; Pistoia, V.; Wei, M.; Hwang, S.; Merlino, A.; et al. Fasting cycles retard growth of tumors and sensitize a range of cancer cell types to chemotherapy. Sci. Transl. Med. 2012, 4, 124ra27. [Google Scholar] [CrossRef] [PubMed]
- Di Biase, S.; Lee, C.; Brandhorst, S.; Manes, B.; Buono, R.; Cheng, C.W.; Cacciottolo, M.; Martin-Montalvo, A.; de Cabo, R.; Wei, M.; et al. Fasting-Mimicking Diet Reduces HO-1 to Promote T Cell-Mediated Tumor Cytotoxicity. Cancer Cell 2016, 30, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Stocks, T.; Rapp, K.; Bjorge, T.; Manjer, J.; Ulmer, H.; Selmer, R.; Lukanova, A.; Johansen, D.; Concin, H.; Tretli, S.; et al. Blood glucose and risk of incident and fatal cancer in the metabolic syndrome and cancer project (me-can): Analysis of six prospective cohorts. PLoS Med. 2009, 6, e1000201. [Google Scholar] [CrossRef]
- Davidson, M.B. The effect of aging on carbohydrate metabolism: A review of the English literature and a practical approach to the diagnosis of diabetes mellitus in the elderly. Metabolism 1979, 28, 688–705. [Google Scholar] [CrossRef]
- Pyorala, K. Relationship of glucose tolerance and plasma insulin to the incidence of coronary heart disease: Results from two population studies in Finland. Diabetes Care 1979, 2, 131–141. [Google Scholar] [CrossRef]
- Stout, R.W. The relationship of abnormal circulating insulin levels to atherosclerosis. Atherosclerosis 1977, 27, 1–13. [Google Scholar] [CrossRef]
- Bjorntorp, P.; De Jounge, K.; Sjostrom, L.; Sullivan, L. The effect of physical training on insulin production in obesity. Metabolism 1970, 19, 631–638. [Google Scholar] [CrossRef]
- LeBlanc, J.; Nadeau, A.; Richard, D.; Tremblay, A. Studies on the sparing effect of exercise on insulin requirements in human subjects. Metabolism 1981, 30, 1119–1124. [Google Scholar] [CrossRef]
- Heath, G.W.; Gavin, J.R., 3rd; Hinderliter, J.M.; Hagberg, J.M.; Bloomfield, S.A.; Holloszy, J.O. Effects of exercise and lack of exercise on glucose tolerance and insulin sensitivity. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1983, 55, 512–517. [Google Scholar] [CrossRef]
- Seals, D.R.; Hagberg, J.M.; Allen, W.K.; Hurley, B.F.; Dalsky, G.P.; Ehsani, A.A.; Holloszy, J.O. Glucose tolerance in young and older athletes and sedentary men. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1984, 56, 1521–1525. [Google Scholar] [CrossRef]
- De Groote, E.; Britto, F.A.; Balan, E.; Warnier, G.; Thissen, J.P.; Nielens, H.; Sylow, L.; Deldicque, L. Effect of hypoxic exercise on glucose tolerance in healthy and prediabetic adults. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E43–E54. [Google Scholar] [CrossRef]
- Holloszy, J.O.; Schultz, J.; Kusnierkiewicz, J.; Hagberg, J.M.; Ehsani, A.A. Effects of exercise on glucose tolerance and insulin resistance. Brief review and some preliminary results. Acta Med. Scand. Suppl. 1986, 711, 55–65. [Google Scholar] [CrossRef]
- Fontana, L.; Meyer, T.E.; Klein, S.; Holloszy, J.O. Long-term low-calorie low-protein vegan diet and endurance exercise are associated with low cardiometabolic risk. Rejuvenation Res. 2007, 10, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.C.; Lee, I.M.; Weiderpass, E.; Campbell, P.T.; Sampson, J.N.; Kitahara, C.M.; Keadle, S.K.; Arem, H.; Berrington de Gonzalez, A.; Hartge, P.; et al. Association of Leisure-Time Physical Activity With Risk of 26 Types of Cancer in 1.44 Million Adults. JAMA Intern. Med. 2016, 176, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Friedenreich, C.M.; Neilson, H.K.; Farris, M.S.; Courneya, K.S. Physical Activity and Cancer Outcomes: A Precision Medicine Approach. Clin. Cancer Res. 2016, 22, 4766–4775. [Google Scholar] [CrossRef] [PubMed]
- Sarfstein, R.; Friedman, Y.; Attias-Geva, Z.; Fishman, A.; Bruchim, I.; Werner, H. Metformin downregulates the insulin/IGF-I signaling pathway and inhibits different uterine serous carcinoma (USC) cells proliferation and migration in p53-dependent or -independent manners. PLoS ONE 2013, 8, e61537. [Google Scholar] [CrossRef]
- Xie, Y.; Wang, J.L.; Ji, M.; Yuan, Z.F.; Peng, Z.; Zhang, Y.; Wen, J.G.; Shi, H.R. Regulation of insulin-like growth factor signaling by metformin in endometrial cancer cells. Oncol. Lett. 2014, 8, 1993–1999. [Google Scholar] [CrossRef]
- Klement, R.J.; Fink, M.K. Dietary and pharmacological modification of the insulin/IGF-1 system: Exploiting the full repertoire against cancer. Oncogenesis 2016, 5, e193. [Google Scholar] [CrossRef]
- Onken, B.; Driscoll, M. Metformin induces a dietary restriction-like state and the oxidative stress response to extend C. elegans Healthspan via AMPK, LKB1, and SKN-1. PLoS ONE 2010, 5, e8758. [Google Scholar] [CrossRef]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.J.; et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 2013, 4, 2192. [Google Scholar] [CrossRef]
- Dowling, R.J.; Zakikhani, M.; Fantus, I.G.; Pollak, M.; Sonenberg, N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007, 67, 10804–10812. [Google Scholar] [CrossRef]
- Kalender, A.; Selvaraj, A.; Kim, S.Y.; Gulati, P.; Brule, S.; Viollet, B.; Kemp, B.E.; Bardeesy, N.; Dennis, P.; Schlager, J.J.; et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010, 11, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Ben Sahra, I.; Regazzetti, C.; Robert, G.; Laurent, K.; Le Marchand-Brustel, Y.; Auberger, P.; Tanti, J.F.; Giorgetti-Peraldi, S.; Bost, F. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011, 71, 4366–4372. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.; Lux, A.; O’Callaghan, F. The journey of metformin from glycaemic control to mTOR inhibition and the suppression of tumour growth. Br. J. Clin. Pharmacol. 2019, 85, 37–46. [Google Scholar] [CrossRef]
- Kane, D.A.; Anderson, E.J.; Price, J.W., 3rd; Woodlief, T.L.; Lin, C.T.; Bikman, B.T.; Cortright, R.N.; Neufer, P.D. Metformin selectively attenuates mitochondrial H2O2 emission without affecting respiratory capacity in skeletal muscle of obese rats. Free Radic. Biol. Med. 2010, 49, 1082–1087. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Cheki, M.; Rezapoor, S.; Geraily, G.; Motevaseli, E.; Carnovale, C.; Clementi, E.; Shirazi, A. Metformin: Prevention of genomic instability and cancer: A review. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2018, 827, 1–8. [Google Scholar] [CrossRef]
- Kelly, B.; Tannahill, G.M.; Murphy, M.P.; O’Neill, L.A. Metformin Inhibits the Production of Reactive Oxygen Species from NADH:Ubiquinone Oxidoreductase to Limit Induction of Interleukin-1beta (IL-1beta) and Boosts Interleukin-10 (IL-10) in Lipopolysaccharide (LPS)-activated Macrophages. J. Biol. Chem. 2015, 290, 20348–20359. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Calorie Restriction (CR) | Metabolic Adaptations | Molecular Adaptations | Cellular Adaptations |
| ↓ IGF-1 ↓ Insulin ↓ Oxidative stress ↓ Inflammation ↑ Cortisol | ↓ PI3K/Akt/S6K ↓ mTOR ↓ Ras/MAPK ↑ Nrf2 ↑ FOXO ↑ PTEN | ↓ Cell proliferation ↓ Oxidative damage ↑ DNA repair ↑ Genome instability |
| Type of Fasting | Schedule | Description | |
|---|---|---|---|
| Intermittent Fasting (IF) | ADF | 24 h fast/ 24 h eating period | Water only fasting every other day |
| 5:2 | 2 days fast or very low calorie consumption (500–700 kcal)/ 5 days eating period | Alternation of 2 days of very low-calorie consumption with a 5 days ad libitum re-feeding period | |
| TRF | 12- to 18 h fast/ 6- to 12 h eating period | Food intake resctricted to 6–12 h per day | |
| Periodic Fasting (PF) | Prolonged fasting | 2–5 days of water fast/ 7 days eating period (or longer) | Water only fasting period followed by an ad libitum re-feeding period |
| Prolonged FMD | 4- to 7 days FMD/ 10- to 25 days eating period | 30–50% of the normal caloric intake using a fasting mimicking diet for 4–7 days followed by an ad libitum re-feeding period |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salvadori, G.; Mirisola, M.G.; Longo, V.D. Intermittent and Periodic Fasting, Hormones, and Cancer Prevention. Cancers 2021, 13, 4587. https://doi.org/10.3390/cancers13184587
Salvadori G, Mirisola MG, Longo VD. Intermittent and Periodic Fasting, Hormones, and Cancer Prevention. Cancers. 2021; 13(18):4587. https://doi.org/10.3390/cancers13184587
Chicago/Turabian StyleSalvadori, Giulia, Mario Giuseppe Mirisola, and Valter D. Longo. 2021. "Intermittent and Periodic Fasting, Hormones, and Cancer Prevention" Cancers 13, no. 18: 4587. https://doi.org/10.3390/cancers13184587
APA StyleSalvadori, G., Mirisola, M. G., & Longo, V. D. (2021). Intermittent and Periodic Fasting, Hormones, and Cancer Prevention. Cancers, 13(18), 4587. https://doi.org/10.3390/cancers13184587