
Harnessing the Immune System to Fight Multiple Myeloma

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
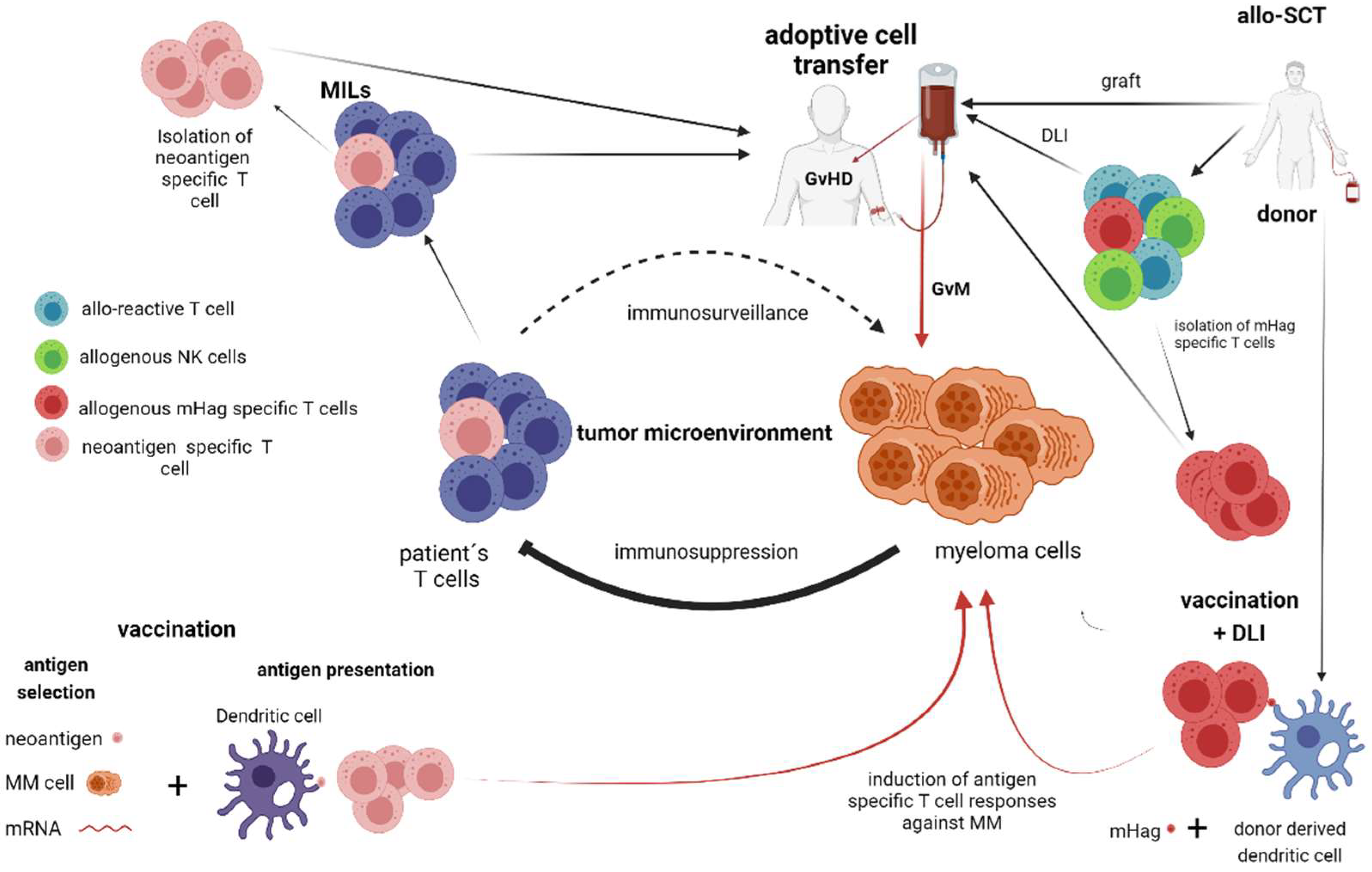
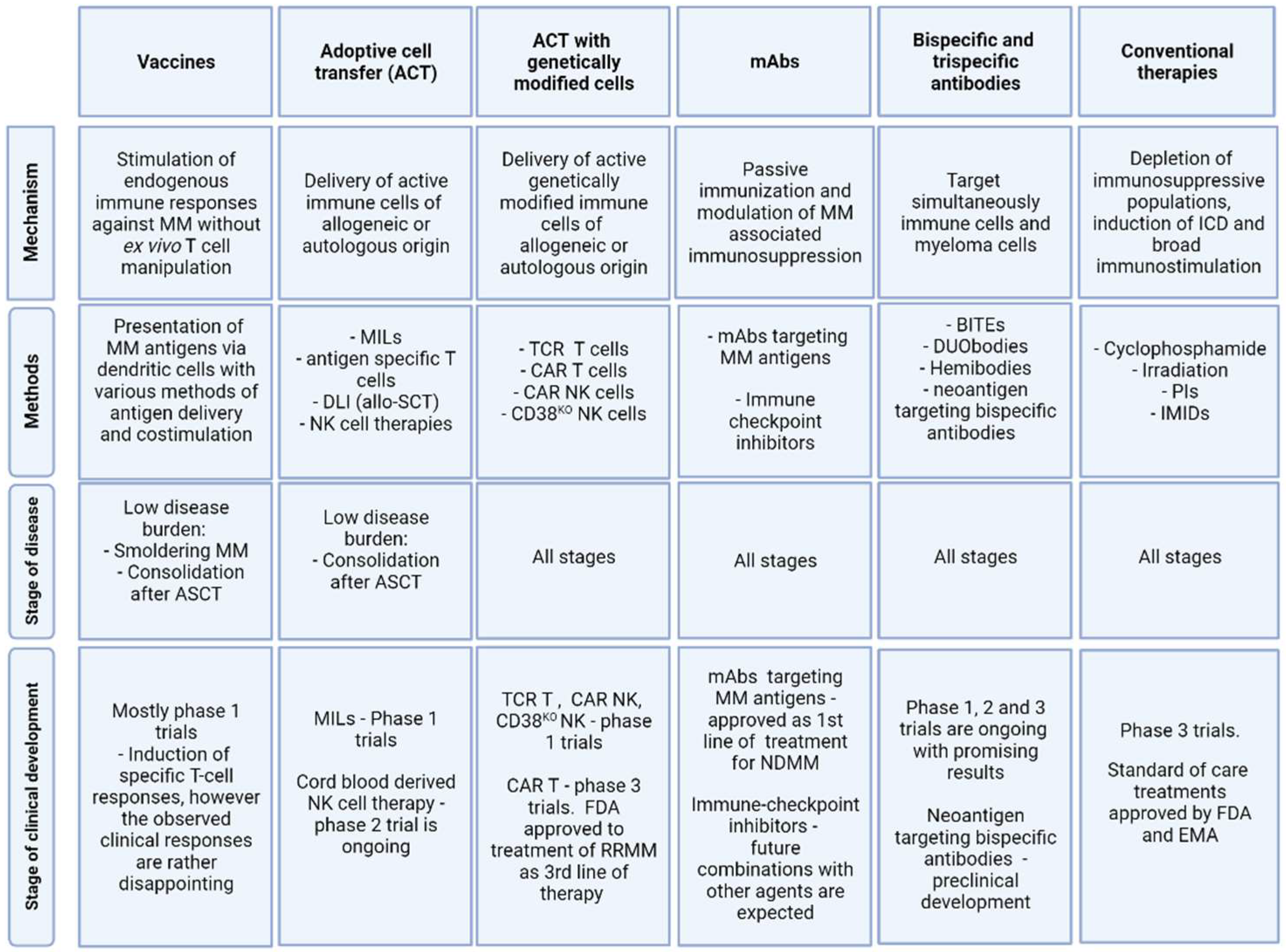
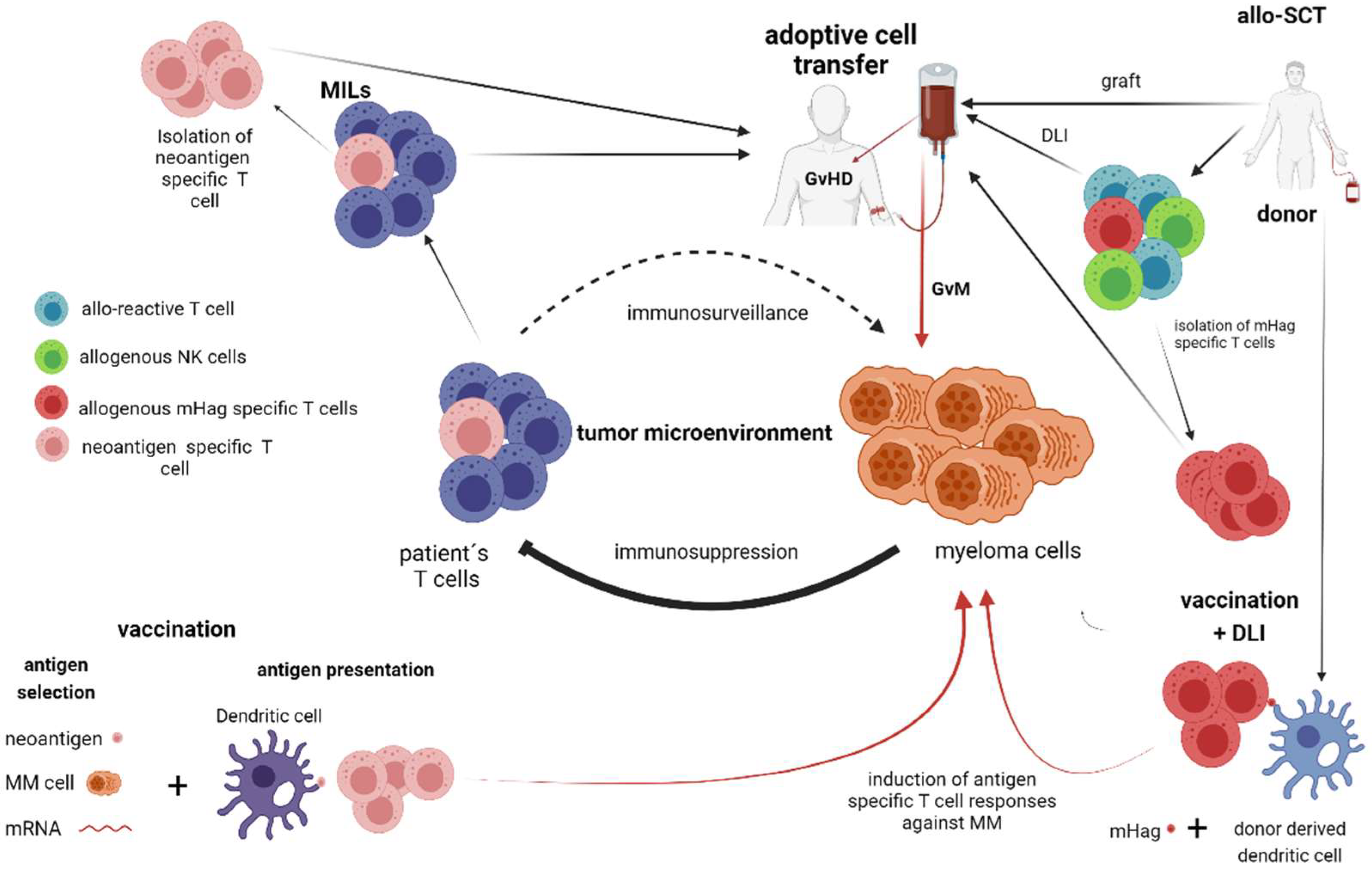
2. T Cell and NK Cell-Dependent Therapies without Genetic Manipulation (Allogeneic Stem Cell Transplantation, Vaccination Strategies)
2.1. Allogeneic Stem Cell Transplantation (Allo-SCT)
2.2. Vaccination Strategies
3. Adaptive Immune Transfer of Autologous and Allogeneic Lymphocytes without Genetic Manipulation—(Marrow-Infiltrating Lymphocytes, Donor Lymphocyte Infusions, Adoptive Transfer of NK-Cells)
3.1. Marrow-Infiltrating Lymphocytes (MILs)
3.2. Donor Lymphocyte Infusions (DLI)
3.3. Adoptive Transfer of NK Cells
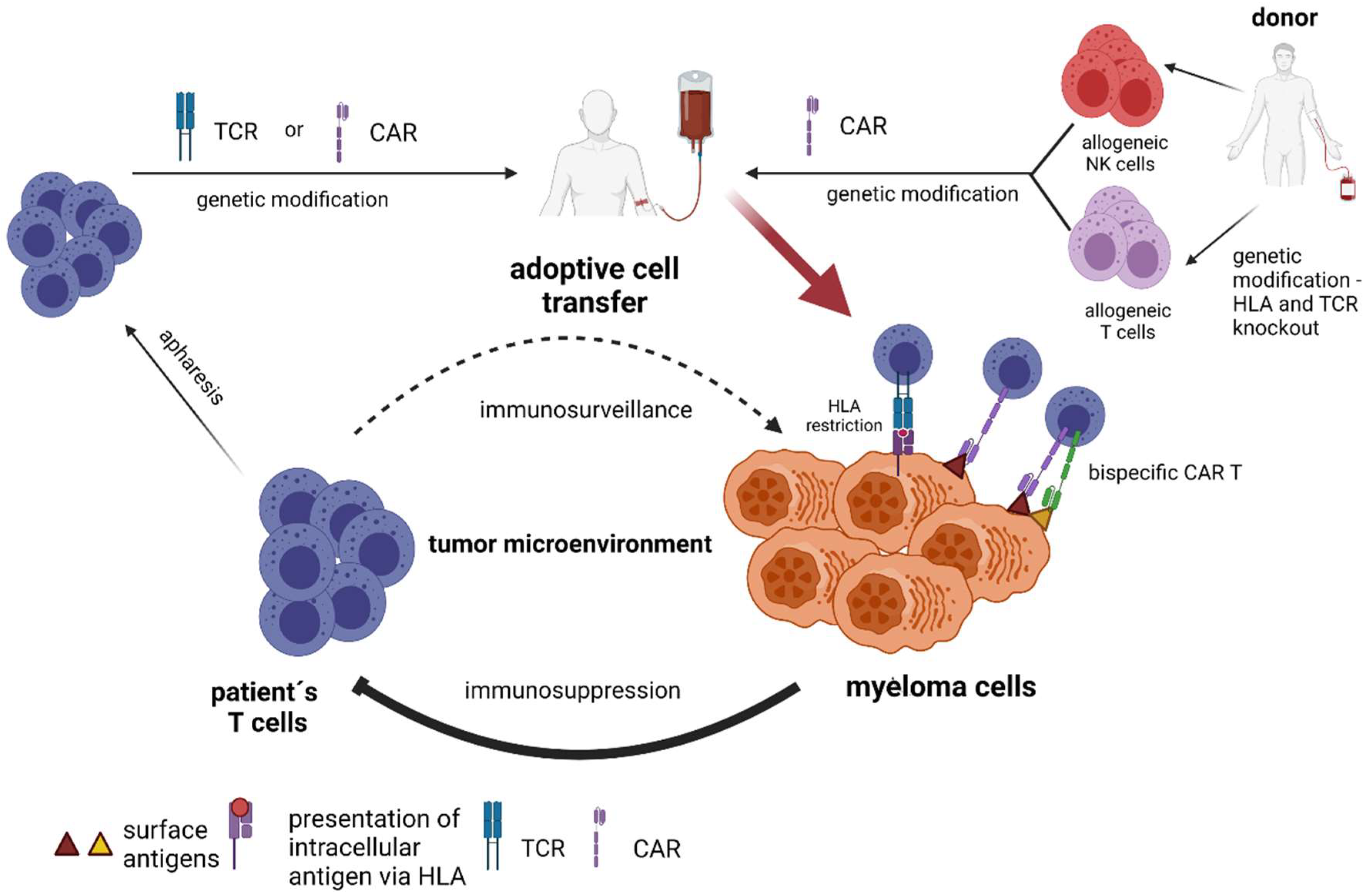
4. Adaptive Immune Transfer of Autologous and Allogeneic Lymphocytes with Genetic Manipulation (TCR–Engineered T cells, Chimeric Antigen Receptor T-Cells and Chimeric Antigen Receptor NK-Cells)
4.1. TCR–Engineered T Cells
4.2. CAR-Engineered T-Cells
4.3. Anti-BCMA CAR T-Cell Therapy
4.4. Resistance to Anti-BCMA CAR T-Cell Therapy
4.5. Allogeneic CAR T-Cells and CAR NK-Cells as ‘off the Shelf’ Adoptive Therapy
5. Strategies Mitigating Immune Resistance—Reviving of Existing Immune Responses and Stimulating Innate Immune System (Standard of Care Treatments, Immune Checkpoints Inhibitors, Bispecific Antibodies)
5.1. Standard of Care Treatments
5.2. Monoclonal Antibodies Targeting CD38
5.3. Immune Checkpoints Inhibitors
5.4. Bispecific Monoclonal Antibodies
6. Oncolytic Virotherapy
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Mel, S.; Lim, S.H.; Tung, M.L.; Chng, W.-J. Implications of Heterogeneity in Multiple Myeloma. BioMed Res. Int. 2014, 2014, 232546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landgren, O.; Kyle, R.A.; Rajkumar, S.V. From Myeloma Precursor Disease to Multiple Myeloma: New Diagnostic Concepts and Opportunities for Early Intervention. Clin. Cancer Res. 2011, 17, 1243–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhodapkar, M.V. MGUS to Myeloma: A Mysterious Gammopathy of Underexplored Significance. Blood 2016, 128, 2599–2606. [Google Scholar] [CrossRef]
- Mikulasova, A.; Wardell, C.P.; Murison, A.; Boyle, E.M.; Jackson, G.H.; Smetana, J.; Kufova, Z.; Pour, L.; Sandecka, V.; Almasi, M.; et al. The Spectrum of Somatic Mutations in Monoclonal Gammopathy of Undetermined Significance Indicates a Less Complex Genomic Landscape than That in Multiple Myeloma. Haematologica 2017, 102, 1617–1625. [Google Scholar] [CrossRef] [Green Version]
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The Genetic Architecture of Multiple Myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef]
- Kyle, R.A.; Larson, D.R.; Therneau, T.M.; Dispenzieri, A.; Kumar, S.; Cerhan, J.R.; Rajkumar, S.V. Long-Term Follow-up of Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 2018, 378, 241–249. [Google Scholar] [CrossRef]
- Dhodapkar, M.V. Harnessing Host Immune Responses to Preneoplasia: Promise and Challenges. Cancer Immunol. Immunother. 2005, 54, 409–413. [Google Scholar] [CrossRef]
- Das, R.; Strowig, T.; Verma, R.; Koduru, S.; Hafemann, A.; Hopf, S.; Kocoglu, M.H.; Borsotti, C.; Zhang, L.; Branagan, A.; et al. Microenvironment-Dependent Growth of Preneoplastic and Malignant Plasma Cells in Humanized Mice. Nat. Med. 2016, 22, 1351–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillerey, C.; Ferrari de Andrade, L.; Vuckovic, S.; Miles, K.; Ngiow, S.F.; Yong, M.C.R.; Teng, M.W.L.; Colonna, M.; Ritchie, D.S.; Chesi, M.; et al. Immunosurveillance and Therapy of Multiple Myeloma Are CD226 Dependent. J. Clin. Investig. 2015, 125, 2077–2089. [Google Scholar] [CrossRef] [Green Version]
- Romano, A.; Conticello, C.; Cavalli, M.; Vetro, C.; La Fauci, A.; Parrinello, N.L.; Di Raimondo, F. Immunological Dysregulation in Multiple Myeloma Microenvironment. BioMed Res. Int. 2014, 2014, 198539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz-Tejedor, A.; Lorenzo-Mohamed, M.; Puig, N.; García-Sanz, R.; Mateos, M.-V.; Garayoa, M.; Paíno, T. Immune System Alterations in Multiple Myeloma: Molecular Mechanisms and Therapeutic Strategies to Reverse Immunosuppression. Cancers 2021, 13, 1353. [Google Scholar] [CrossRef] [PubMed]
- Leblay, N.; Maity, R.; Hasan, F.; Neri, P. Deregulation of Adaptive T Cell Immunity in Multiple Myeloma: Insights Into Mechanisms and Therapeutic Opportunities. Front. Oncol. 2020, 10, 636. [Google Scholar] [CrossRef] [PubMed]
- Prabhala, R.H.; Pelluru, D.; Fulciniti, M.; Prabhala, H.K.; Nanjappa, P.; Song, W.; Pai, C.; Amin, S.; Tai, Y.-T.; Richardson, P.G.; et al. Elevated IL-17 Produced by Th17 Cells Promotes Myeloma Cell Growth and Inhibits Immune Function in Multiple Myeloma. Blood 2010, 115, 5385–5392. [Google Scholar] [CrossRef]
- Brown, R.; Murray, A.; Pope, B.; Sze, D.M.; Gibson, J.; Joy Ho, P.; Hart, D.; Joshua, D. Either Interleukin-12 or Interferon-γ Can Correct the Dendritic Cell Defect Induced by Transforming Growth Factor Β1 in Patients with Myeloma. Br. J. Haematol. 2004, 125, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Moschetta, M.; Kokubun, K.; Lukyanchykov, P.; Sula Karreci, E.; Manier, S.; Tsukamoto, S.; Takagi, S.; Shi, J.; Reagan, M.R.; et al. Characterization of the Role of Regulatory T Cells (Tregs) in Inducing Progression of Multiple Myeloma. Blood 2015, 126, 502. [Google Scholar] [CrossRef]
- Muthu Raja, K.R.; Rihova, L.; Zahradova, L.; Klincova, M.; Penka, M.; Hajek, R. Increased T Regulatory Cells Are Associated with Adverse Clinical Features and Predict Progression in Multiple Myeloma. PLoS ONE 2012, 7, e47077. [Google Scholar] [CrossRef]
- Perez, C.; Botta, C.; Zabaleta, A.; Puig, N.; Cedena, M.-T.; Goicoechea, I.; Alameda, D.; San José-Eneriz, E.; Merino, J.; Rodríguez-Otero, P.; et al. Immunogenomic Identification and Characterization of Granulocytic Myeloid-Derived Suppressor Cells in Multiple Myeloma. Blood 2020, 136, 199–209. [Google Scholar] [CrossRef]
- Andersen, M.N.; Andersen, N.F.; Lauridsen, K.L.; Etzerodt, A.; Sorensen, B.S.; Abildgaard, N.; Plesner, T.; Hokland, M.; Møller, H.J. STAT3 Is Over-Activated within CD163pos Bone Marrow Macrophages in Both Multiple Myeloma and the Benign Pre-Condition MGUS. Cancer Immunol. Immunother. 2021. [Google Scholar] [CrossRef]
- Asimakopoulos, F.; Kim, J.; Denu, R.A.; Hope, C.; Jensen, J.L.; Ollar, S.J.; Hebron, E.; Flanagan, C.; Callander, N.; Hematti, P. Macrophages in Multiple Myeloma: Emerging Concepts and Therapeutic Implications. Leuk Lymphoma 2013, 54, 2112–2121. [Google Scholar] [CrossRef] [Green Version]
- Jelinek, T.; Paiva, B.; Hajek, R. Update on PD-1/PD-L1 Inhibitors in Multiple Myeloma. Front. Immunol 2018, 9, 2431. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Muz, B.; Alhallak, K.; Markovic, M.; Gurley, S.; Wang, Z.; Guenthner, N.; Wasden, K.; Fiala, M.; King, J.; et al. Targeting CD47 as a Novel Immunotherapy for Multiple Myeloma. Cancers (Basel) 2020, 12, 305. [Google Scholar] [CrossRef] [Green Version]
- Guillerey, C.; Harjunpää, H.; Carrié, N.; Kassem, S.; Teo, T.; Miles, K.; Krumeich, S.; Weulersse, M.; Cuisinier, M.; Stannard, K.; et al. TIGIT Immune Checkpoint Blockade Restores CD8+ T-Cell Immunity against Multiple Myeloma. Blood 2018, 132, 1689–1694. [Google Scholar] [CrossRef] [Green Version]
- Häusler, S.F.M.; Montalbán del Barrio, I.; Strohschein, J.; Anoop Chandran, P.; Engel, J.B.; Hönig, A.; Ossadnik, M.; Horn, E.; Fischer, B.; Krockenberger, M.; et al. Ectonucleotidases CD39 and CD73 on OvCA Cells Are Potent Adenosine-Generating Enzymes Responsible for Adenosine Receptor 2A-Dependent Suppression of T Cell Function and NK Cell Cytotoxicity. Cancer Immunol. Immunother. 2011, 60, 1405. [Google Scholar] [CrossRef]
- Horenstein, A.L.; Quarona, V.; Toscani, D.; Costa, F.; Chillemi, A.; Pistoia, V.; Giuliani, N.; Malavasi, F. Adenosine Generated in the Bone Marrow Niche Through a CD38-Mediated Pathway Correlates With Progression of Human Myeloma. Mol. Med. 2016, 22, 694–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponzetta, A.; Benigni, G.; Antonangeli, F.; Sciumè, G.; Sanseviero, E.; Zingoni, A.; Ricciardi, M.R.; Petrucci, M.T.; Santoni, A.; Bernardini, G. Multiple Myeloma Impairs Bone Marrow Localization of Effector Natural Killer Cells by Altering the Chemokine Microenvironment. Cancer Res. 2015, 75, 4766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Sherbiny, Y.M.; Meade, J.L.; Holmes, T.D.; McGonagle, D.; Mackie, S.L.; Morgan, A.W.; Cook, G.; Feyler, S.; Richards, S.J.; Davies, F.E.; et al. The Requirement for DNAM-1, NKG2D, and NKp46 in the Natural Killer Cell-Mediated Killing of Myeloma Cells. Cancer Res. 2007, 67, 8444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbone, E.; Neri, P.; Mesuraca, M.; Fulciniti, M.T.; Otsuki, T.; Pende, D.; Groh, V.; Spies, T.; Pollio, G.; Cosman, D.; et al. HLA Class I, NKG2D, and Natural Cytotoxicity Receptors Regulate Multiple Myeloma Cell Recognition by Natural Killer Cells. Blood 2005, 105, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Pazina, T.; MacFarlane, A.W., 4th; Bernabei, L.; Dulaimi, E.; Kotcher, R.; Yam, C.; Bezman, N.A.; Robbins, M.D.; Ross, E.A.; Campbell, K.S.; et al. Alterations of NK Cell Phenotype in the Disease Course of Multiple Myeloma. Cancers 2021, 13, 226. [Google Scholar] [CrossRef]
- Pittari, G.; Vago, L.; Festuccia, M.; Bonini, C.; Mudawi, D.; Giaccone, L.; Bruno, B. Restoring Natural Killer Cell Immunity against Multiple Myeloma in the Era of New Drugs. Front. Immunol. 2017, 8, 1444. [Google Scholar] [CrossRef] [Green Version]
- Leone, P.; Solimando, A.G.; Malerba, E.; Fasano, R.; Buonavoglia, A.; Pappagallo, F.; De Re, V.; Argentiero, A.; Silvestris, N.; Vacca, A.; et al. Actors on the Scene: Immune Cells in the Myeloma Niche. Front. Oncol. 2020, 10, 599098. [Google Scholar] [CrossRef] [PubMed]
- Greil, C.; Engelhardt, M.; Ihorst, G.; Schoeller, K.; Bertz, H.; Marks, R.; Zeiser, R.; Duyster, J.; Einsele, H.; Finke, J.; et al. Allogeneic Transplantation of Multiple Myeloma Patients May Allow Long-Term Survival in Carefully Selected Patients with Acceptable Toxicity and Preserved Quality of Life. Haematologica 2019, 104, 370–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Copelan, E.A. Hematopoietic Stem-Cell Transplantation. N. Engl. J. Med. 2006, 354, 1813–1826. [Google Scholar] [CrossRef] [PubMed]
- Spierings, E.; Kim, Y.-H.; Hendriks, M.; Borst, E.; Sergeant, R.; Canossi, A.; Oudshoorn, M.; Loiseau, P.; Dolstra, H.; Markiewicz, M.; et al. Multicenter Analyses Demonstrate Significant Clinical Effects of Minor Histocompatibility Antigens on GvHD and GvL after HLA-Matched Related and Unrelated Hematopoietic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2013, 19, 1244–1253. [Google Scholar] [CrossRef] [PubMed]
- Goulmy, E. Minor Histocompatibility Antigens: From Transplantation Problems to Therapy of Cancer. Hum. Immunol. 2006, 67, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Oostvogels, R.; Lokhorst, H.M.; Mutis, T. Minor Histocompatibility Ags: Identification Strategies, Clinical Results and Translational Perspectives. Bone Marrow Transplant. 2016, 51, 163–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warren, E.H.; Fujii, N.; Akatsuka, Y.; Chaney, C.N.; Mito, J.K.; Loeb, K.R.; Gooley, T.A.; Brown, M.L.; Koo, K.K.W.; Rosinski, K.V.; et al. Therapy of Relapsed Leukemia after Allogeneic Hematopoietic Cell Transplantation with T Cells Specific for Minor Histocompatibility Antigens. Blood 2010, 115, 3869–3878. [Google Scholar] [CrossRef] [Green Version]
- Franssen, L.E.; Roeven, M.W.H.; Hobo, W.; Doorn, R.; Oostvogels, R.; Falkenburg, J.H.F.; van de Donk, N.W.; Kester, M.G.D.; Fredrix, H.; Westinga, K.; et al. A Phase I/II Minor Histocompatibility Antigen-Loaded Dendritic Cell Vaccination Trial to Safely Improve the Efficacy of Donor Lymphocyte Infusions in Myeloma. Bone Marrow Transplant. 2017, 52, 1378–1383. [Google Scholar] [CrossRef]
- Oostvogels, R.; Kneppers, E.; Minnema, M.C.; Doorn, R.C.; Franssen, L.E.; Aarts, T.; Emmelot, M.E.; Spierings, E.; Slaper-Cortenbach, I.; Westinga, K.; et al. Efficacy of Host-Dendritic Cell Vaccinations with or without Minor Histocompatibility Antigen Loading, Combined with Donor Lymphocyte Infusion in Multiple Myeloma Patients. Bone Marrow Transplant. 2017, 52, 228–237. [Google Scholar] [CrossRef]
- Garfall, A.L.; Stadtmauer, E.A. Cellular and Vaccine Immunotherapy for Multiple Myeloma. Hematol. Am. Soc. Hematol Educ. Program. 2016, 2016, 521–527. [Google Scholar] [CrossRef] [Green Version]
- Rosenblatt, J.; Avivi, I.; Vasir, B.; Uhl, L.; Munshi, N.C.; Katz, T.; Dey, B.R.; Somaiya, P.; Mills, H.; Campigotto, F.; et al. Vaccination with Dendritic Cell/Tumor Fusions Following Autologous Stem Cell Transplant Induces Immunologic and Clinical Responses in Multiple Myeloma Patients. Clin. Cancer Res. 2013, 19, 3640. [Google Scholar] [CrossRef] [Green Version]
- Hobo, W.; Strobbe, L.; Maas, F.; Fredrix, H.; Greupink-Draaisma, A.; Esendam, B.; de Witte, T.; Preijers, F.; Levenga, H.; van Rees, B.; et al. Immunogenicity of Dendritic Cells Pulsed with MAGE3, Survivin and B-Cell Maturation Antigen MRNA for Vaccination of Multiple Myeloma Patients. Cancer Immunol. Immunother. 2013, 62, 1381–1392. [Google Scholar] [CrossRef]
- Palucka, K.; Banchereau, J. Dendritic-Cell-Based Therapeutic Cancer Vaccines. Immunity 2013, 39, 38–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dranoff, G. GM-CSF-Based Cancer Vaccines. Immunol. Rev. 2002, 188, 147–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrello, I.M.; Noonan, K.; Huff, C.A.; Ferguson, A.; Sidorski, A.; Rudraraju, L.; Cimbro, R.; Marchionni, L. Allogeneic Myeloma GVAX with Lenalidomide Enhances Progression Free Survival through the Generation of Tumor Specific Immunity in Patients in Near Complete Remission. Blood 2015, 126, 4238. [Google Scholar] [CrossRef]
- Weinstock, M.; Rosenblatt, J.; Avigan, D. Dendritic Cell Therapies for Hematologic Malignancies. Mol. Ther. - Methods Clin. Dev. 2017, 5, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Wong, K.K.; Hassan, R.; Yaacob, N.S. Hypomethylating Agents and Immunotherapy: Therapeutic Synergism in Acute Myeloid Leukemia and Myelodysplastic Syndromes. Front. Oncol. 2021, 11, 72. [Google Scholar] [CrossRef] [PubMed]
- Nooka, A.K.; Wang, M. (Luhua); Yee, A.J.; Kaufman, J.L.; Bae, J.; Peterkin, D.; Richardson, P.G.; Raje, N.S. Assessment of Safety and Immunogenicity of PVX-410 Vaccine With or Without Lenalidomide in Patients With Smoldering Multiple Myeloma: A Nonrandomized Clinical Trial. JAMA Oncol. 2018, 4, e183267. [Google Scholar] [CrossRef]
- Blass, E.; Ott, P.A. Advances in the Development of Personalized Neoantigen-Based Therapeutic Cancer Vaccines. Nat. Rev. Clin. Oncol. 2021, 18, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An Immunogenic Personal Neoantigen Vaccine for Patients with Melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen Vaccine Generates Intratumoral T Cell Responses in Phase Ib Glioblastoma Trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef]
- Perumal, D.; Imai, N.; Laganà, A.; Finnigan, J.; Melnekoff, D.; Leshchenko, V.V.; Solovyov, A.; Madduri, D.; Chari, A.; Cho, H.J.; et al. Mutation-Derived Neoantigen-Specific T-Cell Responses in Multiple Myeloma. Clin. Cancer Res. 2020, 26, 450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, S.A. Cell Transfer Immunotherapy for Metastatic Solid Cancer—What Clinicians Need to Know. Nat. Rev. Clin. Oncol 2011, 8, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable Complete Responses in Heavily Pretreated Patients with Metastatic Melanoma Using T-Cell Transfer Immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhodapkar, M.V.; Krasovsky, J.; Olson, K. T Cells from the Tumor Microenvironment of Patients with Progressive Myeloma Can Generate Strong, Tumor-Specific Cytolytic Responses to Autologous, Tumor-Loaded Dendritic Cells. Proc. Natl. Acad. Sci. USA 2002, 99, 13009–13013. [Google Scholar] [CrossRef] [Green Version]
- Noonan, K.; Matsui, W.; Serafini, P.; Carbley, R.; Tan, G.; Khalili, J.; Bonyhadi, M.; Levitsky, H.; Whartenby, K.; Borrello, I. Activated Marrow-Infiltrating Lymphocytes Effectively Target Plasma Cells and Their Clonogenic Precursors. Cancer Res. 2005, 65, 2026. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.-C.; Yao, X.; Crystal, J.S.; Li, Y.F.; El-Gamil, M.; Gross, C.; Davis, L.; Dudley, M.E.; Yang, J.C.; Samuels, Y.; et al. Efficient Identification of Mutated Cancer Antigens Recognized by T Cells Associated with Durable Tumor Regressions. Clin. Cancer Res. 2014, 20, 3401. [Google Scholar] [CrossRef] [Green Version]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.-C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer Immunotherapy Based on Mutation-Specific CD4+ T Cells in a Patient with Epithelial Cancer. Science 2014, 344, 641. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Lu, Y.-C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational Heterogeneity in Cancer and the Search for New Cancer-Associated Genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Noonan, K.A.; Huff, C.A.; Davis, J.; Lemas, M.V.; Fiorino, S.; Bitzan, J.; Ferguson, A.; Emerling, A.; Luznik, L.; Matsui, W.; et al. Adoptive Transfer of Activated Marrow-Infiltrating Lymphocytes Induces Measurable Antitumor Immunity in the Bone Marrow in Multiple Myeloma. Sci Transl Med. 2015, 7, 288ra78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheper, W.; Kelderman, S.; Fanchi, L.F.; Linnemann, C.; Bendle, G.; de Rooij, M.A.J.; Hirt, C.; Mezzadra, R.; Slagter, M.; Dijkstra, K.; et al. Low and Variable Tumor Reactivity of the Intratumoral TCR Repertoire in Human Cancers. Nat. Med. 2019, 25, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Ding, Z.-Y. The Ways of Isolating Neoantigen-Specific T Cells. Front. Oncol 2020, 10, 1347. [Google Scholar] [CrossRef] [PubMed]
- Lokhorst, H.M.; Schattenberg, A.; Cornelissen, J.J.; van Oers, M.H.J.; Fibbe, W.; Russell, I.; Donk, N.W.C.J.v.d.; Verdonck, L.F. Donor Lymphocyte Infusions for Relapsed Multiple Myeloma After Allogeneic Stem-Cell Transplantation: Predictive Factors for Response and Long-Term Outcome. JCO 2000, 18, 3031–3037. [Google Scholar] [CrossRef]
- Beitinjaneh, A.M.; Saliba, R.; Bashir, Q.; Shah, N.; Parmar, S.; Hosing, C.; Popat, U.; Anderlini, P.; Dinh, Y.; Qureshi, S.; et al. Durable Responses after Donor Lymphocyte Infusion for Patients with Residual Multiple Myeloma Following Non-Myeloablative Allogeneic Stem Cell Transplant. null 2012, 53, 1525–1529. [Google Scholar] [CrossRef]
- Simonetta, F.; Alvarez, M.; Negrin, R.S. Natural Killer Cells in Graft-versus-Host-Disease after Allogeneic Hematopoietic Cell Transplantation. Front. Immunol 2017, 8, 465. [Google Scholar] [CrossRef]
- Caligiuri, M.A. Human Natural Killer Cells. Blood 2008, 112, 461–469. [Google Scholar] [CrossRef]
- Jurisic, V.; Srdic, T.; Konjevic, G.; Markovic, O.; Colovic, M. Clinical Stage-Depending Decrease of NK Cell Activity in Multiple Myeloma Patients. Med. Oncol. 2007, 24, 312–317. [Google Scholar] [CrossRef]
- Fauriat, C.; Mallet, F.; Olive, D.; Costello, R.T. Impaired Activating Receptor Expression Pattern in Natural Killer Cells from Patients with Multiple Myeloma. Leukemia 2006, 20, 732–733. [Google Scholar] [CrossRef]
- Costello, R.T.; Boehrer, A.; Sanchez, C.; Mercier, D.; Baier, C.; Le Treut, T.; Sébahoun, G. Differential Expression of Natural Killer Cell Activating Receptors in Blood versus Bone Marrow in Patients with Monoclonal Gammopathy. Immunology 2013, 139, 338–341. [Google Scholar] [CrossRef]
- Shi, J.; Tricot, G.; Szmania, S.; Rosen, N.; Garg, T.K.; Malaviarachchi, P.A.; Moreno, A.; Dupont, B.; Hsu, K.C.; Baxter-Lowe, L.A.; et al. Infusion of Haplo-Identical Killer Immunoglobulin-like Receptor Ligand Mismatched NK Cells for Relapsed Myeloma in the Setting of Autologous Stem Cell Transplantation. Br. J. Haematol 2008, 143, 641–653. [Google Scholar] [CrossRef] [Green Version]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of Donor Natural Killer Cell Alloreactivity in Mismatched Hematopoietic Transplants. Science 2002, 295, 2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Single, R.M.; Martin, M.P.; Gao, X.; Meyer, D.; Yeager, M.; Kidd, J.R.; Kidd, K.K.; Carrington, M. Global Diversity and Evidence for Coevolution of KIR and HLA. Nat. Genet. 2007, 39, 1114–1119. [Google Scholar] [CrossRef]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; Defor, T.E.; Burns, L.J.; et al. Successful Adoptive Transfer and in Vivo Expansion of Human Haploidentical NK Cells in Patients with Cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tricot, G.; Vesole, D.H.; Jagannath, S.; Hilton, J.; Munshi, N.; Barlogie, B. Graft-Versus-Myeloma Effect: Proof of Principle. Blood 1996, 87, 1196–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, N.; Li, L.; McCarty, J.; Kaur, I.; Yvon, E.; Shaim, H.; Muftuoglu, M.; Liu, E.; Orlowski, R.Z.; Cooper, L.; et al. Phase I Study of Cord Blood-Derived Natural Killer Cells Combined with Autologous Stem Cell Transplantation in Multiple Myeloma. Br. J. Haematol 2017, 177, 457–466. [Google Scholar] [CrossRef]
- Shah, N.; Mehta, R.; Li, L.; Mccarty, J.; Kaur, I.; Orlowski, R.Z.; Cooper, L.; Lee, D.A.; Cao, K.; Parmar, S.; et al. Phase II Study of Ex Vivo Expanded Cord Blood Natural Killer Cells for Multiple Myeloma. JCO 2018, 36, 8006. [Google Scholar] [CrossRef]
- Hagner, P.R.; Chiu, H.; Ortiz, M.; Apollonio, B.; Wang, M.; Couto, S.; Waldman, M.F.; Flynt, E.; Ramsay, A.G.; Trotter, M.; et al. Activity of Lenalidomide in Mantle Cell Lymphoma Can Be Explained by NK Cell-Mediated Cytotoxicity. Br. J. Haematol. 2017, 179, 399–409. [Google Scholar] [CrossRef] [Green Version]
- Hsu, A.K.; Quach, H.; Tai, T.; Prince, H.M.; Harrison, S.J.; Trapani, J.A.; Smyth, M.J.; Neeson, P.; Ritchie, D.S. The Immunostimulatory Effect of Lenalidomide on NK-Cell Function Is Profoundly Inhibited by Concurrent Dexamethasone Therapy. Blood 2011, 117, 1605–1613. [Google Scholar] [CrossRef] [Green Version]
- Lokhorst, H.M.; Plesner, T.; Laubach, J.P.; Nahi, H.; Gimsing, P.; Hansson, M.; Minnema, M.C.; Lassen, U.; Krejcik, J.; Palumbo, A.; et al. Targeting CD38 with Daratumumab Monotherapy in Multiple Myeloma. N. Engl. J. Med. 2015, 373, 1207–1219. [Google Scholar] [CrossRef]
- Casneuf, T.; Xu, X.S.; Adams, H.C., 3rd; Axel, A.E.; Chiu, C.; Khan, I.; Ahmadi, T.; Yan, X.; Lonial, S.; Plesner, T.; et al. Effects of Daratumumab on Natural Killer Cells and Impact on Clinical Outcomes in Relapsed or Refractory Multiple Myeloma. Blood Adv. 2017, 1, 2105–2114. [Google Scholar] [CrossRef]
- Naeimi Kararoudi, M.; Nagai, Y.; Elmas, E.; de Souza Fernandes Pereira, M.; Ali, S.A.; Imus, P.H.; Wethington, D.; Borrello, I.M.; Lee, D.A.; Ghiaur, G. CD38 Deletion of Human Primary NK Cells Eliminates Daratumumab-Induced Fratricide and Boosts Their Effector Activity. Blood 2020, 136, 2416–2427. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P. Adoptive Cell Transfer as Personalized Immunotherapy for Human Cancer. Science 2015, 348, 62. [Google Scholar] [CrossRef] [Green Version]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- Albinger, N.; Hartmann, J.; Ullrich, E. Current Status and Perspective of CAR-T and CAR-NK Cell Therapy Trials in Germany. Gene Ther. 2021. [Google Scholar] [CrossRef]
- Bishop, D.C.; Clancy, L.E.; Simms, R.; Burgess, J.; Mathew, G.; Moezzi, L.; Street, J.A.; Sutrave, G.; Atkins, E.; McGuire, H.M.; et al. Development of CAR T-Cell Lymphoma in Two of Ten Patients Effectively Treated with PiggyBac Modified CD19 CAR T-Cells. Blood 2021. [Google Scholar] [CrossRef]
- Johnson, L.A.; Morgan, R.A.; Dudley, M.E.; Cassard, L.; Yang, J.C.; Hughes, M.S.; Kammula, U.S.; Royal, R.E.; Sherry, R.M.; Wunderlich, J.R.; et al. Gene Therapy with Human and Mouse T-Cell Receptors Mediates Cancer Regression and Targets Normal Tissues Expressing Cognate Antigen. Blood 2009, 114, 535–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Donk, N.W.C.J.; Richardson, P.G.; Malavasi, F. CD38 Antibodies in Multiple Myeloma: Back to the Future. Blood 2018, 131, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Feng, K.; Tong, C.; Jia, H.; Liu, Y.; Wang, Y.; Ti, D.; Yang, Q.; Wu, Z.; Han, W. Efficiency and Side Effects of Anti-CD38 CAR T Cells in an Adult Patient with Relapsed B-ALL after Failure of Bi-Specific CD19/CD22 CAR T Cell Treatment. Cell. Mol. Immunol. 2020, 17, 430–432. [Google Scholar] [CrossRef]
- Rapoport, A.P.; Stadtmauer, E.A.; Binder-Scholl, G.K.; Goloubeva, O.; Vogl, D.T.; Lacey, S.F.; Badros, A.Z.; Garfall, A.; Weiss, B.; Finklestein, J.; et al. NY-ESO-1–Specific TCR–Engineered T Cells Mediate Sustained Antigen-Specific Antitumor Effects in Myeloma. Nat. Med. 2015, 21, 914–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-Engineered T Cells in Patients with Refractory Cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-Cell Therapy. Cancer Discov. 2018, 8, 1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, B.P.; Raman, V.S.; Erickson, L.D.; Cook, W.J.; Weaver, L.K.; Ahonen, C.; Lin, L.-L.; Mantchev, G.T.; Bram, R.J.; Noelle, R.J. BCMA Is Essential for the Survival of Long-Lived Bone Marrow Plasma Cells. J. Exp. Med. 2004, 199, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Drent, E.; Themeli, M.; Poels, R.; de Jong-Korlaar, R.; Yuan, H.; de Bruijn, J.; Martens, A.C.M.; Zweegman, S.; van de Donk, N.W.C.J.; Groen, R.W.J.; et al. A Rational Strategy for Reducing On-Target Off-Tumor Effects of CD38-Chimeric Antigen Receptors by Affinity Optimization. Mol. Ther. 2017, 25, 1946–1958. [Google Scholar] [CrossRef]
- Van de Donk, N.W.C.J.; Usmani, S.Z.; Yong, K. CAR T-Cell Therapy for Multiple Myeloma: State of the Art and Prospects. Lancet Haematol. 2021, 8, e446–e461. [Google Scholar] [CrossRef]
- Teoh, P.J.; Chng, W.J. CAR T-Cell Therapy in Multiple Myeloma: More Room for Improvement. Blood Cancer J. 2021, 11, 84. [Google Scholar] [CrossRef]
- First CAR-T Therapy to Target BCMA Gets FDA Nod. Nat. Biotechnol. 2021, 39, 531. [CrossRef]
- Munshi, N.C.; Anderson, L.D.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene Autoleucel, a B-Cell Maturation Antigen-Directed Chimeric Antigen Receptor T-Cell Therapy in Patients with Relapsed or Refractory Multiple Myeloma (CARTITUDE-1): A Phase 1b/2 Open-Label Study. The Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef]
- Da Vià, M.C.; Dietrich, O.; Truger, M.; Arampatzi, P.; Duell, J.; Heidemeier, A.; Zhou, X.; Danhof, S.; Kraus, S.; Chatterjee, M.; et al. Homozygous BCMA Gene Deletion in Response to Anti-BCMA CAR T Cells in a Patient with Multiple Myeloma. Nat. Med. 2021, 27, 616–619. [Google Scholar] [CrossRef]
- Samur, M.K.; Fulciniti, M.; Aktas-Samur, A.; Bazarbachi, A.H.; Tai, Y.-T.; Campbell, T.B.; Petrocca, F.; Hege, K.; Kaiser, S.; Anderson, K.; et al. Biallelic Loss of BCMA Triggers Resistance to Anti-BCMA CAR T Cell Therapy in Multiple Myeloma. Blood 2020, 136, 14. [Google Scholar] [CrossRef]
- Shah, N.N.; Fry, T.J. Mechanisms of Resistance to CAR T Cell Therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef]
- McLellan, A.D.; Ali Hosseini Rad, S.M. Chimeric Antigen Receptor T Cell Persistence and Memory Cell Formation. Immunol. Cell Biol. 2019, 97, 664–674. [Google Scholar] [CrossRef]
- Zah, E. Systematically Optimized BCMA/CS1 Bispecific CAR-T Cells Robustly Control Heterogeneous Multiple Myeloma. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Fernández de Larrea, C.; Staehr, M.; Lopez, A.V.; Ng, K.Y.; Chen, Y.; Godfrey, W.D.; Purdon, T.J.; Ponomarev, V.; Wendel, H.-G.; Brentjens, R.J.; et al. Defining an Optimal Dual-Targeted CAR T-Cell Therapy Approach Simultaneously Targeting BCMA and GPRC5D to Prevent BCMA Escape–Driven Relapse in Multiple Myeloma. Blood Cancer Discov 2020, 1, 146. [Google Scholar] [CrossRef]
- Schmidts, A.; Ormhøj, M.; Choi, B.D.; Taylor, A.O.; Bouffard, A.A.; Scarfò, I.; Larson, R.C.; Frigault, M.J.; Gallagher, K.; Castano, A.P.; et al. Rational Design of a Trimeric APRIL-Based CAR-Binding Domain Enables Efficient Targeting of Multiple Myeloma. Blood Adv. 2019, 3, 3248–3260. [Google Scholar] [CrossRef]
- Ayuk, F.; Fehse, B.; Janson, D.; Berger, C.; Riecken, K.; Kröger, N. Excellent Proliferation and Persistence of Allogeneic Donor-Derived 41-BB Based CAR-T Cells despite Immunosuppression with Cyclosporine A. Haematologica 2020, 105, 322–324. [Google Scholar] [CrossRef]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-Shelf’ Allogeneic CAR T Cells: Development and Challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Zhang, C.; Oberoi, P.; Oelsner, S.; Waldmann, A.; Lindner, A.; Tonn, T.; Wels, W.S. Chimeric Antigen Receptor-Engineered NK-92 Cells: An Off-the-Shelf Cellular Therapeutic for Targeted Elimination of Cancer Cells and Induction of Protective Antitumor Immunity. Front. Immunol. 2017, 8, 533. [Google Scholar] [CrossRef]
- Wrona, E.; Borowiec, M.; Potemski, P. CAR-NK Cells in the Treatment of Solid Tumors. Int. J. Mol. Sci. 2021, 22, 5899. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.X.; Braggio, E.; Shi, C.-X.; Kortuem, K.M.; Bruins, L.A.; Schmidt, J.E.; Chang, X.-B.; Langlais, P.; Luo, M.; Jedlowski, P.; et al. Identification of Cereblon-Binding Proteins and Relationship with Response and Survival after IMiDs in Multiple Myeloma. Blood 2014, 124, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.-K.; Bradner, J.E.; Kaelin, W.G., Jr. The Myeloma Drug Lenalidomide Promotes the Cereblon-Dependent Destruction of Ikaros Proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalia, V.; Sarkar, S. Regulation of Effector and Memory CD8 T Cell Differentiation by IL-2—A Balancing Act. Front. Immunol. 2018, 9, 2987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nijhof, I.S.; Franssen, L.E.; Levin, M.-D.; Bos, G.M.J.; Broijl, A.; Klein, S.K.; Koene, H.R.; Bloem, A.C.; Beeker, A.; Faber, L.M.; et al. Phase 1/2 Study of Lenalidomide Combined with Low-Dose Cyclophosphamide and Prednisone in Lenalidomide-Refractory Multiple Myeloma. Blood 2016, 128, 2297–2306. [Google Scholar] [CrossRef] [Green Version]
- Pasquier, E.; Kavallaris, M.; André, N. Metronomic Chemotherapy: New Rationale for New Directions. Nat. Rev. Clin. Oncol. 2010, 7, 455–465. [Google Scholar] [CrossRef]
- Cerullo, V.; Diaconu, I.; Kangasniemi, L.; Rajecki, M.; Escutenaire, S.; Koski, A.; Romano, V.; Rouvinen, N.; Tuuminen, T.; Laasonen, L.; et al. Immunological Effects of Low-Dose Cyclophosphamide in Cancer Patients Treated With Oncolytic Adenovirus. Mol. Ther. 2011, 19, 1737–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Y.; Domschke, C.; Stoiber, N.; Schott, S.; Heil, J.; Rom, J.; Blumenstein, M.; Thum, J.; Sohn, C.; Schneeweiss, A.; et al. Metronomic Cyclophosphamide Treatment in Metastasized Breast Cancer Patients: Immunological Effects and Clinical Outcome. Cancer Immunol. Immunother. 2012, 61, 353–362. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Larmonier, N.; Schmitt, E.; Parcellier, A.; Cathelin, D.; Garrido, C.; Chauffert, B.; Solary, E.; Bonnotte, B.; Martin, F. CD4+CD25+ Regulatory T Cells Suppress Tumor Immunity but Are Sensitive to Cyclophosphamide Which Allows Immunotherapy of Established Tumors to Be Curative. Eur. J. Immunol. 2004, 34, 336–344. [Google Scholar] [CrossRef]
- Zhao, J.; Cao, Y.; Lei, Z.; Yang, Z.; Zhang, B.; Huang, B. Selective Depletion of CD4+CD25+Foxp3+ Regulatory T Cells by Low-Dose Cyclophosphamide Is Explained by Reduced Intracellular ATP Levels. Cancer Res. 2010, 70, 4850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Del Valle, A.; Anel, A.; Naval, J.; Marzo, I. Immunogenic Cell Death and Immunotherapy of Multiple Myeloma. Front. Cell Dev. Biol. 2019, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Maes, K.; Breckpot, K. Commentary: Immunogenic Cell Death and Immunotherapy of Multiple Myeloma. Front. Cell Dev. Biol 2019, 7, 149. [Google Scholar] [CrossRef]
- Gulla, A.; Morelli, E.; Samur, M.K.; Botta, C.; Hideshima, T.; Bianchi, G.; Fulciniti, M.; Malvestiti, S.; Prabhala, R.H.; Talluri, S.; et al. Bortezomib Induces Anti–Multiple Myeloma Immune Response Mediated by CGAS/STING Pathway Activation. Blood Cancer Discov. 2021. [Google Scholar] [CrossRef]
- Saba, R.; Saleem, N.; Peace, D. Long-Term Survival Consequent on the Abscopal Effect in a Patient with Multiple Myeloma. BMJ Case Rep. 2016, 2016, bcr2016215237. [Google Scholar] [CrossRef] [Green Version]
- Craig, D.J.; Nanavaty, N.S.; Devanaboyina, M.; Stanbery, L.; Hamouda, D.; Edelman, G.; Dworkin, L.; Nemunaitis, J.J. The Abscopal Effect of Radiation Therapy. Future Oncol. 2021, 17, 1683–1694. [Google Scholar] [CrossRef] [PubMed]
- Nijhof, I.S.; Casneuf, T.; van Velzen, J.; van Kessel, B.; Axel, A.E.; Syed, K.; Groen, R.W.J.; van Duin, M.; Sonneveld, P.; Minnema, M.C.; et al. CD38 Expression and Complement Inhibitors Affect Response and Resistance to Daratumumab Therapy in Myeloma. Blood 2016, 128, 959–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krejcik, J.; Frerichs, K.A.; Nijhof, I.S.; van Kessel, B.; van Velzen, J.F.; Bloem, A.C.; Broekmans, M.E.C.; Zweegman, S.; van Meerloo, J.; Musters, R.J.P.; et al. Monocytes and Granulocytes Reduce CD38 Expression Levels on Myeloma Cells in Patients Treated with Daratumumab. Clin. Cancer Res. 2017, 23, 7498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krejcik, J.; van de Donk, N.W.C.J. Trogocytosis Represents a Novel Mechanism of Action of Daratumumab in Multiple Myeloma. Oncotarget 2018, 9, 33621–33622. [Google Scholar] [CrossRef]
- Plesner, T.; van de Donk, N.; Richardson, P.G. Controversy in the Use of CD38 Antibody for Treatment of Myeloma: Is High CD38 Expression Good or Bad? Cells 2020, 9, 378. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Diao, L.; Yang, Y.; Yi, X.; Rodriguez, B.L.; Li, Y.; Villalobos, P.A.; Cascone, T.; Liu, X.; Tan, L.; et al. CD38-Mediated Immunosuppression as a Mechanism of Tumor Cell Escape from PD-1/PD-L1 Blockade. Cancer Discov. 2018, 8, 1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quarona, V.; Ferri, V.; Chillemi, A.; Bolzoni, M.; Mancini, C.; Zaccarello, G.; Roato, I.; Morandi, F.; Marimpietri, D.; Faccani, G.; et al. Unraveling the Contribution of Ectoenzymes to Myeloma Life and Survival in the Bone Marrow Niche. Ann. N. Y. Acad. Sci. 2015, 1335, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.C.J.; Weiss, B.M.; et al. Daratumumab Depletes CD38+ Immune Regulatory Cells, Promotes T-Cell Expansion, and Skews T-Cell Repertoire in Multiple Myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimopoulos, M.; Bringhen, S.; Anttila, P.; Capra, M.; Cavo, M.; Cole, C.; Gasparetto, C.; Hungria, V.; Jenner, M.; Vorobyev, V.; et al. Isatuximab as Monotherapy and Combined with Dexamethasone in Patients with Relapsed/Refractory Multiple Myeloma. Blood 2021, 137, 1154–1165. [Google Scholar] [CrossRef]
- Atanackovic, D.; Yousef, S.; Shorter, C.; Tantravahi, S.K.; Steinbach, M.; Iglesias, F.; Sborov, D.; Radhakrishnan, S.V.; Chiron, M.; Miles, R.; et al. In Vivo Vaccination Effect in Multiple Myeloma Patients Treated with the Monoclonal Antibody Isatuximab. Leukemia 2020, 34, 317–321. [Google Scholar] [CrossRef]
- Costa, F.; Marchica, V.; Storti, P.; Malavasi, F.; Giuliani, N. PD-L1/PD-1 Axis in Multiple Myeloma Microenvironment and a Possible Link with CD38-Mediated Immune-Suppression. Cancers 2021, 13, 164. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.M., Jr.; Bakan, C.E.; Mishra, A.; Hofmeister, C.C.; Efebera, Y.; Becknell, B.; Baiocchi, R.A.; Zhang, J.; Yu, J.; Smith, M.K.; et al. The PD-1/PD-L1 Axis Modulates the Natural Killer Cell versus Multiple Myeloma Effect: A Therapeutic Target for CT-011, a Novel Monoclonal Anti–PD-1 Antibody. Blood 2010, 116, 2286–2294. [Google Scholar] [CrossRef]
- Görgün, G.; Samur, M.K.; Cowens, K.B.; Paula, S.; Bianchi, G.; Anderson, J.E.; White, R.E.; Singh, A.; Ohguchi, H.; Suzuki, R.; et al. Lenalidomide Enhances Immune Checkpoint Blockade-Induced Immune Response in Multiple Myeloma. Clin. Cancer Res. 2015, 21, 4607–4618. [Google Scholar] [CrossRef] [Green Version]
- Rosenblatt, J.; Avigan, D. Targeting the PD-1/PD-L1 Axis in Multiple Myeloma: A Dream or a Reality? Blood 2017, 129, 275–279. [Google Scholar] [CrossRef] [Green Version]
- Verkleij, C.P.M.; Jhatakia, A.; Broekmans, M.E.C.; Frerichs, K.A.; Zweegman, S.; Mutis, T.; Bezman, N.A.; van de Donk, N.W.C.J. Preclinical Rationale for Targeting the PD-1/PD-L1 Axis in Combination with a CD38 Antibody in Multiple Myeloma and Other CD38-Positive Malignancies. Cancers (Basel) 2020, 12, 3713. [Google Scholar] [CrossRef]
- Cohen, Y.C.; Oriol, A.; Wu, K.L.; Lavi, N.; Vlummens, P.; Jackson, C.; Garvin, W.; Carson, R.; Crist, W.; Fu, J.; et al. Daratumumab With Cetrelimab, an Anti–PD-1 Monoclonal Antibody, in Relapsed/Refractory Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2021, 21, 46–54.e4. [Google Scholar] [CrossRef]
- Mateos, M.-V.; Orlowski, R.Z.; Siegel, D.S.D.; Reece, D.E.; Moreau, P.; Ocio, E.M.; Shah, J.J.; Rodríguez-Otero, P.; Munshi, N.C.; Avigan, D.; et al. Pembrolizumab in Combination with Lenalidomide and Low-Dose Dexamethasone for Relapsed/Refractory Multiple Myeloma (RRMM): Final Efficacy and Safety Analysis. JCO 2016, 34, 8010. [Google Scholar] [CrossRef]
- Zanwar, S.; Nandakumar, B.; Kumar, S. Immune-Based Therapies in the Management of Multiple Myeloma. Blood Cancer J. 2020, 10, 84. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.-V.; Blacklock, H.; Schjesvold, F.; Oriol, A.; Simpson, D.; George, A.; Goldschmidt, H.; Larocca, A.; Chanan-Khan, A.; Sherbenou, D.; et al. Pembrolizumab plus Pomalidomide and Dexamethasone for Patients with Relapsed or Refractory Multiple Myeloma (KEYNOTE-183): A Randomised, Open-Label, Phase 3 Trial. Lancet Haematol. 2019, 6, e459–e469. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Schjesvold, F.; Oriol, A.; Karlin, L.; Cavo, M.; Rifkin, R.M.; Yimer, H.A.; LeBlanc, R.; Takezako, N.; McCroskey, R.D.; et al. Pembrolizumab plus Lenalidomide and Dexamethasone for Patients with Treatment-Naive Multiple Myeloma (KEYNOTE-185): A Randomised, Open-Label, Phase 3 Trial. Lancet Haematol. 2019, 6, e448–e458. [Google Scholar] [CrossRef]
- Genmab Announces That Janssen Will Stop Studies of Daratumumab in Combination with Anti-PD-(L)1 - Genmab A/S. Available online: https://ir.genmab.com/news-releases/news-release-details/genmab-announces-janssen-will-stop-studies-daratumumab (accessed on 23 June 2021).
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.-T.; et al. Determinants of Response and Resistance to CD19 Chimeric Antigen Receptor (CAR) T Cell Therapy of Chronic Lymphocytic Leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Zolov, S.N.; Rietberg, S.P.; Bonifant, C.L. Programmed Cell Death Protein 1 Activation Preferentially Inhibits CD28.CAR–T Cells. Cytotherapy 2018, 20, 1259–1266. [Google Scholar] [CrossRef]
- Galon, J.; Rossi, J.; Turcan, S.; Danan, C.; Locke, F.L.; Neelapu, S.S.; Miklos, D.B.; Bartlett, N.L.; Jacobson, C.A.; Braunschweig, I.; et al. Characterization of Anti-CD19 Chimeric Antigen Receptor (CAR) T Cell-Mediated Tumor Microenvironment Immune Gene Profile in a Multicenter Trial (ZUMA-1) with Axicabtagene Ciloleucel (Axi-Cel, KTE-C19). JCO 2017, 35, 3025. [Google Scholar] [CrossRef]
- Bernabei, L.; Garfall, A.L.; Melenhorst, J.J.; Lacey, S.F.; Stadtmauer, E.A.; Vogl, D.T.; Gonzalez, V.; Plesa, G.; Young, R.M.; Waxman, A.; et al. PD-1 Inhibitor Combinations As Salvage Therapy for Relapsed/Refractory Multiple Myeloma (MM) Patients Progressing after Bcma-Directed CAR T Cells. Blood 2018, 132, 1973. [Google Scholar] [CrossRef]
- Wudhikarn, K.; Mailankody, S.; Smith, E.L. Future of CAR T Cells in Multiple Myeloma. Hematol. Am. Soc. Hematol Educ Program. 2020, 2020, 272–279. [Google Scholar] [CrossRef]
- Suarez, E.R.; Chang, D.K.; Sun, J.; Sui, J.; Freeman, G.J.; Signoretti, S.; Zhu, Q.; Marasco, W.A. Chimeric Antigen Receptor T Cells Secreting Anti-PD-L1 Antibodies More Effectively Regress Renal Cell Carcinoma in a Humanized Mouse Model. Oncotarget 2016, 7, 34341–34355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted Delivery of a PD-1-Blocking ScFv by CAR-T Cells Enhances Anti-Tumor Efficacy in Vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef]
- Zhang, C.; Peng, Y.; Hublitz, P.; Zhang, H.; Dong, T. Genetic Abrogation of Immune Checkpoints in Antigen-Specific Cytotoxic T-Lymphocyte as a Potential Alternative to Blockade Immunotherapy. Sci. Rep. 2018, 8, 5549. [Google Scholar] [CrossRef]
- Advani, R.; Flinn, I.; Popplewell, L.; Forero, A.; Bartlett, N.L.; Ghosh, N.; Kline, J.; Roschewski, M.; LaCasce, A.; Collins, G.P.; et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 379, 1711–1721. [Google Scholar] [CrossRef]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Myklebust, J.H.; Varghese, B.; Gill, S.; Jan, M.; Cha, A.C.; Chan, C.K.; Tan, B.T.; et al. Anti-CD47 Antibody Synergizes with Rituximab to Promote Phagocytosis and Eradicate Non-Hodgkin Lymphoma. Cell 2010, 142, 699–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, D.; Volkmer, J.-P.; Willingham, S.B.; Contreras-Trujillo, H.; Fathman, J.W.; Fernhoff, N.B.; Seita, J.; Inlay, M.A.; Weiskopf, K.; Miyanishi, M.; et al. Anti-CD47 Antibody–Mediated Phagocytosis of Cancer by Macrophages Primes an Effective Antitumor T-Cell Response. Proc. Natl. Acad. Sci. USA 2013, 110, 11103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storti, P.; Vescovini, R.; Costa, F.; Marchica, V.; Toscani, D.; Dalla Palma, B.; Craviotto, L.; Malavasi, F.; Giuliani, N. CD14+CD16+ Monocytes Are Involved in Daratumumab-Mediated Myeloma Cells Killing and in Anti-CD47 Therapeutic Strategy. Br. J. Haematol. 2020, 190, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Khair, D.O.; Bax, H.J.; Mele, S.; Crescioli, S.; Pellizzari, G.; Khiabany, A.; Nakamura, M.; Harris, R.J.; French, E.; Hoffmann, R.M.; et al. Combining Immune Checkpoint Inhibitors: Established and Emerging Targets and Strategies to Improve Outcomes in Melanoma. Front. Immunol. 2019, 10, 453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Einsele, H.; Danhof, S. Bispecific Antibodies: A New Era of Treatment for Multiple Myeloma. J. Clin. Med. 2020, 9, 2166. [Google Scholar] [CrossRef]
- Brinkmann, U.; Kontermann, R.E. The Making of Bispecific Antibodies. MAbs 2017, 9, 182–212. [Google Scholar] [CrossRef]
- Topp, M.S.; Duell, J.; Zugmaier, G.; Attal, M.; Moreau, P.; Langer, C.; Krönke, J.; Facon, T.; Salnikov, A.V.; Lesley, R.; et al. Anti–B-Cell Maturation Antigen BiTE Molecule AMG 420 Induces Responses in Multiple Myeloma. JCO 2020, 38, 775–783. [Google Scholar] [CrossRef]
- Verkleij, C.P.M.; Frerichs, K.A.; Broekmans, M.; Absalah, S.; Maas-Bosman, P.W.C.; Kruyswijk, S.; Nijhof, I.S.; Mutis, T.; Zweegman, S.; van de Donk, N.W.C.J. T-Cell Redirecting Bispecific Antibodies Targeting BCMA for the Treatment of Multiple Myeloma. Oncotarget 2020, 11, 4076–4081. [Google Scholar] [CrossRef]
- Frerichs, K.A.; Broekmans, M.E.C.; Marin Soto, J.A.; van Kessel, B.; Heymans, M.W.; Holthof, L.C.; Verkleij, C.P.M.; Boominathan, R.; Vaidya, B.; Sendecki, J.; et al. Preclinical Activity of JNJ-7957, a Novel BCMA×CD3 Bispecific Antibody for the Treatment of Multiple Myeloma, Is Potentiated by Daratumumab. Clin. Cancer Res. 2020, 26, 2203. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.-F.; Lin, L.; Xing, L.; Liu, J.; Yu, T.; Wen, K.; Hsieh, P.; Munshi, N.; Anderson, K.; Tai, Y.-T. Anti-BCMA BiTE® AMG 701 Potently Induces Specific T Cell Lysis of Human Multiple Myeloma (MM) Cells and Immunomodulation in the Bone Marrow Microenvironment. Blood 2018, 132, 592. [Google Scholar] [CrossRef]
- Li, J.; Stagg, N.J.; Johnston, J.; Harris, M.J.; Menzies, S.A.; DiCara, D.; Clark, V.; Hristopoulos, M.; Cook, R.; Slaga, D.; et al. Membrane-Proximal Epitope Facilitates Efficient T Cell Synapse Formation by Anti-FcRH5/CD3 and Is a Requirement for Myeloma Cell Killing. Cancer Cell 2017, 31, 383–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, M.; Damon, L.E.; Jeyakumar, D.; Costello, C.L.; Tzachanis, D.; Schiller, G.J.; Reiner, J.; Wieduwilt, M.J. Blinatumomab in Combination with Pembrolizumab Is Safe for Adults with Relapsed or Refractory B-Lineage Acute Lymphoblastic Leukemia: University of California Hematologic Malignancies Consortium Study 1504. Blood 2019, 134, 3880. [Google Scholar] [CrossRef]
- Correnti, C.E.; Laszlo, G.S.; de van der Schueren, W.J.; Godwin, C.D.; Bandaranayake, A.; Busch, M.A.; Gudgeon, C.J.; Bates, O.M.; Olson, J.M.; Mehlin, C.; et al. Simultaneous Multiple Interaction T-Cell Engaging (SMITE) Bispecific Antibodies Overcome Bispecific T-Cell Engager (BiTE) Resistance via CD28 Co-Stimulation. Leukemia 2018, 32, 1239–1243. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Seung, E.; Xu, L.; Rao, E.; Lord, D.M.; Wei, R.R.; Cortez-Retamozo, V.; Ospina, B.; Posternak, V.; Ulinski, G.; et al. Trispecific Antibodies Enhance the Therapeutic Efficacy of Tumor-Directed T Cells through T Cell Receptor Co-Stimulation. Nat. Cancer 2020, 1, 86–98. [Google Scholar] [CrossRef]
- El-Murr, N.; Henry, C.; Francesconi, E.; Attenot, F.; Virone-Oddos, A.; Vidard, L.; Wu, L.; Yang, Z.-Y.; Chiron, M. Abstract 5641: CD28 Expression on Multiple Myeloma Cells Enhances the Cytotoxic Activity of CD38/CD28xCD3 Trispecific T Cell Engager. Cancer Res. 2020, 80, 5641. [Google Scholar] [CrossRef]
- Banaszek, A.; Bumm, T.G.P.; Nowotny, B.; Geis, M.; Jacob, K.; Wölfl, M.; Trebing, J.; Kucka, K.; Kouhestani, D.; Gogishvili, T.; et al. On-Target Restoration of a Split T Cell-Engaging Antibody for Precision Immunotherapy. Nat. Commun. 2019, 10, 5387. [Google Scholar] [CrossRef]
- Weidanz, J. Targeting Cancer with Bispecific Antibodies. Science 2021, eabg5568. [Google Scholar] [CrossRef]
- Hsiue, E.H.-C.; Wright, K.M.; Douglass, J.; Hwang, M.S.; Mog, B.J.; Pearlman, A.H.; Paul, S.; DiNapoli, S.R.; Konig, M.F.; Wang, Q.; et al. Targeting a Neoantigen Derived from a Common TP53 Mutation. Science 2021, eabc8697. [Google Scholar] [CrossRef]
- Gantke, T.; Reusch, U.; Kellner, C.; Ellwanger, K.; Fucek, I.; Weichel, M.; Kerber, A.; Peipp, M.; Treder, M. AFM26 Is a Novel, Highly Potent BCMA/CD16A-Directed Bispecific Antibody for High Affinity NK-Cell Engagement in Multiple Myeloma. JCO 2017, 35, 8045. [Google Scholar] [CrossRef]
- Gantke, T.; Weichel, M.; Herbrecht, C.; Reusch, U.; Ellwanger, K.; Fucek, I.; Eser, M.; Müller, T.; Griep, R.; Molkenthin, V.; et al. Trispecific Antibodies for CD16A-Directed NK Cell Engagement and Dual-Targeting of Tumor Cells. Protein Eng. Des. Sel. 2017, 30, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Bluming, A.Z.; Ziegler, J.L. REGRESSION OF BURKITT’S LYMPHOMA IN ASSOCIATION WITH MEASLES INFECTION. Lancet 1971, 298, 105–106. [Google Scholar] [CrossRef]
- Taqi, A.M.; Abdurrahman, M.B.; Yakubu, A.M.; Fleming, A.F. Regression of Hodgkin’s Disease After Measles. Lancet 1981, 317, 1112. [Google Scholar] [CrossRef]
- Coffey, M.C.; Strong, J.E.; Forsyth, P.A.; Lee, P.W.K. Reovirus Therapy of Tumors with Activated Ras Pathway. Science 1998, 282, 1332. [Google Scholar] [CrossRef]
- Ong, H.T.; Timm, M.M.; Greipp, P.R.; Witzig, T.E.; Dispenzieri, A.; Russell, S.J.; Peng, K.-W. Oncolytic Measles Virus Targets High CD46 Expression on Multiple Myeloma Cells. Exp. Hematol. 2006, 34, 713–720. [Google Scholar] [CrossRef]
- Meyers, D.E.; Thakur, S.; Thirukkumaran, C.M.; Morris, D.G. Oncolytic Virotherapy as an Immunotherapeutic Strategy for Multiple Myeloma. Blood Cancer J. 2017, 7, 640. [Google Scholar] [CrossRef] [Green Version]
- Calton, C.M.; Kelly, K.R.; Anwer, F.; Carew, J.S.; Nawrocki, S.T. Oncolytic Viruses for Multiple Myeloma Therapy. Cancers 2018, 10, 198. [Google Scholar] [CrossRef] [Green Version]
- Packiriswamy, N.; Upreti, D.; Zhou, Y.; Khan, R.; Miller, A.; Diaz, R.M.; Rooney, C.M.; Dispenzieri, A.; Peng, K.-W.; Russell, S.J. Oncolytic Measles Virus Therapy Enhances Tumor Antigen-Specific T-Cell Responses in Patients with Multiple Myeloma. Leukemia 2020, 34, 3310–3322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dingli, D.; Peng, K.-W.; Harvey, M.E.; Greipp, P.R.; O’Connor, M.K.; Cattaneo, R.; Morris, J.C.; Russell, S.J. Image-Guided Radiovirotherapy for Multiple Myeloma Using a Recombinant Measles Virus Expressing the Thyroidal Sodium Iodide Symporter. Blood 2004, 103, 1641–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, S.K.; Classic, K.L.; Hadac, E.M.; Dingli, D.; Bender, C.E.; Kemp, B.J.; Russell, S.J. Quantitative Molecular Imaging of Viral Therapy for Pancreatic Cancer Using an Engineered Measles Virus Expressing the Sodium-Iodide Symporter Reporter Gene. AJR Am. J. Roentgenol 2009, 192, 279–287. [Google Scholar] [CrossRef] [Green Version]
- Dispenzieri, A.; Tong, C.; LaPlant, B.; Lacy, M.Q.; Laumann, K.; Dingli, D.; Zhou, Y.; Federspiel, M.J.; Gertz, M.A.; Hayman, S.; et al. Phase I Trial of Systemic Administration of Edmonston Strain of Measles Virus Genetically Engineered to Express the Sodium Iodide Symporter in Patients with Recurrent or Refractory Multiple Myeloma. Leukemia 2017, 31, 2791–2798. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.; Peng, K.W.; Geyer, S.M.; Ginos, B.F.; Dueck, A.C.; Packiriswamy, N.; Zhang, L.; Brunton, B.; Balakrishnan, B.; Witzig, T.E.; et al. Clinical Activity of Systemic VSV-IFNβ-NIS Oncolytic Virotherapy in Patients with Relapsed Refractory T-Cell Lymphoma. JCO 2021, 39, 2500. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krejcik, J.; Barnkob, M.B.; Nyvold, C.G.; Larsen, T.S.; Barington, T.; Abildgaard, N. Harnessing the Immune System to Fight Multiple Myeloma. Cancers 2021, 13, 4546. https://doi.org/10.3390/cancers13184546
Krejcik J, Barnkob MB, Nyvold CG, Larsen TS, Barington T, Abildgaard N. Harnessing the Immune System to Fight Multiple Myeloma. Cancers. 2021; 13(18):4546. https://doi.org/10.3390/cancers13184546
Chicago/Turabian StyleKrejcik, Jakub, Mike Bogetofte Barnkob, Charlotte Guldborg Nyvold, Thomas Stauffer Larsen, Torben Barington, and Niels Abildgaard. 2021. "Harnessing the Immune System to Fight Multiple Myeloma" Cancers 13, no. 18: 4546. https://doi.org/10.3390/cancers13184546
APA StyleKrejcik, J., Barnkob, M. B., Nyvold, C. G., Larsen, T. S., Barington, T., & Abildgaard, N. (2021). Harnessing the Immune System to Fight Multiple Myeloma. Cancers, 13(18), 4546. https://doi.org/10.3390/cancers13184546