
Darwinian Approaches for Cancer Treatment: Benefits of Mathematical Modeling



Abstract
Simple Summary
Abstract
1. Introduction
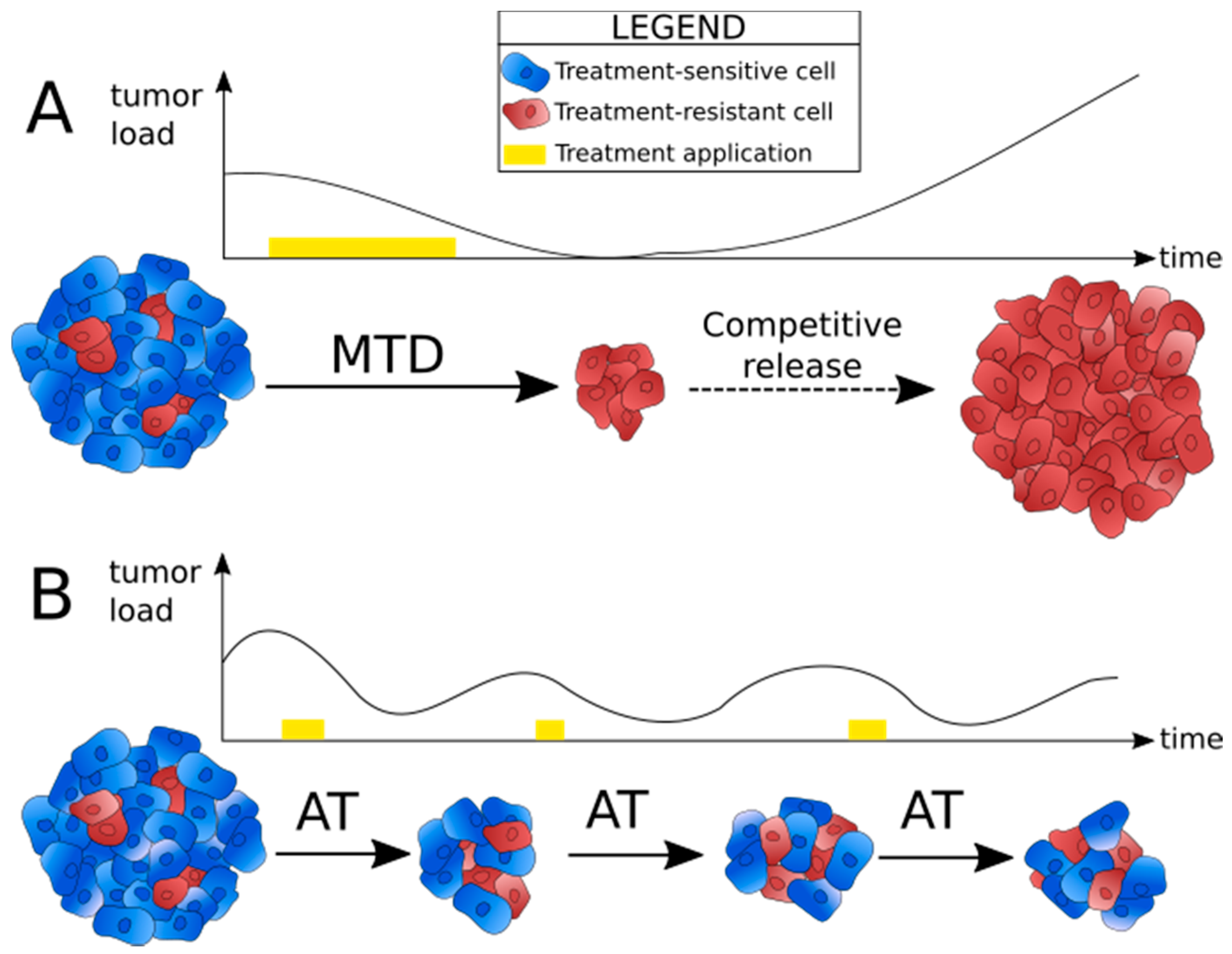
2. Adaptive Therapy: Principle and First Models
2.1. Identifying the Reasons for Failure of Conventional Treatment Strategies
2.2. Principle of Adaptive Therapy
3. Main Types of Models
3.1. Lotka–Volterra Models
3.1.1. Identifying the Patients Who Could Benefit from Adaptive Therapy
3.1.2. Designing Efficient Adaptive Therapy Schedules
3.1.3. Improving the Models
3.1.4. Advantages and Limitations of Lotka–Volterra Models
3.2. Agent-Based Models
3.2.1. Applications of ABMs to Model Adaptive Therapy
3.2.2. Advantages and Limitations of ABMs
4. From Models to Clinical Applications and Vice Versa
5. Conclusions and Perspectives
Funding
Conflicts of Interest
Appendix A. Lotka–Volterra Models

{kind=link}
{kind=link}
| T+ | TP | T− | |
|---|---|---|---|
| T+ | α11 | α12 | α13 |
| TP | α21 | α22 | α23 |
| T− | α31 | α32 | α33 |
Appendix B. Agent-Based Models
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Brown, J.S. Integrating Evolutionary Dynamics into Cancer Therapy. Nat. Rev. Clin. Oncol. 2020, 17, 675–686. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour Heterogeneity and Resistance to Cancer Therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Zahreddine, H.; Borden, K.L.B. Mechanisms and Insights into Drug Resistance in Cancer. Front Pharm. 2013, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Jiang, J.; Lu, Y.; Nice, E.C.; Huang, C.; Zhang, J.; He, W. Emerging Role of Tumor Cell Plasticity in Modifying Therapeutic Response. Signal. Transduct. Target. Ther. 2020, 5, 228. [Google Scholar] [CrossRef]
- Marine, J.-C.; Dawson, S.-J.; Dawson, M.A. Non-Genetic Mechanisms of Therapeutic Resistance in Cancer. Nat. Rev. Cancer 2020, 20, 743–756. [Google Scholar] [CrossRef]
- Rehman, S.K.; Haynes, J.; Collignon, E.; Brown, K.R.; Wang, Y.; Nixon, A.M.L.; Bruce, J.P.; Wintersinger, J.A.; Singh Mer, A.; Lo, E.B.L.; et al. Colorectal Cancer Cells Enter a Diapause-like DTP State to Survive Chemotherapy. Cell 2021, 184, 226–242.e21. [Google Scholar] [CrossRef]
- Cunningham, J.J. A Call for Integrated Metastatic Management. Nat. Ecol. Evol. 2019, 3, 996–998. [Google Scholar] [CrossRef]
- Stanková, K.; Brown, J.S.; Dalton, W.S.; Gatenby, R.A. Optimizing Cancer Treatment Using Game Theory: A Review. JAMA Oncol. 2019, 5, 96. [Google Scholar] [CrossRef]
- Aktipis, C.A.; Nesse, R.M. Evolutionary Foundations for Cancer Biology. Evol. Appl. 2013, 6, 144–159. [Google Scholar] [CrossRef]
- Basanta, D.; Anderson, A.R.A. Exploiting Ecological Principles to Better Understand Cancer Progression and Treatment. Interface Focus 2013, 3, 20130020. [Google Scholar] [CrossRef]
- Basanta, D.; Anderson, A.R.A. Homeostasis Back and Forth: An Ecoevolutionary Perspective of Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a028332. [Google Scholar] [CrossRef] [PubMed]
- Thomas, F.; Fisher, D.; Fort, P.; Marie, J.-P.; Daoust, S.; Roche, B.; Grunau, C.; Cosseau, C.; Mitta, G.; Baghdiguian, S.; et al. Applying Ecological and Evolutionary Theory to Cancer: A Long and Winding Road. Evol. Appl. 2013, 6, 1–10. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Silva, A.S.; Gillies, R.J.; Frieden, B.R. Adaptive Therapy. Cancer Res. 2009, 69, 4894–4903. [Google Scholar] [CrossRef]
- Strobl, M.A.R.; Gallaher, J.; West, J.; Robertson-Tessi, M.; Maini, P.K.; Anderson, A.R.A. Spatial Structure Impacts Adaptive Therapy by Shaping Intra-Tumoral Competition. bioRxiv. 2021. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Frieden, B.R. Inducing Catastrophe in Malignant Growth. Math. Med. Biol. 2008, 25, 267–283. [Google Scholar] [CrossRef]
- Cunningham, J.J.; Brown, J.S.; Gatenby, R.A.; Staňková, K. Optimal Control to Develop Therapeutic Strategies for Metastatic Castrate Resistant Prostate Cancer. J. Theor. Biol. 2018, 459, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Gallaher, J.A.; Enriquez-Navas, P.M.; Luddy, K.A.; Gatenby, R.A.; Anderson, A.R.A. Spatial Heterogeneity and Evolutionary Dynamics Modulate Time to Recurrence in Continuous and Adaptive Cancer Therapies. Cancer Res. 2018, 78, 2127–2139. [Google Scholar] [CrossRef]
- Zhang, J.; Cunningham, J.J.; Brown, J.S.; Gatenby, R.A. Integrating Evolutionary Dynamics into Treatment of Metastatic Castrate-Resistant Prostate Cancer. Nat. Commun. 2017, 8, 1816. [Google Scholar] [CrossRef] [PubMed]
- Gluzman, M.; Scott, J.G.; Vladimirsky, A. Optimizing Adaptive Cancer Therapy: Dynamic Programming and Evolutionary Game Theory. Proc. R. Soc. B Biol. Sci. 2020, 287, 20192454. [Google Scholar] [CrossRef]
- You, L.; Brown, J.S.; Thuijsman, F.; Cunningham, J.J.; Gatenby, R.A.; Zhang, J.; Staňková, K. Spatial vs. Non-Spatial Eco-Evolutionary Dynamics in a Tumor Growth Model. J. Theor. Biol. 2017, 435, 78–97. [Google Scholar] [CrossRef]
- Altrock, P.M.; Liu, L.L.; Michor, F. The Mathematics of Cancer: Integrating Quantitative Models. Nat. Rev. Cancer 2015, 15, 730–745. [Google Scholar] [CrossRef] [PubMed]
- Swierniak, A.; Kimmel, M.; Smieja, J. Mathematical Modeling as a Tool for Planning Anticancer Therapy. Eur. J. Pharmacol. 2009, 625, 108–121. [Google Scholar] [CrossRef]
- Enderling, H.; Chaplain, M. Mathematical Modeling of Tumor Growth and Treatment. CPD 2014, 20, 4934–4940. [Google Scholar] [CrossRef]
- Foo, J.; Michor, F. Evolution of Acquired Resistance to Anti-Cancer Therapy. J. Theor. Biol. 2014, 355, 10–20. [Google Scholar] [CrossRef]
- Ledzewicz, U.; Schättler, H. Drug Resistance in Cancer Chemotherapy as an Optimal Control Problem. Discret. Contin. Dyn. Syst. B 2006, 6, 129. [Google Scholar] [CrossRef]
- Pasquier, E.; Kavallaris, M.; André, N. Metronomic Chemotherapy: New Rationale for New Directions. Nat. Rev. Clin. Oncol. 2010, 7, 455–465. [Google Scholar] [CrossRef]
- Benzekry, S.; Hahnfeldt, P. Maximum Tolerated Dose versus Metronomic Scheduling in the Treatment of Metastatic Cancers. J. Theor. Biol. 2013, 335, 235–244. [Google Scholar] [CrossRef]
- André, N.; Barbolosi, D.; Billy, F.; Chapuisat, G.; Hubert, F.; Grenier, E.; Rovini, A. Mathematical Model of Cancer Growth Controled by Metronomic Chemotherapies. ESAIM Proc. 2013, 41, 77–94. [Google Scholar] [CrossRef]
- Algazi, A.P.; Othus, M.; Daud, A.I.; Lo, R.S.; Mehnert, J.M.; Truong, T.-G.; Conry, R.; Kendra, K.; Doolittle, G.C.; Clark, J.I.; et al. Continuous versus Intermittent BRAF and MEK Inhibition in Patients with BRAF-Mutated Melanoma: A Randomized Phase 2 Trial. Nat. Med. 2020, 26, 1564–1568. [Google Scholar] [CrossRef]
- de Moura, C.R.; Vercellino, L.; Jouenne, F.; Baroudjian, B.; Sadoux, A.; Louveau, B.; Delyon, J.; Serror, K.; Goldwirt, L.; Merlet, P.; et al. Intermittent Versus Continuous Dosing of MAPK Inhibitors in the Treatment of BRAF-Mutated Melanoma. Transl. Oncol. 2020, 13, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, J.J.; Gatenby, R.A.; Brown, J.S. Evolutionary Dynamics in Cancer Therapy. Mol. Pharm. 2011, 8, 2094. [Google Scholar] [CrossRef] [PubMed]
- Viossat, Y.; Noble, R. The Logic of Containing Tumors. bioRxiv 2020. [Google Scholar] [CrossRef]
- Cunningham, J.; Thuijsman, F.; Peeters, R.; Viossat, Y.; Brown, J.; Gatenby, R.; Staňková, K. Optimal Control to Reach Eco-Evolutionary Stability in Metastatic Castrate-Resistant Prostate Cancer. PLoS ONE 2020, 15, e0243386. [Google Scholar] [CrossRef]
- Kim, E.; Brown, J.S.; Eroglu, Z.; Anderson, A.R.A. Adaptive Therapy for Metastatic Melanoma: Predictions from Patient Calibrated Mathematical Models. Cancers 2021, 13, 823. [Google Scholar] [CrossRef]
- Ma, Y.; Newton, P.K. On the Design of Treatment Schedules That Avoid Chemotherapeutic Resistance. bioRxiv 2018, 325381. [Google Scholar] [CrossRef]
- Ma, Y.; Newton, P.K. Role of Synergy and Antagonism in Designing Multidrug Adaptive Chemotherapy Schedules. Phys. Rev. E 2021, 103, 032408. [Google Scholar] [CrossRef]
- Silva, A.S.; Khin, Z.; Shain, K.H.; Baz, R.; Hazlehurst, L.A.; Durand, A.; Gatenby, R. Personalizing Myeloma Chemotherapy Using Evolutionary Computational Models and Ex Vivo Assays. Blood 2012, 120, 2960. [Google Scholar] [CrossRef]
- Silva, A.S.; Kam, Y.; Khin, Z.P.; Minton, S.E.; Gillies, R.J.; Gatenby, R.A. Evolutionary Approaches to Prolong Progression-Free Survival in Breast Cancer. Cancer Res. 2012, 72, 6362–6370. [Google Scholar] [CrossRef]
- Silva, A.S.; Durand, A.; Ribeiro, M.C.; Alsina, M.; Shain, K.; Baz, R. An Evolutionary Approach for Personalized Therapy in Multiple Myeloma. Appl. Math. 2016, 7, 159–169. [Google Scholar] [CrossRef][Green Version]
- Smalley, I.; Kim, E.; Li, J.; Spence, P.; Wyatt, C.J.; Eroglu, Z.; Sondak, V.K.; Messina, J.L.; Babacan, N.A.; Maria-Engler, S.S.; et al. Leveraging Transcriptional Dynamics to Improve BRAF Inhibitor Responses in Melanoma. EBioMedicine 2019, 48, 178–190. [Google Scholar] [CrossRef]
- Strobl, M.A.R.; West, J.; Viossat, Y.; Damaghi, M.; Robertson-Tessi, M.; Brown, J.S.; Gatenby, R.A.; Maini, P.K.; Anderson, A.R.A. Turnover Modulates the Need for a Cost of Resistance in Adaptive Therapy. Cancer Res. 2021, 81, 1135–1147. [Google Scholar] [CrossRef]
- Viossat, Y.; Noble, R. A Theoretical Analysis of Tumour Containment. Nat. Ecol. Evol. 2021, 5, 826–835. [Google Scholar] [CrossRef]
- West, J.; Ma, Y.; Newton, P.K. Capitalizing on Competition: An Evolutionary Model of Competitive Release in Metastatic Castration Resistant Prostate Cancer Treatment. J. Theor. Biol. 2018, 455, 249–260. [Google Scholar] [CrossRef]
- West, J.; You, L.; Zhang, J.; Gatenby, R.A.; Brown, J.; Newton, P.K.; Anderson, A.R.A. Towards Multi-Drug Adaptive Therapy. bioRxiv 2019, 476507. [Google Scholar] [CrossRef]
- West, J.B.; Dinh, M.N.; Brown, J.S.; Zhang, J.; Anderson, A.R.; Gatenby, R.A. Multidrug Cancer Therapy in Metastatic Castrate-Resistant Prostate Cancer: An Evolution-Based Strategy. Clin. Cancer Res. 2019, 25, 4413–4421. [Google Scholar] [CrossRef] [PubMed]
- Bacevic, K.; Noble, R.; Soffar, A.; Wael Ammar, O.; Boszonyik, B.; Prieto, S.; Vincent, C.; Hochberg, M.E.; Krasinska, L.; Fisher, D. Spatial Competition Constrains Resistance to Targeted Cancer Therapy. Nat. Commun. 2017, 8, 1995. [Google Scholar] [CrossRef]
- Hansen, E.; Read, A.F. Cancer Therapy: Attempt Cure or Manage Drug Resistance? Evol. Appl. 2020, 13, 1660–1672. [Google Scholar] [CrossRef]
- Hansen, E.; Woods, R.J.; Read, A.F. How to Use a Chemotherapeutic Agent When Resistance to It Threatens the Patient. PLoS Biol. 2017, 15, e2001110. [Google Scholar] [CrossRef]
- Hansen, E.; Read, A.F. Modifying Adaptive Therapy to Enhance Competitive Suppression. Cancers 2020, 12, 3556. [Google Scholar] [CrossRef] [PubMed]
- Enriquez-Navas, P.M.; Kam, Y.; Das, T.; Hassan, S.; Silva, A.; Foroutan, P.; Ruiz, E.; Martinez, G.; Minton, S.; Gillies, R.J.; et al. Exploiting Evolutionary Principles to Prolong Tumor Control in Preclinical Models of Breast Cancer. Sci. Transl. Med. 2016, 8, 327ra24. [Google Scholar] [CrossRef] [PubMed]
- Mistry, H.B. Evolutionary Based Adaptive Dosing Algorithms: Beware the Cost of Cumulative Risk. bioRxiv 2020. [Google Scholar] [CrossRef]
- Farrokhian, N.; Maltas, J.; Ellsworth, P.; Durmaz, A.; Dinh, M.; Hitomi, M.; Kaznatcheev, A.; Marusyk, A.; Scott, J.G. Dose Dependent Evolutionary Game Dynamics Modulate Competitive Release in Cancer Therapy. bioRxiv 2020. [Google Scholar] [CrossRef]
- Persi, E.; Wolf, Y.I.; Horn, D.; Ruppin, E.; Demichelis, F.; Gatenby, R.A.; Gillies, R.J.; Koonin, E.V. Mutation–Selection Balance and Compensatory Mechanisms in Tumour Evolution. Nat. Rev. Genet. 2020, 22, 251–262. [Google Scholar] [CrossRef]
- Chisholm, R.H.; Lorenzi, T.; Clairambault, J. Cell Population Heterogeneity and Evolution towards Drug Resistance in Cancer: Biological and Mathematical Assessment, Theoretical Treatment Optimisation. Biochim. Biophys. Acta-Gen. Subj. 2016, 1860, 2627–2645. [Google Scholar] [CrossRef]
- Muros, F.J.; Maestre, J.M.; You, L.; Stankova, K. Model Predictive Control for Optimal Treatment in a Spatial Cancer Game. In Proceedings of the 2017 IEEE 56th Annual Conference on Decision and Control (CDC), Melbourne, VIC, Australia, 11–14 December 2017; pp. 5539–5544. [Google Scholar]
- Wang, Z.; Butner, J.D.; Kerketta, R.; Cristini, V.; Deisboeck, T.S. Simulating Cancer Growth with Multiscale Agent-Based Modeling. Semin. Cancer Biol. 2015, 30, 70–78. [Google Scholar] [CrossRef]
- Osborne, J.M.; Fletcher, A.G.; Pitt-Francis, J.M.; Maini, P.K.; Gavaghan, D.J. Comparing Individual-Based Approaches to Modelling the Self-Organization of Multicellular Tissues. PLoS Comput. Biol. 2017, 13, e1005387. [Google Scholar] [CrossRef]
- Anderson, A.R.A.; Quaranta, V. Integrative Mathematical Oncology. Nat. Rev. Cancer 2008, 8, 227–234. [Google Scholar] [CrossRef]
- Driessens, G.; Beck, B.; Caauwe, A.; Simons, B.D.; Blanpain, C. Defining the Mode of Tumour Growth by Clonal Analysis. Nature 2012, 488, 527–530. [Google Scholar] [CrossRef]
- Parker, T.M.; Gupta, K.; Palma, A.M.; Yekelchyk, M.; Fisher, P.B.; Grossman, S.R.; Won, K.J.; Madan, E.; Moreno, E.; Gogna, R. Cell Competition in Intratumoral and Tumor Microenvironment Interactions. EMBO J. 2021, 40, e107271. [Google Scholar] [CrossRef]
- Maley, C.C.; Reid, B.J.; Forrest, S. Cancer Prevention Strategies That Address the Evolutionary Dynamics of Neoplastic Cells: Simulating Benign Cell Boosters and Selection for Chemosensitivity. Cancer Epidemiol Biomark. Prev. 2004, 13, 1375. [Google Scholar]
- Kam, Y.; Das, T.; Tian, H.; Foroutan, P.; Ruiz, E.; Martinez, G.; Minton, S.; Gillies, R.J.; Gatenby, R.A. Sweat but No Gain: Inhibiting Proliferation of Multidrug Resistant Cancer Cells with “Ersatzdroges”. Int. J. Cancer 2015, 136, E188–E196. [Google Scholar] [CrossRef]
- Lin, K.H.; Rutter, J.C.; Xie, A.; Pardieu, B.; Winn, E.T.; Bello, R.D.; Forget, A.; Itzykson, R.; Ahn, Y.-R.; Dai, Z.; et al. Using Antagonistic Pleiotropy to Design a Chemotherapy-Induced Evolutionary Trap to Target Drug Resistance in Cancer. Nat. Genet. 2020, 52, 408–417. [Google Scholar] [CrossRef]
- Acar, A.; Nichol, D.; Fernandez-Mateos, J.; Cresswell, G.D.; Barozzi, I.; Hong, S.P.; Trahearn, N.; Spiteri, I.; Stubbs, M.; Burke, R.; et al. Exploiting Evolutionary Steering to Induce Collateral Drug Sensitivity in Cancer. Nat. Commun. 2020, 11, 1923. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Levy, D. The Impact of Competition between Cancer Cells and Healthy Cells on Optimal Drug Delivery. Math. Model. Nat. Phenom. 2020, 15, 42. [Google Scholar] [CrossRef]
- Fernandez-Antoran, D.; Piedrafita, G.; Murai, K.; Ong, S.H.; Herms, A.; Frezza, C.; Jones, P.H. Outcompeting P53-Mutant Cells in the Normal Esophagus by Redox Manipulation. Cell Stem Cell 2019, 25, 329–341.e6. [Google Scholar] [CrossRef] [PubMed]
- Archetti, M.; Pienta, K.J. Cooperation among cancer cells: Applying game theory to cancer. Nat. Rev. Cancer 2019, 19, 110–117. [Google Scholar] [CrossRef] [PubMed]


| Reference | Model Type | Cancer Type | Stage | Number of Cell Types | Parameters of Interest 1 | Predicted Outcomes |
|---|---|---|---|---|---|---|
| Cunningham et al. (2018) [17] | Lotka–Volterra | mCRPC | Advanced | 3 (TP, T+, T−) | N/A | Optimized AT strategy outperforms MTD and metronomic strategies. |
| Cunningham et al. (2020) [34] | Lotka–Volterra | mCRPC | Advanced | 3 (TP, T+, T−) | N/A | AT can delay competitive release compared to MTD, but it provides permanent control only for a small subset of initial tumor compositions. Other treatment schedules such as dose titration could be more successful. |
| Gatenby et al. (2009) [14] | Lotka–Volterra | Ovarian | Early (300mm3) | 5 (different fitness and resistance) | Phenotypic sensitivity of population i to therapy (σi)/Environmental sensitivity (β) | AT prolongs survival compared to MTD or metronomic strategies. |
| Gluzman et al. (2020) [20] | Lotka–Volterra | Lung | N/A | 3 (GLY, DEF, VOP) | Constants representing the benefit per unit of acidification, the benefit from the oxygen per unit of vascularization, and the cost of production of Vascular Endothelial Growth Factor respectively. | Optimized AT strategy outperforms MTD strategies and reduces the amount of drugs used. |
| Kim et al. (2021) [35] | Lotka–Volterra | Melanoma | Advanced | 2 (sensitive and resistant) | Phenotype switching between drug-sensitive and resistant cell types (α and β rates) | AT delays time to progression, and even more when phenotypic switching is included in the model. A smaller burden reduction criterion may also result in better outcomes. |
| Ma et Newton (2018) [36] | Lotka–Volterra | N/A | N/A | 3 (healthy, sensitive, resistant) | N/A | Optimized AT strategy controls the tumor for longer than MTD and metronomic strategies and could indefinitely balance the subpopulations of cells. |
| Ma et Newton (2021) [37] | Lotka–Volterra | N/A | N/A | 3 (healthy, sensitive, resistant) | Nature of multi-drug interaction (additive, synergistic, antagonistic) (e) | AT strategy delays with two drugs delays recurrence for longer when the drugs are antagonistic. |
| Silva et al. (2012a) [38] | Lotka–Volterra | Multiple myeloma | Advanced | 2 (sensitive and resistant with varying drug resistance levels) | N/A | Some patients could benefit from AT, but not all, hence the need to classify patients and determine which tumor compositions are suitable for AT. |
| Silva et al. (2012b) [39] | Lotka–Volterra | Breast | Advanced | 2 (sensitive PGP- and resistant PGP+) | N/A | Compared to MTD, AT increases progression-free survival, and AT combined with drugs that increase the fitness disadvantage of resistant cells yields even better results. |
| Silva et al. (2016) [40] | Lotka–Volterra | Multiple myeloma | Advanced | 2 subpopulations with different levels of sensitivity to each of the chemotherapeutic agents or combinations. | N/A | AT could prolong control of the disease, and combining drugs under AT regimes can be even more beneficial. |
| Smalley et al. (2019) [41] | Lotka–Volterra | Melanoma | Advanced | Transcriptional heterogeneity resulting in different levels of resistance | Phenotype switching between drug-sensitive and resistant cell types (α and β rates) | AT leads to delayed time to resistance and better therapeutic responses than MTD and metronomic strategies (confirmed in vivo). |
| Strobl et al. (2021) [42] | Lotka–Volterra | mCRPC | Advanced | 2 (sensitive and resistant) | Cell turnover (density-independent death rate dt) | Adaptive therapy can prolong time to progression even without a resistance cost. |
| Viossat et Noble (2020) [33] | Lotka–Volterra | N/A | N/A | 2 (sensitive and resistant) | N/A | AT containing the tumor at its initial size maximizes time to progression, while containment at the maximal tolerable size maximizes time to treatment failure. In some cases where resistant cells are sufficiently sensitive, MTD could be superior to AT. |
| Viossat et Noble (2021) [43] | Lotka–Volterra | N/A | N/A | 2 (sensitive and resistant) | N/A | AT is likely to be optimal in a broad range of cases, including if there is no explicit cost of resistance. Clinical gains strongly depend on competition intensity and on a few key patient-specific factors. |
| West et al. (2018) [44] | Lotka–Volterra | mCRPC | Advanced | 3 (healthy, sensitive, resistant) | N/A | Assuming a cost of resistance for chemo-resistant cells, AT achieves better time to relapse than MTD. AT with longer treatment-on periods but equal time off perform better. |
| West et al. (2019a) [45] | Lotka–Volterra | mCRPC | Advanced | 3 (TP, T+, T−) | Weighting term to adapt payoff matrix and carrying capacity for each drug (wi) | Multi-drug AT could delay emergence of resistance for even longer than single-drug AT. |
| West et al. (2019b) [46] | Lotka–Volterra | mCRPC | Advanced | 4 (T+, TP, T−, and doubly resistant T−/−) | N/A | Single drug AT increases time to progression compared with conventional strategies, and primary-secondary multi-drug AT is even more beneficial. |
| Zhang et al. (2017) [19] | Lotka–Volterra | mCRPC | Advanced | 3 (TP, T+, T−) | N/A | AT prolongs time to progression compared to MTD and metronomic strategies. |
| Bacevic et al. (2017) [47] | Lotka–Volterra and ABM | Colorectal | Early (first detection—1.3 cm3) | 2 (sensitive and resistant) | Micro-environmental feedback (constants that represent how strongly the tumor stimulates and inhibits vascularization for Lotka–Volterra models, constants regarding oxygen and cyclin-dependent kinase inhibitor’s diffusion, consumption and effects on cell proliferation and/or death for ABMs)/death rate (Mi) | AT outperforms MTD in the condition that there is a fitness cost of resistance. |
| Gallaher et al. (2018) [18] | ABM | Breast | Early | Continuous resistance % | Phenotypic drift, migration (angle of movement and persistence time) | In heterogeneous tumors with resistant cells, AT significantly delays time to progression. |
| Strobl et al. (2021) [15] | ABM | mCRPC | Advanced | 2 (sensitive and resistant) | Cell turnover (density-independent death rate dt) | AT is on average superior to MTD in incurable and resistant tumors. High initial tumor density and low initial resistance fractions maximize its benefits, and a cost of resistance is not necessary. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belkhir, S.; Thomas, F.; Roche, B. Darwinian Approaches for Cancer Treatment: Benefits of Mathematical Modeling. Cancers 2021, 13, 4448. https://doi.org/10.3390/cancers13174448
Belkhir S, Thomas F, Roche B. Darwinian Approaches for Cancer Treatment: Benefits of Mathematical Modeling. Cancers. 2021; 13(17):4448. https://doi.org/10.3390/cancers13174448
Chicago/Turabian StyleBelkhir, Sophia, Frederic Thomas, and Benjamin Roche. 2021. "Darwinian Approaches for Cancer Treatment: Benefits of Mathematical Modeling" Cancers 13, no. 17: 4448. https://doi.org/10.3390/cancers13174448
APA StyleBelkhir, S., Thomas, F., & Roche, B. (2021). Darwinian Approaches for Cancer Treatment: Benefits of Mathematical Modeling. Cancers, 13(17), 4448. https://doi.org/10.3390/cancers13174448