
Myelodysplastic Syndromes in the Postgenomic Era and Future Perspectives for Precision Medicine

 , , , , , ,
, , , , , ,  and
and  on behalf of the Swiss MDS Study Group
on behalf of the Swiss MDS Study Group
Abstract
:Simple Summary
Abstract
1. Epidemiology
2. Pathophysiology
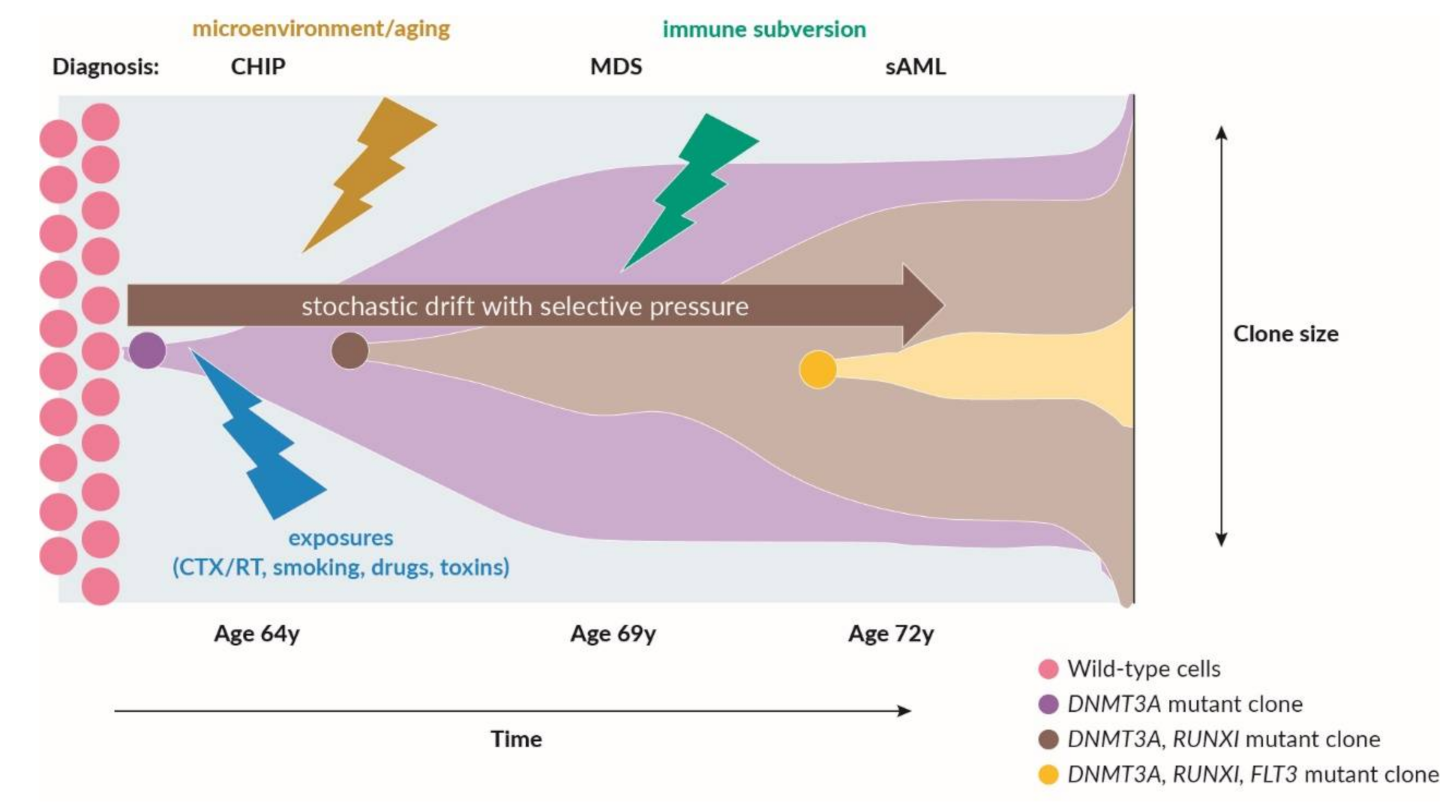
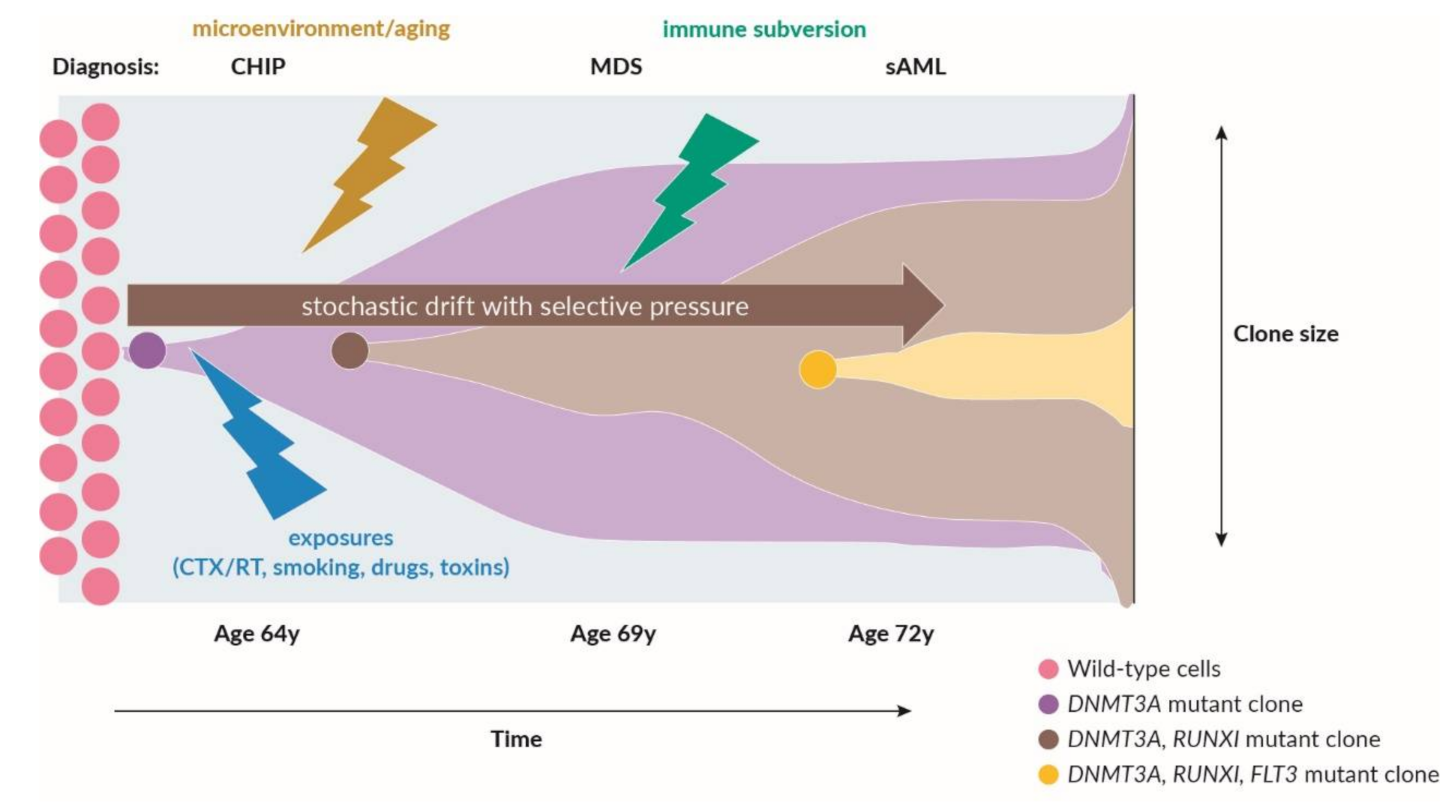
2.1. Recurrent Somatic Leukemia-Associated Driver Mutations and Clonal Hematopoiesis
2.2. The Role of Adaptive and Innate Immunity in MDS
3. General Aspects of MDS Patient Management
4. Diagnostic Approach and Risk-Stratification
4.1. WHO Classification and Minimal Diagnostic Criteria for MDS
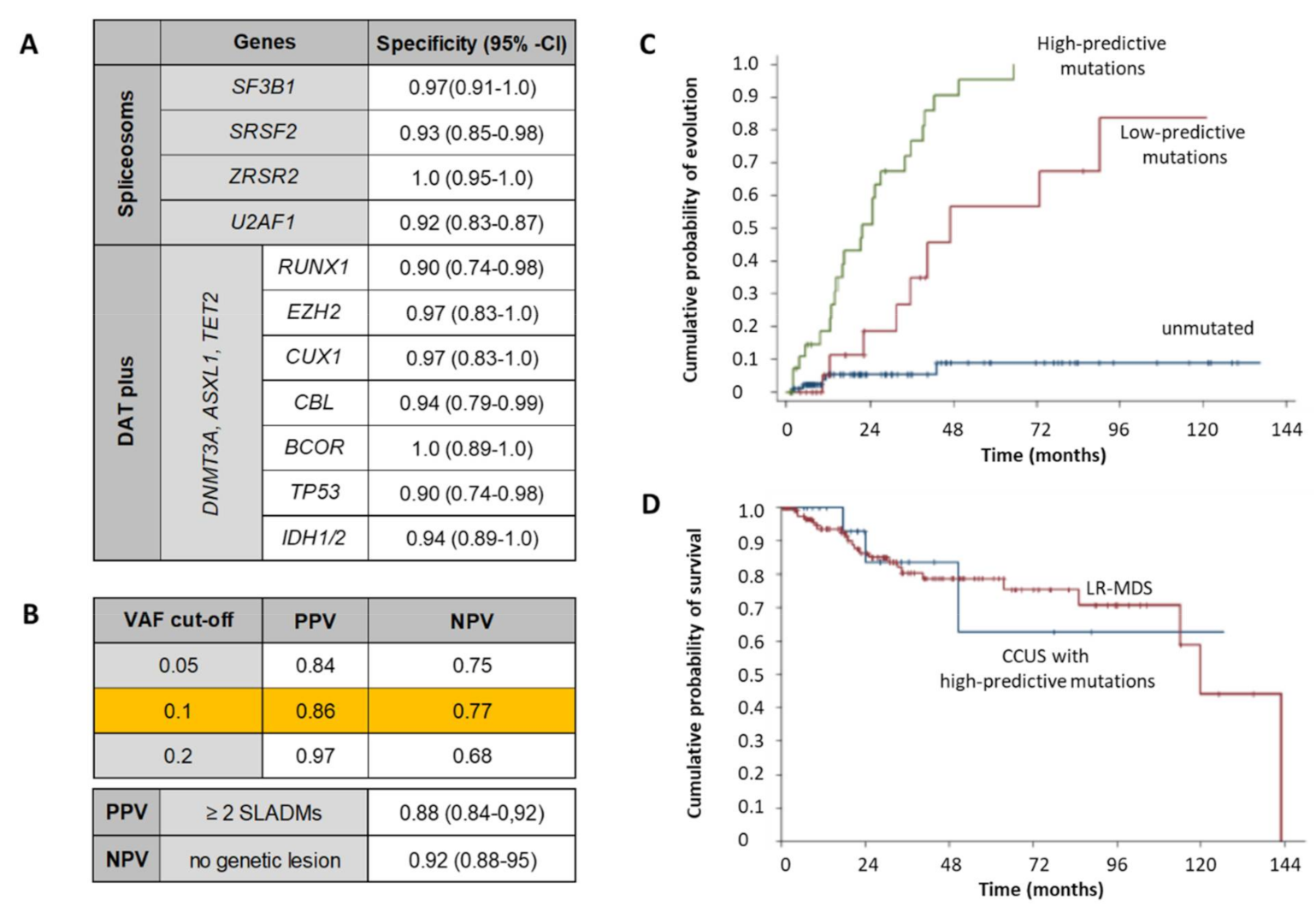
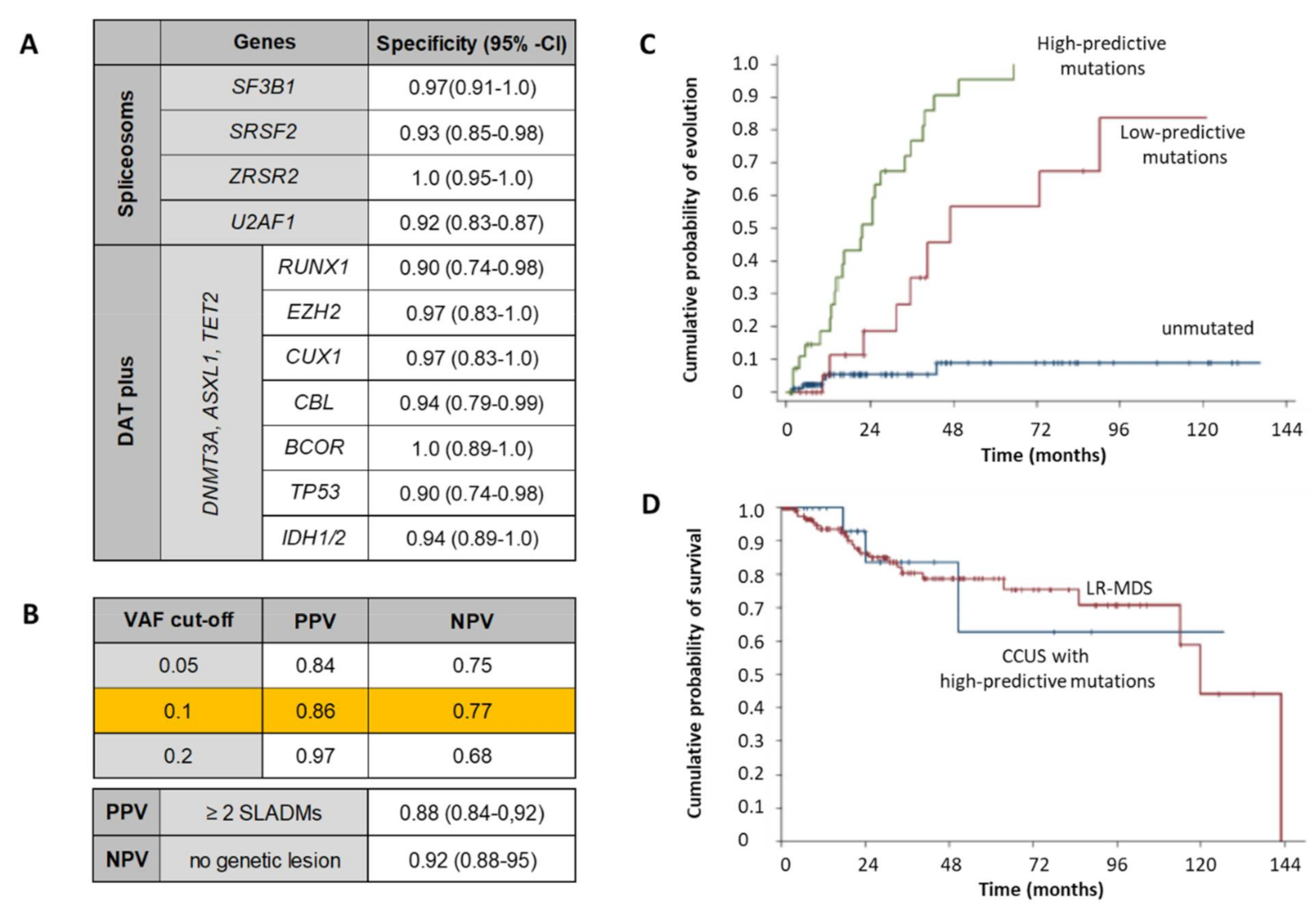
4.2. Role of NGS in MDS Diagnosis, Follow-Up and Risk-Stratification
4.3. Hypoplastic MDS and Aplastic Anemia
4.4. Disease-Based Risk Stratification
4.5. Patient-Based Risk Stratification
4.6. Patient-Reported Outcomes (PROs)
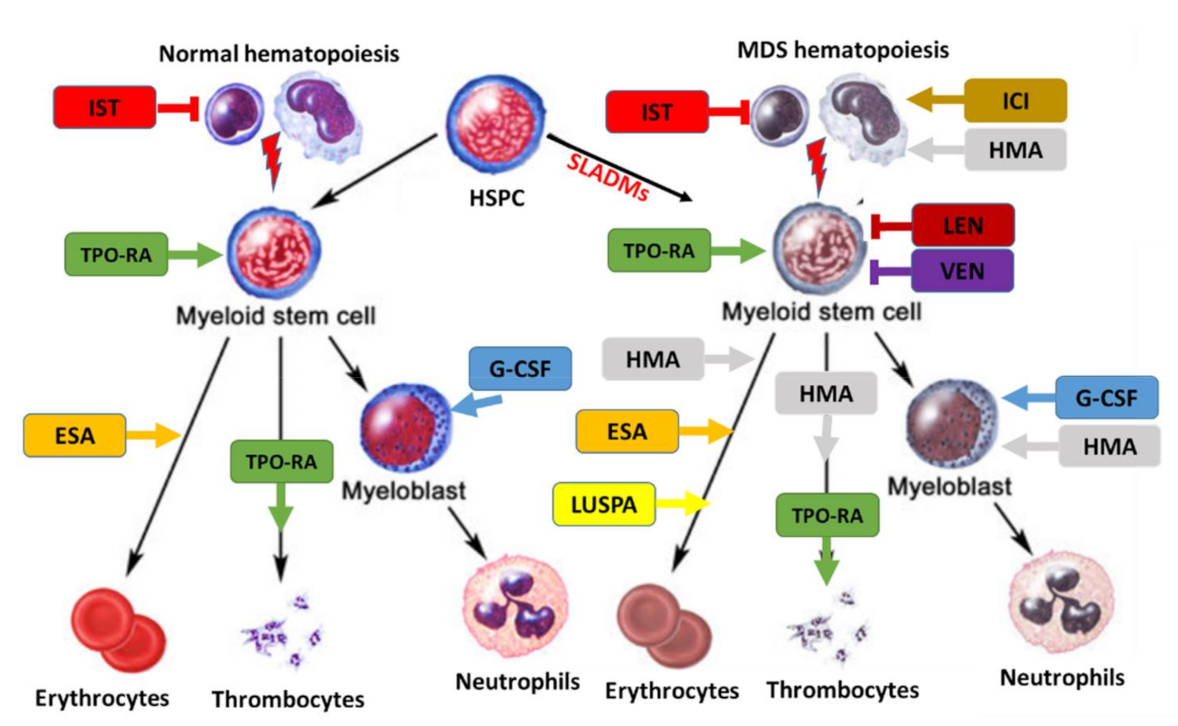
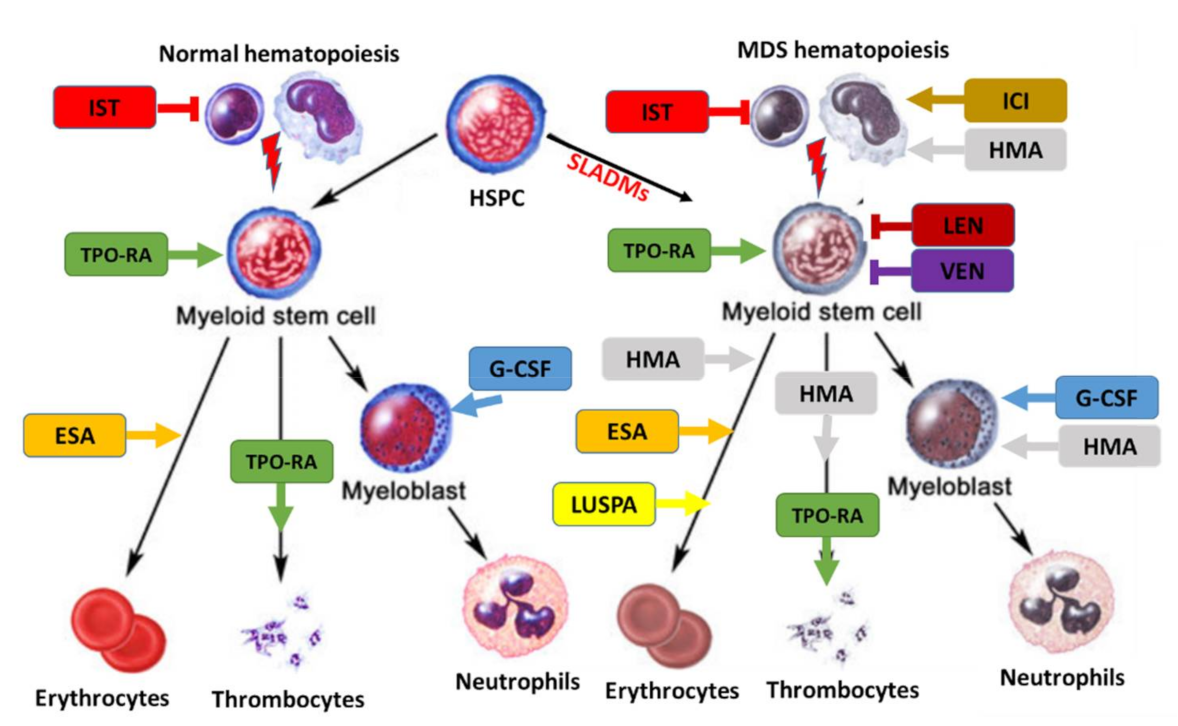
5. Therapeutic Approach
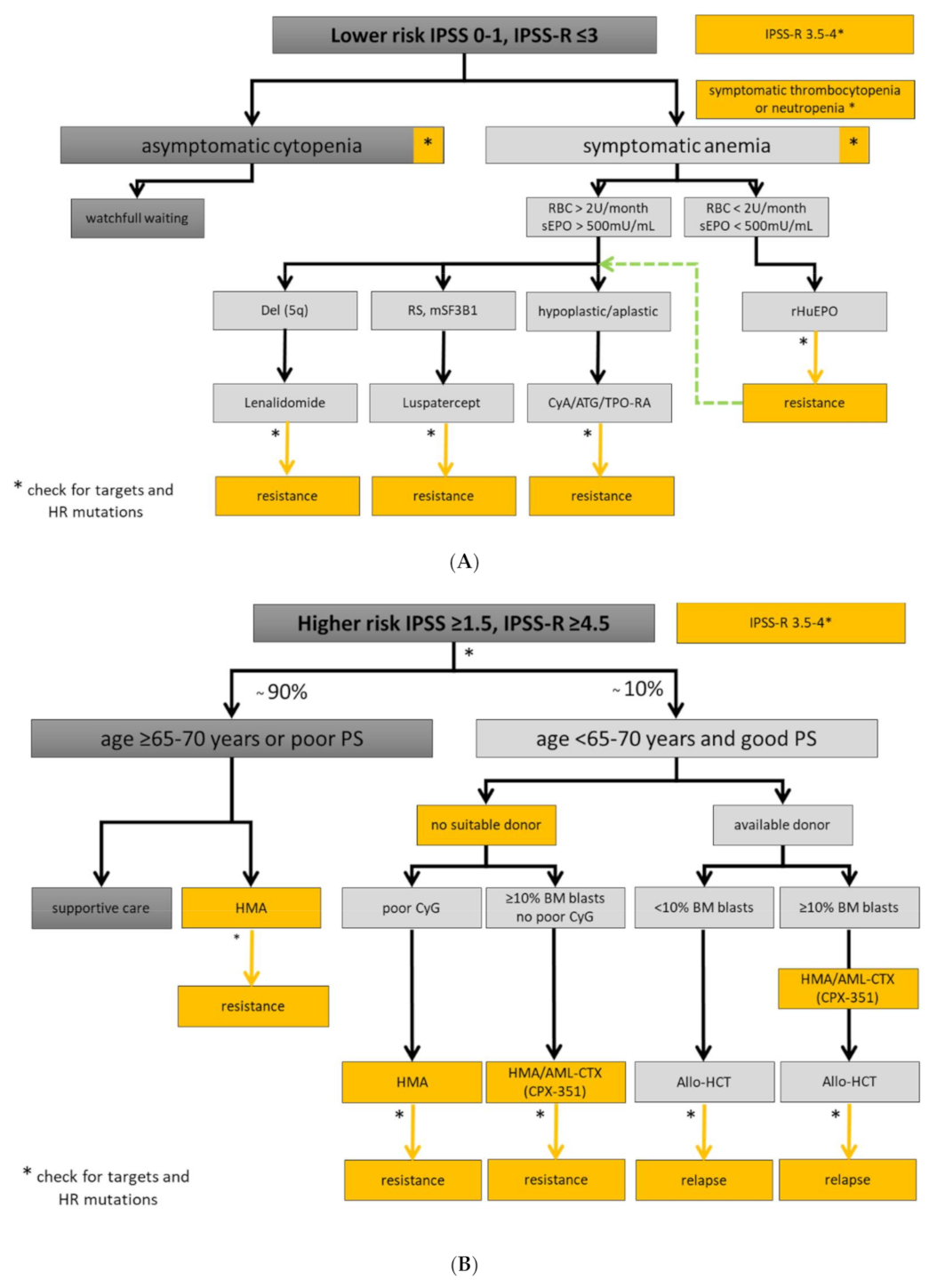
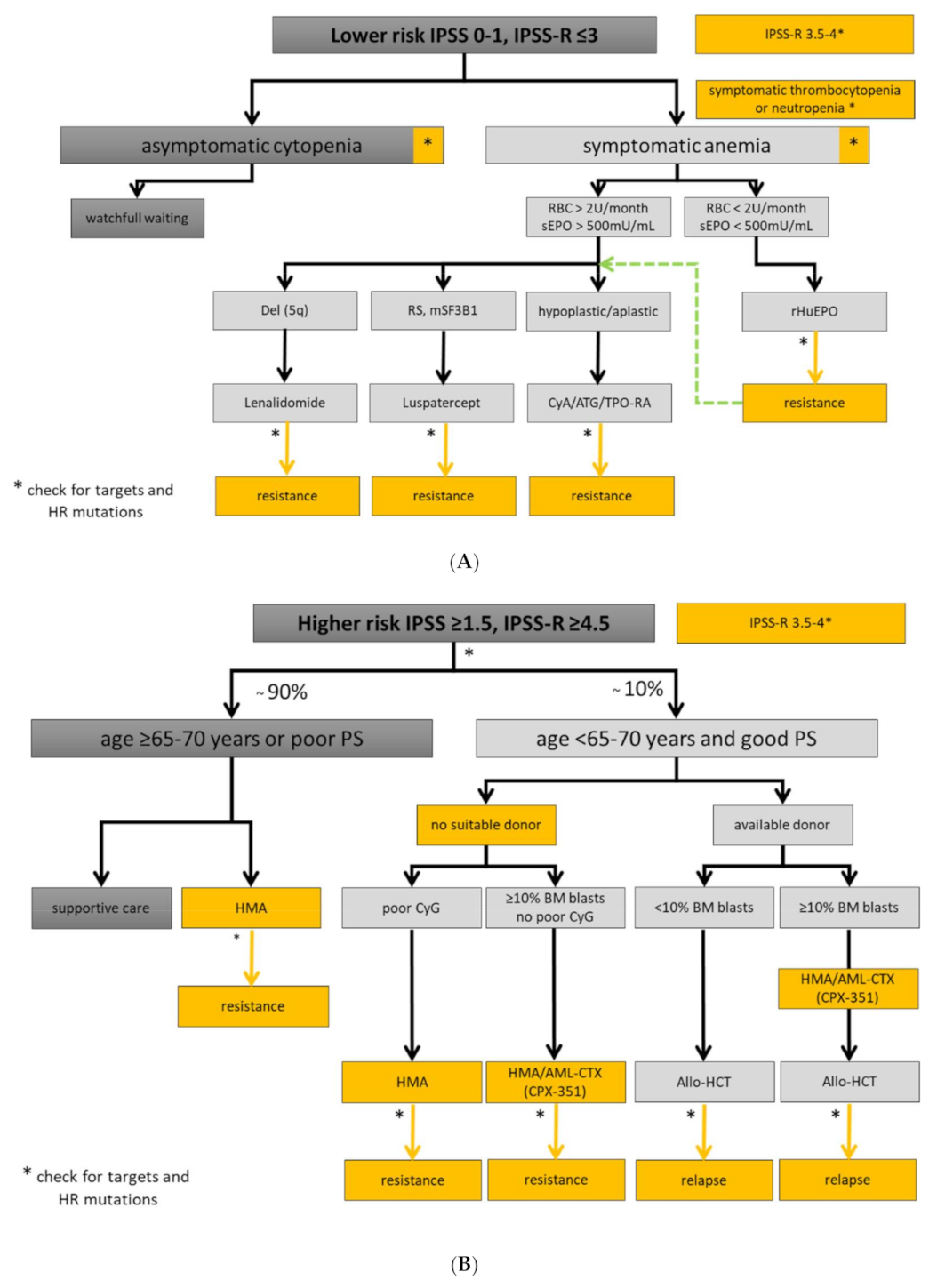
5.1. Lower-Risk MDS Patients
5.1.1. Watchful Observation and General Supportive Treatment
5.1.2. Treatment of Anemia
5.1.3. Treatment of Thrombocytopenia
5.1.4. Treatment of Neutropenia and Infection Prophylaxis
5.1.5. Disease Modifying Treatments in Specific Subsets of Lower-Risk MDS
5.1.6. Hypomethylating Agents
5.2. Higher-Risk MDS Patients
5.2.1. Hypomethylating Agents
5.2.2. Induction Chemotherapy
5.2.3. Allogeneic Hematopoietic Stem Cell Transplantation
6. Ongoing Clinical Trials with Targeting Compounds
6.1. Lower-Risk MDS
6.2. Higher-Risk MDS
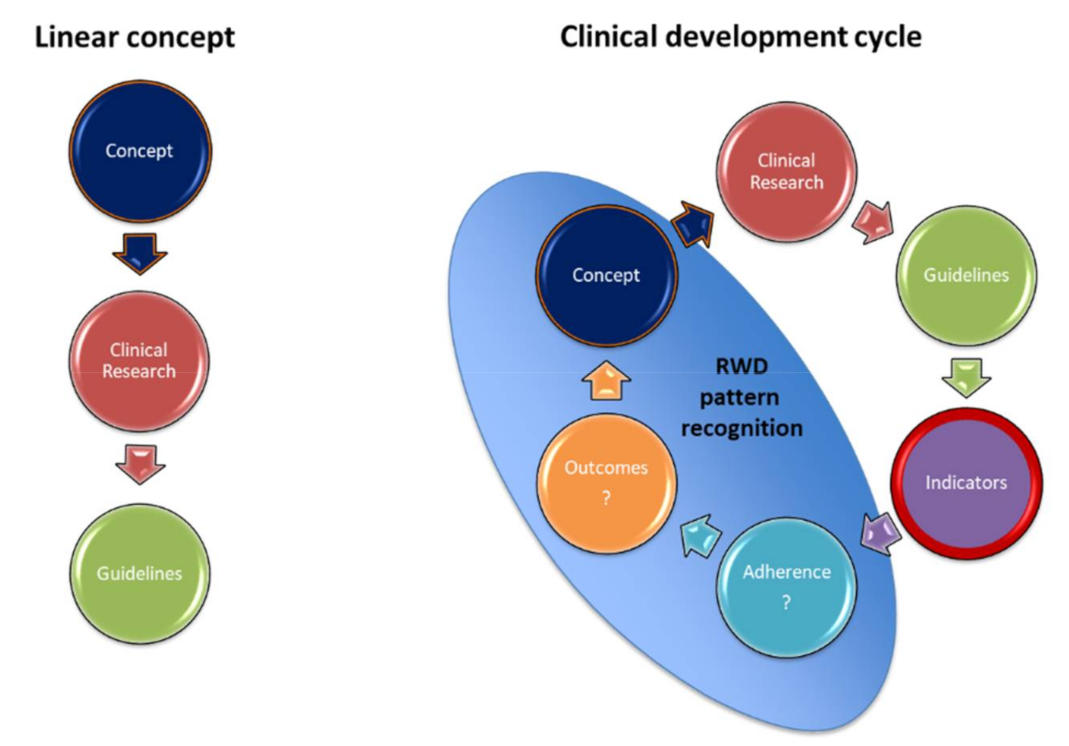
7. Future Perspectives for Precision Medicine
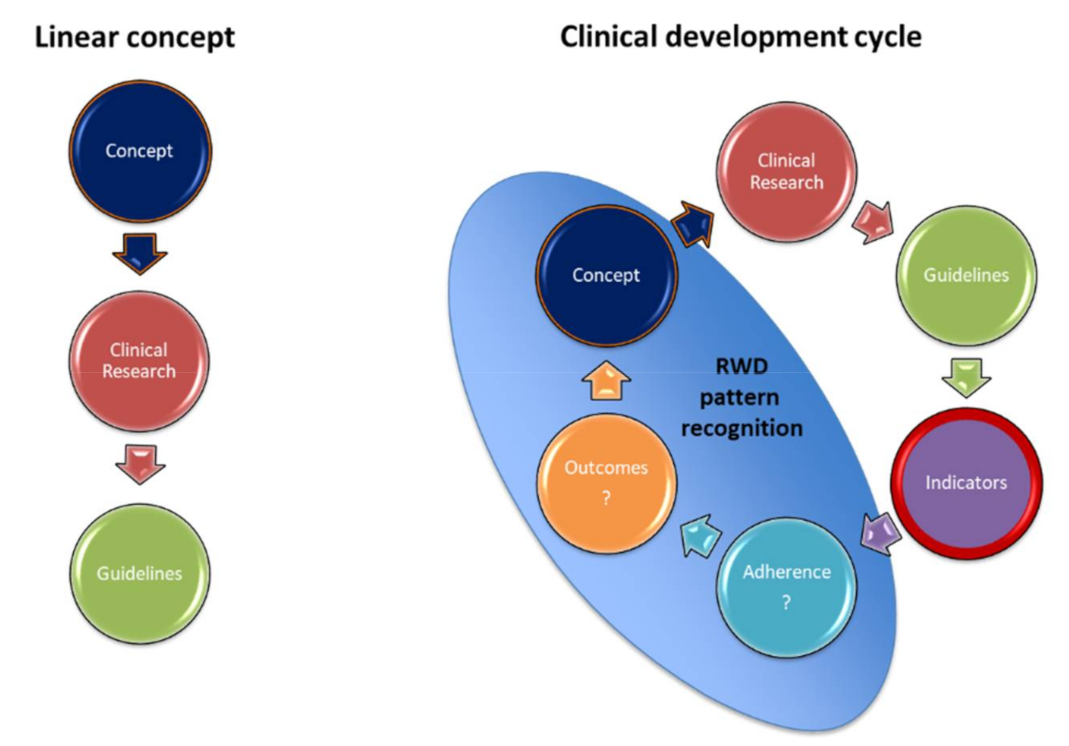
7.1. Clinical Management Using Guideline-Based Indicators (GBIs)
7.2. Diagnosis and Risk-Assessment
7.3. Patient Selection for Targeted Therapies
7.4. Understanding Clonal Heterogeneity at Single Cell Resolution
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACMG | American College of Medical Genetics |
| Allo-HCT | Allogeneic hematopoietic stem cell transplantation |
| ARCH | Age-related clonal hematopoiesis |
| ATG | Antithymocyte globulin |
| AML | Acute myeloid leukemia |
| BM | Bone marrow |
| CCUS | Clonal cytopenia of undetermined significance |
| CGH | Comparative Genomic Hybridization |
| CHIP | Clonal hematopoiesis of indeterminate potential |
| CsA | Cyclosporine A |
| EMA | Erythroid Maturation Agents |
| EPO | Erythropoietin |
| ESA | Erythropoietin Stimulating Agents |
| FISH | Fluorescent in situ hybridization |
| GBIs | Guideline-based indicators |
| G-CSF | Granulocyte Colony-Stimulating Factor |
| G/Rs | Guidelines and recommendations |
| HCT-CI | Hematopoietic Cell Transplantation Comorbidity Index |
| HI | Hematological improvement |
| HMA | Hypomethylating agents |
| HSPC | Hematopoietic stem and progenitor cells |
| HU | Hydroxyurea |
| ICI | Immune checkpoint inhibitor |
| ICUS | Idiopathic cytopenia of undetermined significance |
| IDUS | Idiopathic dysplasia of undetermined significance |
| IP | Immunophenotyping |
| IPSS | International Prognostic Scoring System |
| IPSS-R | Revised International Prognostic Scoring System |
| IST | Immunosuppressive treatment |
| LD-AraC | Low-dose cytarabine |
| LEN | Lenalidomid |
| LUSPA | Luspatercept |
| MDS | Myelodysplastic syndromes |
| MRD | Measurable residual disease |
| NGS | Next generation sequencing |
| OS | Overall survival |
| PB | Peripheral blood |
| RBC | Red blood cell concentrates |
| sAML | Secondary AML |
| SLADMs | Somatic leukemia-associated driver mutations |
| TC | Thrombocyte concentrates |
| TLR | Toll-like receptor |
| TPO-RA | Thrombopoietin receptor agonists |
| QoL | Quality of life |
| VAF | Variant allele frequency |
| WHO | World Health Organization |
| WPSS | WHO Prognostic Scoring System |
References
- Shastri, A.; Will, B.; Steidl, U.; Verma, A. Stem and progenitor cell alterations in myelodysplastic syndromes. Blood 2017, 129, 1586–1594. [Google Scholar] [PubMed]
- da Silva-Coelho, P.; Kroeze, L.I.; Yoshida, K.; Koorenhof-Scheele, T.N.; Knops, R.; Van De Locht, L.T.; De Graaf, A.O.; Massop, M.; Sandmann, S.; Dugas, M.; et al. Clonal evolution in myelodysplastic syndromes. Nat. Commun. 2017, 8, 15099. [Google Scholar] [PubMed]
- Zeidan, A.M.; Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X. Epidemiology of myelodysplastic syndromes: Why characterizing the beast is a prerequisite to taming it. Blood Rev. 2019, 34, 1–15. [Google Scholar] [PubMed]
- Bonadies, N.; Feller, A.; Rovo, A.; Ruefer, A.; Blum, S.; Gerber, B.; Stuessi, G.; Benz, R.; Cantoni, N.; Holbro, A.; et al. Trends of classification, incidence, mortality, and survival of MDS patients in Switzerland between 2001 and 2012. Cancer Epidemiol. 2017, 46, 85–92. [Google Scholar]
- Candelaria, M.; Dueñas-Gonzalez, A. Therapy-related myelodysplastic syndrome. Expert Opin. Drug Saf. 2015, 14, 655–665. [Google Scholar] [CrossRef]
- Abou Zahr, A.; Kavi, A.M.; Mukherjee, S.; Zeidan, A.M. Therapy-related myelodysplastic syndromes, or are they? Blood Rev. 2017, 31, 119–128. [Google Scholar]
- Locatelli, F.; Strahm, B. How I treat myelodysplastic syndromes of childhood. Blood 2018, 131, 1406–1414. [Google Scholar] [PubMed]
- Stauder, R.; Yu, G.; Koinig, K.A.; Bagguley, T.; Fenaux, P.; Symeonidis, A.; Sanz, G.; Cermak, J.; Mittelman, M.; Hellström-Lindberg, E.; et al. Health-related quality of life in lower-risk MDS patients compared with age- and sex-matched reference populations: A European LeukemiaNet study. Leukemia 2018, 32, 1380–1392. [Google Scholar]
- Corey, S.J.; Minden, M.D.; Barber, D.L.; Kantarjian, H.; Wang, J.C.Y.; Schimmer, A.D. Myelodysplastic syndromes: The complexity of stem-cell diseases. Nat. Rev. Cancer 2007, 7, 118–129. [Google Scholar]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, O.; Figueroa, M.E. Interpreting new molecular genetics in myelodysplastic syndromes. Hematol. Am. Soc. Hematol. Educ. Program 2012, 2012, 56–64. [Google Scholar] [CrossRef] [Green Version]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef]
- Kon, A.; Shih, L.-Y.; Minamino, M.; Sanada, M.; Shiraishi, Y.; Nagata, Y.; Yoshida, K.; Okuno, Y.; Bando, M.; Nakato, R.; et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat. Genet. 2013, 45, 1232–1237. [Google Scholar] [CrossRef]
- Steensma, D.P. Clinical implications of clonal hematopoiesis. Mayo Clin. Proc. 2018, 93, 1122–1130. [Google Scholar] [CrossRef] [Green Version]
- Bowman, R.L.; Busque, L.; Levine, R.L. Clonal hematopoiesis and evolution to hematopoietic malignancies. Cell Stem Cell 2018, 22, 157–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heuser, M.; Thol, F.; Ganser, A. Clonal hematopoiesis of indeterminate potential. Dtsch. Aerzteblatt Online 2016, 113, 317–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef]
- Chanias, I.; Bonadies, N. Current Standard of Care in Patients with Myelodysplastic Syndromes and Future Perspectives. Heal. Online Med. J. 2020, 10–22. [Google Scholar] [CrossRef]
- Valent, P.; Orazi, A.; Steensma, D.P.; Ebert, B.L.; Haase, D.; Malcovati, L.; Van De Loosdrecht, A.A.; Haferlach, T.; Westers, T.M.; Wells, D.A.; et al. Proposed minimal diagnostic criteria for myelodysplastic syndromes (MDS) and potential pre-MDS conditions. Oncotarget 2017, 8, 73483–73500. [Google Scholar] [CrossRef] [Green Version]
- Kulasekararaj, A.G.; Jiang, J.; Smith, A.E.; Mohamedali, A.M.; Mian, S.A.; Gandhi, S.; Gaken, J.; Czepulkowski, B.; Marsh, J.C.W.; Mufti, G.J. Somatic mutations identify a subgroup of aplastic anemia patients who progress to myelodysplastic syndrome. Blood 2014, 124, 2698–2704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshizato, T.; Dumitriu, B.; Hosokawa, K.; Makishima, H.; Yoshida, K.; Townsley, D.; Sato-Otsubo, A.; Sato, Y.; Liu, D.; Suzuki, H.; et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N. Engl. J. Med. 2015, 373, 35–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yura, Y.; Sano, S.; Walsh, K. Clonal hematopoiesis: A new step linking inflammation to heart failure. JACC Basic Transl. Sci. 2020, 5, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Mekinian, A.; Grignano, E.; Braun, T.; Decaux, O.; Liozon, E.; Costedoat-Chalumeau, N.; Kahn, J.E.; Hamidou, M.; Park, S.; Puéchal, X.; et al. Systemic inflammatory and autoimmune manifestations associated with myelodysplastic syndromes and chronic myelomonocytic leukaemia: A French multicentre retrospective study. Rheumatology 2016, 55, 291–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristinsson, S.Y.; Björkholm, M.; Hultcrantz, M.; Derolf Åsa, R.; Landgren, O.; Goldin, L.R. Chronic immune stimulation might act as a trigger for the development of acute myeloid leukemia or myelodysplastic syndromes. J. Clin. Oncol. 2011, 29, 2897–2903. [Google Scholar] [CrossRef] [Green Version]
- Winter, S.; Shoaie, S.; Kordasti, S.Y.; Platzbecker, U. Integrating the “immunome” in the stratification of myelodysplastic syndromes and Future Clinical Trial Design. J. Clin. Oncol. 2020, 38, 1723–1735. [Google Scholar] [CrossRef]
- Gañán-Gómez, I.; Wei, Y.; Starczynowski, D.T.; Colla, S.; Yang, H.; Cabrero-Calvo, M.; Bohannan, Z.; Verma, A.; Steidl, U.; Garciamanero, G. Deregulation of innate immune and inflammatory signaling in myelodysplastic syndromes. Leukemia 2015, 29, 1458–1469. [Google Scholar] [CrossRef]
- Shetty, V.; Mundle, S.; Alvi, S.; Showel, M.; Broady-Robinson, L.; Dar, S.; Borok, R.; Showel, J.; Gregory, S.; Rifkin, S.; et al. Measurement of apoptosis, proliferation and three cytokines in 46 patients with myelodysplastic syndromes. Leuk. Res. 1996, 20, 891–900. [Google Scholar] [CrossRef]
- Allampallam, K.; Shetty, V.; Hussaini, S.; Mazzoran, L.; Zorat, F.; Huang, R.; Raza, A. Measurement of mRNA expression for a variety of cytokines and its receptors in bone marrows of patients with myelodysplastic syndromes. Anticancer Res. 2000, 19, 5323–5328. [Google Scholar]
- Kitagawa, M.; Saito, I.; Kuwata, T.; Yoshida, S.; Yamaguchi, S.; Takahashi, M.; Tanizawa, T.; Kamiyama, R.; Hirokawa, K. Overexpression of tumor necrosis factor (TNF)-α and interferon (IFN)-γ by bone marrow cells from patients with myelodysplastic syndromes. Leukemia 1997, 11, 2049–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalamaga, M.; Petridou, E.; Cook, F.E.; Trichopoulos, D. Risk factors for myelodysplastic syndromes: A case–control study in Greece. Cancer Causes Control 2002, 13, 603–608. [Google Scholar] [CrossRef]
- Anderson, L.A.; Pfeiffer, R.M.; Landgren, O.; Gadalla, S.; Berndt, S.I.; Engels, E.A. Risks of myeloid malignancies in patients with autoimmune conditions. Br. J. Cancer 2009, 100, 822–828. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, X.; Zhang, F.; Yin, H. Toll-like receptors as therapeutic targets for autoimmune connective tissue diseases. Pharmacol. Ther. 2013, 138, 441–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maratheftis, C.I.; Andreakos, E.; Moutsopoulos, H.M.; Voulgarelis, M. Toll-like Receptor-4 is up-regulated in hematopoietic progenitor cells and contributes to increased apoptosis in myelodysplastic syndromes. Clin. Cancer Res. 2007, 13, 1154–1160. [Google Scholar] [CrossRef] [Green Version]
- Basiorka, A.A.; McGraw, K.L.; Eksioglu, E.A.; Chen, X.; Johnson, J.; Zhang, L.; Zhang, Q.; Irvine, B.A.; Cluzeau, T.; Sallman, D.A.; et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood 2016, 128, 2960–2975. [Google Scholar] [CrossRef]
- Ambinder, A.J.; Miller, J.; DeZern, A.E. Autoimmune disease in CMML-the chicken or the egg? Best Pract. Res. Clin. Haematol. 2020, 33, 101136. [Google Scholar] [CrossRef]
- Kipfer, B.; Daikeler, T.; Kuchen, S.; Hallal, M.; Andina, N.; Allam, R.; Bonadies, N. Increased cardiovascular comorbidities in patients with myelodysplastic syndromes and chronic myelomonocytic leukemia presenting with systemic inflammatory and autoimmune manifestations. Semin. Hematol. 2018, 55, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Santini, V. Treatment of low-risk myelodysplastic syndromes. Hematology 2016, 2016, 462–469. [Google Scholar] [CrossRef] [Green Version]
- Fenaux, P.; Ades, L. How we treat lower-risk myelodysplastic syndromes. Blood 2013, 121, 4280–4286. [Google Scholar] [CrossRef]
- Komrokji, R.S. Current state of the art: Management of higher risk myelodysplastic syndromes. Clin. Lymphoma Myeloma Leuk. 2016, 16, S39–S43. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G. Myelodysplastic syndromes: 2015 Update on diagnosis, risk-stratification and management. Am. J. Hematol. 2015, 90, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Malcovati, L.; Hellström-Lindberg, E.; Bowen, D.; Adès, L.; Cermak, J.; Del Cañizo, C.; Della Porta, M.G.; Fenaux, P.; Gattermann, N.; Germing, U.; et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: Recommendations from the European LeukemiaNet. Blood 2013, 122, 2943–2964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stojkov, K.; Silzle, T.; Stussi, G.; Schwappach, D.; Bernhard, J.; Bowen, D.; Čermák, J.; Dinmohamed, A.G.; Eeltink, C.; Eggmann, S.; et al. Guideline-based indicators for adult patients with myelodysplastic syndromes. Blood Adv. 2020, 4, 4029–4044. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Swerdlow, S.H.C.E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Arber, D.A.; Hasserjian, R.P.; Le Beau, M.M.; Orazi, A.; et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2017.
- Valent, P.; Horny, H.-P. Minimal diagnostic criteria for myelodysplastic syndromes and separation from ICUS and IDUS: Update and open questions. Eur. J. Clin. Investig. 2009, 39, 548–553. [Google Scholar] [CrossRef]
- Braggio, E.; Egan, J.B.; Fonseca, R.; Stewart, A.K. Lessons from next-generation sequencing analysis in hematological malignancies. Blood Cancer J. 2013, 3, e127. [Google Scholar] [CrossRef] [Green Version]
- Merker, J.D.; Valouev, A.; Gotlib, J. Next-generation sequencing in hematologic malignancies: What will be the dividends? Ther. Adv. Hematol. 2012, 3, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Malcovati, L.; Gallì, A.; Travaglino, E.; Ambaglio, I.; Rizzo, E.; Molteni, E.; Elena, C.; Ferretti, V.V.; Catricalà, S.; Bono, E.; et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood 2017, 129, 3371–3378. [Google Scholar] [CrossRef]
- Yu, J.; Li, Y.; Li, T.; Li, Y.; Xing, H.; Sun, H.; Sun, L.; Wan, D.; Liu, Y.; Xie, X.; et al. Gene mutational analysis by NGS and its clinical significance in patients with myelodysplastic syndrome and acute myeloid leukemia. Exp. Hematol. Oncol. 2020, 9, 1–11. [Google Scholar] [CrossRef]
- Bonadies, N.; Bacher, V.U. What role can next-generation sequencing play in myelodysplastic syndrome care? Expert Rev. Hematol. 2019, 12, 379–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz, G.F.; Ibañez, M.; Such, E. Do next-generation sequencing results drive diagnostic and therapeutic decisions in MDS? Blood Adv. 2019, 3, 3454–3460. [Google Scholar] [CrossRef] [Green Version]
- Thol, F.; Platzbecker, U. Do next-generation sequencing results drive diagnostic and therapeutic decisions in MDS? Blood Adv. 2019, 3, 3449–3453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bersanelli, M.; Travaglino, E.; Meggendorfer, M.; Matteuzzi, T.; Sala, C.; Mosca, E.; Chiereghin, C.; Di Nanni, N.; Gnocchi, M.; Zampini, M.; et al. Classification and personalized prognostic assessment on the basis of clinical and genomic features in myelodysplastic syndromes. J. Clin. Oncol. 2021, 39, 1223–1233. [Google Scholar] [CrossRef]
- Bono, E.; McLornan, D.; Travaglino, E.; Gandhi, S.; Gallì, A.; Khan, A.A.; Kulasekararaj, A.G.; Boveri, E.; Raj, K.; Elena, C.; et al. Clinical, histopathological and molecular characterization of hypoplastic myelodysplastic syndrome. Leukemia 2019, 33, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, P.; Cox, C.; Lebeau, M.M.; Fenaux, P.; Morel, P.; Sanz, G.; Sanz, M.; Vallespi, T.; Hamblin, T.; Oscier, D.; et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997, 89, 2079–2088. [Google Scholar] [CrossRef]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef] [PubMed]
- Malcovati, L.; Germing, U.; Kuendgen, A.; Della Porta, M.G.; Pascutto, C.; Invernizzi, R.; Giagounidis, A.; Hildebrandt, B.; Bernasconi, P.; Knipp, S.; et al. Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. J. Clin. Oncol. 2007, 25, 3503–3510. [Google Scholar] [CrossRef]
- Sorror, M.L.; Storb, R.F.; Sandmaier, B.M.; Maziarz, R.T.; Pulsipher, M.A.; Maris, M.B.; Bhatia, S.; Ostronoff, F.; Deeg, H.J.; Syrjala, K.L.; et al. Comorbidity-age index: A clinical measure of biologic age before allogeneic hematopoietic cell transplantation. J. Clin. Oncol. 2014, 32, 3249–3256. [Google Scholar] [CrossRef]
- Della Porta, M.G.; Malcovati, L.; Strupp, C.; Ambaglio, I.; Kuendgen, A.; Zipperer, E.; Travaglino, E.; Invernizzi, R.; Pascutto, C.; Lazzarino, M.; et al. Risk stratification based on both disease status and extra-hematologic comorbidities in patients with myelodysplastic syndrome. Haematologica 2010, 96, 441–449. [Google Scholar] [CrossRef] [Green Version]
- Starkman, R.; Alibhai, S.; Wells, R.A.; Geddes, M.; Zhu, N.; Keating, M.M.; Leber, B.; Chodirker, L.; Sabloff, M.; Christou, G.; et al. An MDS-specific frailty index based on cumulative deficits adds independent prognostic information to clinical prognostic scoring. Leukemia 2019, 34, 1394–1406. [Google Scholar] [CrossRef] [PubMed]
- Buckstein, R.; Wells, R.A.; Zhu, N.; Leitch, H.A.; Nevill, T.J.; Yee, K.W.L.; Leber, B.; Sabloff, M.; Hilaire, E.S.; Kumar, R.; et al. Patient-related factors independently impact overall survival in patients with myelodysplastic syndromes: An MDS-CAN prospective study. Br. J. Haematol. 2016, 174, 88–101. [Google Scholar] [CrossRef]
- Efficace, F.; Gaidano, G.; Breccia, M.; Voso, M.T.; Cottone, F.; Angelucci, E.; Caocci, G.; Stauder, R.; Selleslag, D.; Sprangers, M.; et al. Prognostic value of self-reported fatigue on overall survival in patients with myelodysplastic syndromes: A multicentre, prospective, observational, cohort study. Lancet Oncol. 2015, 16, 1506–1514. [Google Scholar] [CrossRef]
- Devlin, N.J.; Krabbe, P.F.M. The development of new research methods for the valuation of EQ-5D-5L. Eur. J. Health Econ. HEPAC Health Econ. Prev. Care 2013, 14, S1–S3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cella, D. The Functional Assessment of Cancer Therapy-Anemia (FACT-An) Scale: A new tool for the assessment of outcomes in cancer anemia and fatigue. Semin. Hematol. 1997, 34, 13–19. [Google Scholar] [PubMed]
- Abel, G.A.; Efficace, F.; Buckstein, R.J.; Tinsley, S.; Jurcic, J.G.; Martins, Y.; Steensma, D.P.; Watts, C.D.; Raza, A.; Lee, S.J.; et al. Prospective international validation of the Quality of Life in Myelodysplasia Scale (QUALMS). Haematologica 2016, 101, 781–788. [Google Scholar] [CrossRef] [Green Version]
- Aaronson, N.K.; Ahmedzai, S.; Bergman, B.; Bullinger, M.; Cull, A.; Duez, N.J.; Filiberti, A.; Flechtner, H.; Fleishman, S.B.; De Haes, J.C.; et al. The European Organization for Research and Treatment of Cancer QLQ-C30: A quality-of-life instrument for use in international clinical trials in oncology. J. Natl. Cancer Inst. 1993, 85, 365–376. [Google Scholar] [CrossRef]
- Wan, B.A.; Nazha, A.; Starkman, R.; Alibhai, S.; Wells, R.A.; Geddes, M.; Zhu, N.; Keating, M.-M.; Leber, B.; Chodirker, L.; et al. Revised 15-item MDS-specific frailty scale maintains prognostic potential. Leukemia 2020, 34, 3434–3438. [Google Scholar] [CrossRef]
- Fenaux, P.; Platzbecker, U.; Ades, L. How we manage adults with myelodysplastic syndrome. Br. J. Haematol. 2020, 189, 1016–1027. [Google Scholar] [CrossRef]
- Malcovati, L.; Papaemmanuil, E.; Ambaglio, I.; Elena, C.; Gallì, A.; Della Porta, M.G.; Travaglino, E.; Pietra, D.; Pascutto, C.; Ubezio, M.; et al. Driver somatic mutations identify distinct disease entities within myeloid neoplasms with myelodysplasia. Blood 2014, 124, 1513–1521. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.E.; Caughey, B.; Lindsley, C.; Mar, B.; Stojanov, P.; Getz, G.; Steensma, D.P.; Ritz, J.; Soiffer, R.; et al. Somatic mutations predict poor outcome in patients with myelodysplastic syndrome after hematopoietic stem-cell transplantation. J. Clin. Oncol. 2014, 32, 2691–2698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakurai, A.; Nakazato, T. The prognostic value of the controlling nutritional status score in patients with myelodysplastic syndrome and acute myeloid leukemia with myelodysplasia related changes treated with azacitidine. Leuk. Lymphoma 2020, 61, 2995–2997. [Google Scholar] [CrossRef] [PubMed]
- Gattermann, N.; Finelli, C.; Della Porta, M.; Fenaux, P.; Stadler, M.; Guerci-Bresler, A.; Schmid, M.; Taylor, K.; Vassilieff, D.; Habr, D.; et al. Hematologic responses to deferasirox therapy in transfusion-dependent patients with myelodysplastic syndromes. Haematologica 2012, 97, 1364–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- List, A.F.; Baer, M.R.; Steensma, D.; Raza, A.; Esposito, J.; Martinez-Lopez, N.; Paley, C.; Feigert, J.; Besa, E. Deferasirox reduces serum ferritin and labile plasma iron in rbc transfusion–dependent patients with myelodysplastic syndrome. J. Clin. Oncol. 2012, 30, 2134–2139. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, E.; Greenberg, P.; Izquierdo, M.; Garcia-Manero, G. Iron chelation in transfusion-dependent patients with low- to intermediate-1–risk myelodysplastic syndromes. Ann. Intern. Med. 2020, 173, 595–596. [Google Scholar] [CrossRef]
- Chanias, I.; Wilk, C.M.; Benz, R.; Daskalakis, M.; Stüssi, G.; Schmidt, A.; Bacher, U.; Bonadies, N.; on behalf of the Swiss MDS Study Group. Survey on recommended health care for adult patients with myelodysplastic syndromes identifies areas for improvement. Int. J. Environ. Res. Public Health 2020, 17, 9562. [Google Scholar] [CrossRef]
- Mitchell, M.; Gore, S.D.; Zeidan, A.M. Iron chelation therapy in myelodysplastic syndromes: Where do we stand? Expert Rev. Hematol. 2013, 6, 397–410. [Google Scholar] [CrossRef] [Green Version]
- Zeidan, A.M.; Griffiths, E.A. To chelate or not to chelate in MDS: That is the question! Blood Rev. 2018, 32, 368–377. [Google Scholar] [CrossRef]
- Jaekel, N.; Lieder, K.; Albrecht, S.; Leismann, O.; Hubert, K.; Bug, G.; Kröger, N.; Platzbecker, U.; Stadler, M.; De Haas, K.; et al. Efficacy and safety of deferasirox in non-thalassemic patients with elevated ferritin levels after allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2015, 51, 89–95. [Google Scholar] [CrossRef]
- Armand, P.; Kim, H.T.; Cutler, C.S.; Ho, V.T.; Koreth, J.; Alyea, E.P.; Soiffer, R.J.; Antin, J.H. Prognostic impact of elevated pretransplantation serum ferritin in patients undergoing myeloablative stem cell transplantation. Blood 2007, 109, 4586–4588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Grabar, S.; Kelaidi, C.; Beyne-Rauzy, O.; Picard, F.; Bardet, V.; Coiteux, V.; Leroux, G.; Lepelley, P.; Daniel, M.-T.; et al. Predictive factors of response and survival in myelodysplastic syndrome treated with erythropoietin and G-CSF: The GFM experience. Blood 2008, 111, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Hellstrom-Lindberg, E.; Negrin, R.; Stein, R.; Krantz, S.; Lindberg, G.; Vardiman, J.; Ost, A.; Greenberg, P. Erythroid response to treatment with G-CSF plus erythropoietin for the anaemia of patients with myelodysplastic syndromes: Proposal for a predictive model. Br. J. Haematol. 1997, 99, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Platzbecker, U.; Symeonidis, A.; Oliva, E.N.; Goede, J.S.; Delforge, M.; Mayer, J.; Slama, B.; Badre, S.; Gasal, E.; Mehta, B.; et al. A phase 3 randomized placebo-controlled trial of darbepoetin alfa in patients with anemia and lower-risk myelodysplastic syndromes. Leukemia 2017, 31, 1944–1950. [Google Scholar] [CrossRef] [Green Version]
- Fenaux, P.; Santini, V.; Spiriti, M.A.A.; Giagounidis, A.; Schlag, R.; Radinoff, A.; Gercheva-Kyuchukova, L.; Anagnostopoulos, A.; Oliva, E.N.; Symeonidis, A.; et al. A phase 3 randomized, placebo-controlled study assessing the efficacy and safety of epoetin-α in anemic patients with low-risk MDS. Leukemia 2018, 32, 2648–2658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Greenberg, P.; Yucel, A.; Farmer, C.; O’Neill, F.; Brandao, C.D.O.; Fenaux, P. Clinical effectiveness and safety of erythropoietin-stimulating agents for the treatment of low- and intermediate-1−risk myelodysplastic syndrome: A systematic literature review. Br. J. Haematol. 2019, 184, 134–160. [Google Scholar] [CrossRef] [Green Version]
- Affentranger, L.; Bohlius, J.; Hallal, M.; Bonadies, N. Efficacy of granulocyte colony stimulating factor in combination with erythropoiesis stimulating agents for treatment of anemia in patients with lower risk myelodysplastic syndromes: A systematic review. Crit. Rev. Oncol. 2019, 136, 37–47. [Google Scholar] [CrossRef]
- Toma, A.; Kosmider, O.; Chevret, S.; Delaunay, J.; Stamatoullas, A.; Rose, C.D.; Beynerauzy, O.; Banos, A.; Guercibresler, A.; Wickenhauser, S.; et al. Lenalidomide with or without erythropoietin in transfusion-dependent erythropoiesis-stimulating agent-refractory lower-risk MDS without 5q deletion. Leukemia 2016, 30, 897–905. [Google Scholar] [CrossRef]
- Platzbecker, U.; Germing, U.; Götze, K.S.; Kiewe, P.; Mayer, K.; Chromik, J.; Radsak, M.; Wolff, T.; Zhang, X.; Laadem, A.; et al. Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS): A multicentre, open-label phase 2 dose-finding study with long-term extension study. Lancet Oncol. 2017, 18, 1338–1347. [Google Scholar] [CrossRef]
- Fenaux, P.; Platzbecker, U.; Mufti, G.J.; Garcia-Manero, G.; Buckstein, R.; Santini, V.; Díez-Campelo, M.; Finelli, C.; Cazzola, M.; Ilhan, O.; et al. Luspatercept in patients with lower-risk myelodysplastic syndromes. N. Engl. J. Med. 2020, 382, 140–151. [Google Scholar] [CrossRef]
- Squires, J.E. Indications for platelet transfusion in patients with thrombocytopenia. Blood Transfus. 2015, 13, 221–226. [Google Scholar]
- Napolitano, M.; Saccullo, G.; Marietta, M.; Carpenedo, M.; Castaman, G.; Cerchiara, E.; Chistolini, A.; Contino, L.; Falanga, A.; Federici, A.B.; et al. Platelet cut-off for anticoagulant therapy in thrombocytopenic patients with blood cancer and venous thromboembolism: An expert consensus. Blood Transfus. 2019, 17, 171–180. [Google Scholar] [PubMed]
- Manoharan, A.; Brighton, T.; Gemmell, R.; Lopez, K.; Moran, S.; Kyle, P. Platelet dysfunction in myelodysplastic syndromes: A clinicopathological study. Int. J. Hematol. 2002, 76, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Giagounidis, A.; Mufti, G.J.; Fenaux, P.; Sekeres, M.; Szer, J.; Platzbecker, U.; Kuendgen, A.; Gaidano, G.; Wiktor-Jedrzejczak, W.; Hu, K.; et al. Results of a randomized, double-blind study of romiplostim versus placebo in patients with low/intermediate-1–risk myelodysplastic syndrome and thrombocytopenia. Cancer 2014, 120, 1838–1846. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; Fenaux, P.; Sekeres, M.; Szer, J.; Platzbecker, U.; Kuendgen, A.; Gaidano, G.; Wiktor-Jedrzejczak, W.; Carpenter, N.; Mehta, B.; et al. Long-term follow-up for up to 5 years on the risk of leukaemic progression in thrombocytopenic patients with lower-risk myelodysplastic syndromes treated with romiplostim or placebo in a randomised double-blind trial. Lancet Haematol. 2018, 5, e117–e126. [Google Scholar] [CrossRef]
- Vicente, A.; Patel, B.A.; Gutierrez-Rodrigues, F.; Groarke, E.M.; Giudice, V.; Lotter, J.; Feng, X.; Kajigaya, S.; Weinstein, B.; Barranta, E.; et al. Eltrombopag monotherapy can improve hematopoiesis in patients with low to intermediate risk-1 myelodysplastic syndrome. Haematologica 2020, 105, 2785–2794. [Google Scholar] [CrossRef]
- Fenaux, P.; Giagounidis, A.; Selleslag, D.; Beyne-Rauzy, O.; Mufti, G.; Mittelman, M.; Muus, P.; Boekhorst, P.T.; Sanz, G.; Del Cañizo, C.; et al. A randomized phase 3 study of lenalidomide versus placebo in RBC transfusion-dependent patients with Low-/Intermediate-1-risk myelodysplastic syndromes with del5q. Blood 2011, 118, 3765–3776. [Google Scholar] [CrossRef] [PubMed]
- Jaedersten, M.; Saft, L.; Smith, A.; Kulasekararaj, A.; Pomplun, S.; Goehring, G.; Hedlund, A.; Hast, R.; Schlegelberger, B.; Porwit, A.; et al. TP53 mutations in low-risk myelodysplastic syndromes with del(5q) predict disease progression. J. Clin. Oncol. 2011, 29, 1971–1979. [Google Scholar] [CrossRef]
- Passweg, J.R.; Giagounidis, A.A.; Simcock, M.; Aul, C.; Dobbelstein, C.; Stadler, M.; Ossenkoppele, G.; Hofmann, W.-K.; Schilling, K.; Tichelli, A.; et al. Immunosuppressive therapy for patients with myelodysplastic syndrome: A prospective randomized multicenter Phase III trial comparing antithymocyte globulin plus cyclosporine with best supportive care—SAKK 33/99. J. Clin. Oncol. 2011, 29, 303–309. [Google Scholar] [CrossRef] [Green Version]
- Jabbour, E.; Short, N.J.; Montalban-Bravo, G.; Huang, X.; Bueso-Ramos, C.; Qiao, W.; Yang, H.; Zhao, C.; Kadia, T.; Borthakur, G.; et al. Randomized phase 2 study of low-dose decitabine vs low-dose azacitidine in lower-risk MDS and MDS/MPN. Blood 2017, 130, 1514–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Finelli, C.; Giagounidis, A.; Schoch, R.; Gattermann, N.; Sanz, G.; List, A.; et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase III study. Lancet Oncol. 2009, 10, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Lübbert, M.; Suciu, S.; Baila, L.; Rüter, B.H.; Platzbecker, U.; Giagounidis, A.; Selleslag, D.; LaBar, B.; Germing, U.; Salih, H.R.; et al. Low-Dose Decitabine Versus Best Supportive Care in Elderly Patients With Intermediate- or High-Risk Myelodysplastic Syndrome (MDS) ineligible for intensive chemotherapy: Final results of the randomized Phase III study of the european organisation for research and treatment of cancer leukemia group and the German MDS study group. J. Clin. Oncol. 2011, 29, 1987–1996. [Google Scholar] [PubMed]
- Nakamura, R.; Rodriguez, R.; Palmer, J.; Stein, A.; Naing, A.; Tsai, N.; Chang, K.; Slovak, M.L.; Bhatia, R.; Spielberger, R.; et al. Reduced-intensity conditioning for allogeneic hematopoietic stem cell transplantation with fludarabine and melphalan is associated with durable disease control in myelodysplastic syndrome. Bone Marrow Transplant. 2007, 40, 843–850. [Google Scholar] [CrossRef] [Green Version]
- Dombret, H.; Seymour, J.F.; Butrym, A.; Wierzbowska, A.; Selleslag, D.; Jang, J.H.; Kumar, R.; Cavenagh, J.D.; Schuh, A.C.; Candoni, A.; et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with 30% blasts. Blood 2015, 126, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itzykson, R.; Thépot, S.; Quesnel, B.; Dreyfus, F.; Beyne-Rauzy, O.; Turlure, P.; Vey, N.; Recher, C.; Dartigeas, C.; Legros, L.; et al. Prognostic factors for response and overall survival in 282 patients with higher-risk myelodysplastic syndromes treated with azacitidine. Blood 2011, 117, 403–411. [Google Scholar] [CrossRef] [Green Version]
- Kulasekararaj, A.G.; Smith, A.E.; Mian, S.A.; Mohamedali, A.M.; Krishnamurthy, P.; Lea, N.C.; Gäken, J.; Pennaneach, C.; Ireland, R.; Czepulkowski, B.; et al. TP53mutations in myelodysplastic syndrome are strongly correlated with aberrations of chromosome 5, and correlate with adverse prognosis. Br. J. Haematol. 2013, 160, 660–672. [Google Scholar] [CrossRef]
- Bejar, R.; Steensma, D.P. Recent developments in myelodysplastic syndromes. Blood 2014, 124, 2793–2803. [Google Scholar] [CrossRef] [Green Version]
- Kuendgen, A.; Müller-Thomas, C.; Lauseker, M.; Haferlach, T.; Urbaniak, P.; Schroeder, T.; Brings, C.; Wulfert, M.; Meggendorfer, M.; Hildebrandt, B.; et al. Efficacy of azacitidine is independent of molecular and clinical characteristics—An analysis of 128 patients with myelodysplastic syndromes or acute myeloid leukemia and a review of the literature. Oncotarget 2018, 9, 27882–27894. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Manero, G.; Griffiths, E.A.; Steensma, D.P.; Roboz, G.J.; Wells, R.A.; McCloskey, J.; Odenike, O.; DeZern, A.E.; Yee, K.; Busque, L.; et al. Oral cedazuridine/decitabine for MDS and CMML: A phase 2 pharmacokinetic/pharmacodynamic randomized crossover study. Blood 2020, 136, 674–683. [Google Scholar] [CrossRef]
- de Witte, T.; Bowen, D.; Robin, M.; Malcovati, L.; Niederwieser, D.; Yakoub-Agha, I.; Mufti, G.J.; Fenaux, P.; Sanz, G.; Martino, R.; et al. Allogeneic hematopoietic stem cell transplantation for MDS and CMML: Recommendations from an international expert panel. Blood 2017, 129, 1753–1762. [Google Scholar] [CrossRef]
- Lancet, J.E.; Cortes, J.; Hogge, D.E.; Tallman, M.S.; Kovacsovics, T.J.; Damon, L.E.; Komrokji, R.S.; Solomon, S.R.; Kolitz, J.E.; Cooper, M.; et al. Phase 2 trial of CPX-351, a fixed 5:1 molar ratio of cytarabine/daunorubicin, vs cytarabine/daunorubicin in older adults with untreated AML. Blood 2014, 123, 3239–3246. [Google Scholar] [CrossRef] [PubMed]
- Ossenkoppele, G.J.; Graveland, W.J.; Sonneveld, P.; Daenen, S.M.G.J.; Biesma, D.H.; Verdonck, L.F.; Schaafsma, M.R.; Westveer, P.H.M.; Peters, G.J.; Noordhuis, P.; et al. The value of fludarabine in addition to ARA-C and G-CSF in the treatment of patients with high-risk myelodysplastic syndromes and AML in elderly patients. Blood 2004, 103, 2908–2913. [Google Scholar] [CrossRef] [Green Version]
- Sorror, M.L.; Maris, M.B.; Storb, R.; Baron, F.; Sandmaier, B.M.; Maloney, D.G.; Storer, B. Hematopoietic cell transplantation (HCT)-specific comorbidity index: A new tool for risk assessment before allogeneic HCT. Blood 2005, 106, 2912–2919. [Google Scholar] [CrossRef] [Green Version]
- Cutler, C.S.; Lee, S.J.; Greenberg, P.; Deeg, H.J.; Pérez, W.S.; Anasetti, C.; Bolwell, B.J.; Cairo, M.S.; Gale, R.P.; Klein, J.P.; et al. A decision analysis of allogeneic bone marrow transplantation for the myelodysplastic syndromes: Delayed transplantation for low-risk myelodysplasia is associated with improved outcome. Blood 2004, 104, 579–585. [Google Scholar] [CrossRef]
- Bejar, R. Clinical and genetic predictors of prognosis in myelodysplastic syndromes. Haematologica 2014, 99, 956–964. [Google Scholar] [CrossRef] [Green Version]
- Della Porta, M.G.; Gallì, A.; Bacigalupo, A.; Zibellini, S.; Bernardi, M.; Rizzo, E.; Allione, B.; Van Lint, M.T.; Pioltelli, P.; Marenco, P.; et al. Clinical effects of driver somatic mutations on the outcomes of patients with myelodysplastic syndromes treated with allogeneic hematopoietic stem-cell transplantation. J. Clin. Oncol. 2016, 34, 3627–3637. [Google Scholar] [CrossRef]
- Lindsley, R.C.; Saber, W.; Mar, B.; Redd, R.; Wang, T.; Haagenson, M.D.; Grauman, P.V.; Hu, Z.-H.; Spellman, S.R.; Lee, S.J.; et al. Prognostic mutations in myelodysplastic syndrome after stem-cell transplantation. N. Engl. J. Med. 2017, 376, 536–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshizato, T.; Nannya, Y.; Atsuta, Y.; Shiozawa, Y.; Iijima-Yamashita, Y.; Yoshida, K.; Shiraishi, Y.; Suzuki, H.; Nagata, Y.; Sato, Y.; et al. Genetic abnormalities in myelodysplasia and secondary acute myeloid leukemia: Impact on outcome of stem cell transplantation. Blood 2017, 129, 2347–2358. [Google Scholar] [CrossRef] [PubMed]
- Gui, G.; Dillon, L.W.; Hourigan, C.S. Measurable residual disease before reduced-intensity allogeneic transplantation in patients with myeloid malignancy. J. Clin. Oncol. 2021, JCO2100255. [Google Scholar] [CrossRef]
- Kröger, N.; Iacobelli, S.; Franke, G.-N.; Platzbecker, U.; Uddin, R.; Hübel, K.; Scheid, C.; Weber, T.; Robin, M.; Stelljes, M.; et al. Dose-reduced versus standard conditioning followed by allogeneic stem-cell transplantation for patients with myelodysplastic syndrome: A prospective randomized phase III Study of the EBMT (RICMAC Trial). J. Clin. Oncol. 2017, 35, 2157–2164. [Google Scholar] [CrossRef]
- Festuccia, M.; Baker, K.; Gooley, T.A.; Sandmaier, B.M.; Deeg, H.J.; Scott, B.L. Hematopoietic cell transplantation in myelodysplastic syndromes after treatment with hypomethylating agents. Biol. Blood Marrow Transplant. 2017, 23, 1509–1514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Della Porta, M.; Platzbecker, U.; Santini, V.; Garcia-Manero, G.; Komrokji, R.S.; Ito, R.; Fenaux, P. The commands trial: A Phase 3 study of the efficacy and safety of luspatercept versus epoetin alfa for the treatment of anemia due to IPSS-R very low-, low-, or intermediate-risk MDS in erythropoiesis stimulating agent-naive patients who require RBC transfusions. Blood 2020, 136, 1–2. [Google Scholar]
- Chen, N.; Hao, C.; Peng, X.; Lin, H.; Yin, A.; Hao, L.; Tao, Y.; Liang, X.; Liu, Z.; Xing, C.; et al. Roxadustat for anemia in patients with kidney disease not receiving dialysis. N. Engl. J. Med. 2019, 381, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Landry, J.-R.; Bonadies, N.; Kinston, S.; Knezevic, K.; Wilson, N.K.; Oram, S.H.; Janes, M.; Piltz, S.; Hammett, M.; Carter, J.; et al. Expression of the leukemia oncogene Lmo2 is controlled by an array of tissue-specific elements dispersed over 100 kb and bound by Tal1/Lmo2, Ets, and Gata factors. Blood 2009, 113, 5783–5792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bock, O.; Serinsöz, E.; Schlue, J.; Kreipe, H. Different expression levels of the telomerase catalytic subunit hTERT in myeloproliferative and myelodysplastic diseases. Leuk. Res. 2004, 28, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Briatore, F.; Barrera, G.; Pizzimenti, S.; Toaldo, C.; Della Casa, C.; Laurora, S.; Pettazzoni, P.; Dianzani, M.U.; Ferrero, D. Increase of telomerase activity and hTERT expression in myelodysplastic syndromes. Cancer Biol. Ther. 2009, 8, 883–889. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.; Qian, Y.; Yang, L. Telomerase, hTERT and splice variants in patients with myelodysplastic syndromes. Leuk. Res. 2014, 38, 830–835. [Google Scholar] [CrossRef]
- Herbert, B.-S.; Gellert, G.C.; Hochreiter, A.E.; Pongracz, K.; Wright, W.E.; Zielinska, D.; Chin, A.C.; Harley, C.B.; Shay, J.W.; Gryaznov, S.M. Lipid modification of GRN163, an N3′ → P5′ thio-phosphoramidate oligonucleotide, enhances the potency of telomerase inhibition. Oncogene 2005, 24, 5262–5268. [Google Scholar] [CrossRef] [Green Version]
- Asai, A.; Oshima, Y.; Yamamoto, Y.; Uochi, T.-A.; Kusaka, H.; Akinaga, S.; Yamashita, Y.; Pongracz, K.; Pruzan, R.; Wunder, E.; et al. A novel telomerase template antagonist (GRN163) as a potential anticancer agent. Cancer Res. 2003, 63, 3931–3939. [Google Scholar]
- Steensma, D.P.; Fenaux, P.; Van Eygen, K.; Raza, A.; Santini, V.; Germing, U.; Font, P.; Diez-Campelo, M.; Thepot, S.; Vellenga, E.; et al. Imetelstat achieves meaningful and durable transfusion independence in high transfusion–burden patients with lower-risk myelodysplastic syndromes in a Phase II study. J. Clin. Oncol. 2021, 39, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P. An RNA bestiary in splicing-mutant MDS. Blood 2018, 132, 1217–1219. [Google Scholar] [CrossRef]
- Steensma, D.P.; Maris, M.B.; Yang, J.; Donnellan, W.B.; Brunner, A.M.; McMasters, M.; Greenberg, P.; Komrokji, R.S.; Klimek, V.M.; Goldberg, J.M.; et al. H3B-8800-G0001-101: A first in human phase I study of a splicing modulator in patients with advanced myeloid malignancies. J. Clin. Oncol. 2017, 35, TPS7075. [Google Scholar] [CrossRef]
- Steensma, D.P.; Wermke, M.; Klimek, V.M.; Greenberg, P.L.; Font, P.; Komrokji, R.S.; Yang, J.; Brunner, A.M.; Carraway, H.E.; Ades, L.; et al. Results of a clinical trial of H3B-8800, a splicing modulator, in patients with Myelodysplastic Syndromes (MDS), Acute Myeloid Leukemia (AML) or Chronic Myelomonocytic Leukemia (CMML). Blood 2019, 134, 673. [Google Scholar] [CrossRef]
- Garcia-Manero, G. Highlights in myelodysplastic syndromes from the 60th American Society of Hematology Annual Meeting: Commentary. Clin. Adv. Hematol. Oncol. HO 2019, 5, 16–19. [Google Scholar]
- Astex and Otsuka Announce Results of Phase 3 ASTRAL-2 and ASTRAL-3 Studies of Guadecitabine (SGI-110) in Patients with Previously Treated Acute Myeloid Leukemia (AML) and Myelodysplastic Syndromes or Chronic Myelomonocytic Leukemia (MDS/CMML). 2020. Available online: https://astx.com/astex-and-otsuka-announce-results-of-phase-3-astral-2-and-astral-3-studies-of-guadecitabine-sgi-110-in-patients-with-previously-treated-acute-myeloid-leukemia-aml-and-myelodysplastic-syndromes-or/ (accessed on 29 April 2021).
- Ramsey, H.E.; Oganesian, A.; Gorska, A.E.; Fuller, L.; Arrate, M.; Boyd, K.; Keer, H.; Azab, M.; Savona, M.R. Oral azacitidine and cedazuridine approximate parenteral azacitidine efficacy in Murine Model. Target. Oncol. 2020, 15, 231–240. [Google Scholar] [CrossRef] [Green Version]
- Saunthararajah, Y. Key clinical observations after 5-azacytidine and decitabine treatment of myelodysplastic syndromes suggest practical solutions for better outcomes. Hematology 2013, 2013, 511–521. [Google Scholar] [CrossRef] [Green Version]
- Savona, M.R.; Kolibaba, K.; Conkling, P.; Kingsley, E.C.; Becerra, C.; Morris, J.C.; Rifkin, R.M.; Laille, E.; Kellerman, A.; Ukrainskyj, S.M.; et al. Extended dosing with CC-486 (oral azacitidine) in patients with myeloid malignancies. Am. J. Hematol. 2018, 93, 1199–1206. [Google Scholar] [CrossRef]
- Sallman, D.A.; Al Malki, M.; Asch, A.S.; Lee, D.J.; Kambhampati, S.; Donnellan, W.B.; Bradley, T.J.; Vyas, P.; Jeyakumar, D.; Marcucci, G.; et al. Tolerability and efficacy of the first-in-class anti-CD47 antibody magrolimab combined with azacitidine in MDS and AML patients: Phase Ib results. J. Clin. Oncol. 2020, 38, 7507. [Google Scholar] [CrossRef]
- Brunner, A.M.; Esteve, J.; Porkka, K.; Knapper, S.; Vey, N.; Scholl, S.; Garcia-Manero, G.; Wermke, M.; Janssen, J.; Traer, E.; et al. Efficacy and Safety of Sabatolimab (MBG453) in Combination with Hypomethylating Agents (HMAs) in Patients with Acute Myeloid Leukemia (AML) and high-risk myelodysplastic syndrome (HR-MDS): Updated results from a Phase 1b study. Blood 2020, 136, 1–2. [Google Scholar] [CrossRef]
- Trudel, G.C.; Howes, A.J.; Jeste, N.; Tryon, J.J.; Xiu, L.; Kane, C.; Nottage, K. CULMINATE: A phase II study of cusatuzumab + azacitidine in patients with newly diagnosed AML, ineligible for intensive chemotherapy. J. Clin. Oncol. 2020, 38, TPS7565. [Google Scholar] [CrossRef]
- Dinardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Ball, B.; Famulare, C.A.; Stein, E.M.; Tallman, M.S.; Derkach, A.; Roshal, M.; Gill, S.I.; Manning, B.M.; Koprivnikar, J.; McCloskey, J.; et al. Venetoclax and hypomethylating agents (HMAs) induce high response rates in MDS, including patients after HMA therapy failure. Blood Adv. 2020, 4, 2866–2870. [Google Scholar] [CrossRef]
- Wei, A.H.; Garcia, J.S.; Borate, U.; Fong, C.Y.; Baer, M.R.; Nolte, F.; Peterlin, P.; Jurcic, J.G.; Garcia-Manero, G.; Hong, W.-J.; et al. A Phase 1b study evaluating the safety and efficacy of venetoclax in combination with azacitidine in treatment-naïve patients with higher-risk myelodysplastic syndrome. Blood 2019, 134, 568. [Google Scholar] [CrossRef]
- Zeidan, A.M.; Pollyea, D.A.; Garcia, J.S.; Brunner, A.; Roncolato, F.; Borate, U.; Odenike, O.; Bajel, A.R.; Watson, A.M.; Götze, K.; et al. A Phase 1b study evaluating the safety and efficacy of venetoclax as monotherapy or in combination with azacitidine for the treatment of relapsed/refractory myelodysplastic syndrome. Blood 2019, 134, 565. [Google Scholar] [CrossRef]
- Ades, L.; Watts, J.M.; Radinoff, A.; Arnan, M.; Cerrano, M.; Lopez, P.F.; Zeidner, J.F.; Diez-Campelo, M.; Graux, C.; Liesveld, J.; et al. Phase II study of pevonedistat (P) + azacitidine (A) versus A in patients (pts) with higher-risk myelodysplastic syndromes (MDS)/chronic myelomonocytic leukemia (CMML), or low-blast acute myelogenous leukemia (LB AML) (NCT02610777). J. Clin. Oncol. 2020, 38, 7506. [Google Scholar] [CrossRef]
- Sekeres, M.A.; Fram, R.J.; Hua, Z.; Ades, L. Phase 3 study of first line pevonedistat (PEV) + azacitidine (AZA) versus single-agent AZA in patients with higher-risk myelodysplastic syndromes (HR MDS), chronic myelomonocytic leukemia (CMML) or low-blast acute myelogenous leukemia (AML). J. Clin. Oncol. 2018, 36, TPS7077. [Google Scholar] [CrossRef]
- Maslah, N.; Salomao, N.; Drevon, L.; Verger, E.; Partouche, N.; Ly, P.; Aubin, P.; Naoui, N.; Schlageter, M.-H.; Bally, C.; et al. Synergistic effects of PRIMA-1Met (APR-246) and 5-azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia. Haematologica 2019, 105, 1539–1551. [Google Scholar] [CrossRef] [Green Version]
- Sallman, D.A. To target the untargetable: Elucidation of synergy of APR-246 and azacitidine in TP53 mutant myelodysplastic syndromes and acute myeloid leukemia. Haematologica 2020, 105, 1470–1472. [Google Scholar] [CrossRef]
- Cluzeau, T.; Sebert, M.; Rahmé, R.; Cuzzubbo, S.; Walter-Petrich, A.; Che, J.L.; Peterlin, P.; Beve, B.; Attalah, H.; Chermat, F.; et al. APR-246 Combined with Azacitidine (AZA) in TP53 Mutated Myelodysplastic Syndrome (MDS) and Acute Myeloid Leukemia (AML). A Phase 2 study by the groupe francophone des myélodysplasies (GFM). Blood 2019, 134, 677. [Google Scholar] [CrossRef]
- Sallman, D.A.; DeZern, A.E.; Steensma, D.P.; Sweet, K.L.; Cluzeau, T.; Sekeres, M.M.A.; Garcia-Manero, G.; Roboz, G.J.; McLemore, M.A.F.; McGraw, K.L.; et al. Phase 1b/2 Combination Study of APR-246 and Azacitidine (AZA) in Patients with TP53 mutant Myelodysplastic Syndromes (MDS) and Acute Myeloid Leukemia (AML). Blood 2018, 132, 3091. [Google Scholar] [CrossRef]
- Dinardo, C.D.; Stein, E.M.; De Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable remissions with ivosidenib inIDH1-mutated relapsed or refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef]
- Stein, E.M.; DiNardo, C.D.; Fathi, A.T.; Mims, A.S.; Pratz, K.W.; Savona, M.R.; Stein, A.S.; Stone, R.M.; Winer, E.S.; Seet, C.S.; et al. Ivosidenib or enasidenib combined with intensive chemotherapy in patients with newly diagnosed AML: A phase 1 study. Blood 2021, 137, 1792–1803. [Google Scholar] [CrossRef] [PubMed]
- Roboz, G.J.; Dinardo, C.D.; Stein, E.M.; De Botton, S.; Mims, A.S.; Prince, G.T.; Altman, J.K.; Arellano, M.L.; Donnellan, W.; Erba, H.P.; et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood 2020, 135, 463–471. [Google Scholar] [CrossRef] [Green Version]
- Stein, E.M.; Fathi, A.T.; DiNardo, C.D.; Pollyea, D.A.; Roboz, G.J.; Collins, R.; Sekeres, M.A.; Stone, R.M.; Attar, E.C.; Frattini, M.G.; et al. Enasidenib in patients with mutant IDH2 myelodysplastic syndromes: A phase 1 subgroup analysis of the multicentre, AG221-C-001 trial. Lancet Haematol. 2020, 7, e309–e319. [Google Scholar] [CrossRef]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Perl, A.E.; Martinelli, G.; Cortes, J.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef]
- Swaminathan, M.; Kantarjian, H.M.; Levis, M.; Guerra, V.; Borthakur, G.; Alvarado, Y.; DiNardo, C.D.; Kadia, T.; Garcia-Manero, G.; Ohanian, M.; et al. A phase I/II study of the combination of quizartinib with azacitidine or low-dose cytarabine for the treatment of patients with acute myeloid leukemia and myelodysplastic syndrome. Haematologica 2021. [Google Scholar] [CrossRef]
- Stevens, B.M.; Zhang, W.; Pollyea, D.A.; Winters, A.; Gutman, J.; Smith, C.; Budde, E.; Forman, S.J.; Jordan, C.T.; Purev, E. CD123 CAR T cells for the treatment of myelodysplastic syndrome. Exp. Hematol. 2019, 74, 52–63. [Google Scholar] [CrossRef] [Green Version]
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduwilt, M.J.; Advani, A.S.; Godwin, J.E.; Arellano, M.L.; Sweet, K.L.; Emadi, A.; et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood 2021, 137, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.N.; Ferrari, V.; Tarke, A.; Fields, H.; Ferrari, L.; Ferrari, F.; McCarthy, C.L.; Sanchez, A.P.; Vitiello, A.; Lane, T.A.; et al. Adoptive transfer of neoantigen-specific T-cell therapy is feasible in older patients with higher-risk myelodysplastic syndrome. Cytotherapy 2021, 23, 236–241. [Google Scholar] [CrossRef]
- Califf, R.M.; Peterson, E.D.; Gibbons, R.J.; Garson, A.; Brindis, R.G.; Beller, G.A.; Smith, S.C. Integrating quality into the cycle of therapeutic development. J. Am. Coll. Cardiol. 2002, 40, 1895–1901. [Google Scholar] [CrossRef]
- Kimura, K.; Tabe, Y.; Ai, T.; Takehara, I.; Fukuda, H.; Takahashi, H.; Naito, T.; Komatsu, N.; Uchihashi, K.; Ohsaka, A. A novel automated image analysis system using deep convolutional neural networks can assist to differentiate MDS and AA. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buoro, S.; Moioli, V.; Seghezzi, M.; Previtali, G.; Alessio, M.G.; Lopez, R.S.; Ortolani, C.; Ottomano, C.; Lippi, G. Evaluation and comparison of automated hematology analyzer, flow cytometry, and digital morphology analyzer for monocyte counting. Int. J. Lab. Hematol. 2018, 40, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Stephens, L.; Bevins, N.J.; Bengtsson, H.-I.; Broome, H.E. Comparison of different small clinical hematology laboratory configurations with focus on remote smear imaging. Arch. Pathol. Lab. Med. 2019, 143, 1234–1245. [Google Scholar] [CrossRef] [Green Version]
- Nazha, A.; Sekeres, M.A.; Bejar, R.; Rauh, M.J.; Othus, M.; Komrokji, R.S.; Barnard, J.; Hilton, C.B.; Kerr, C.M.; Steensma, D.P.; et al. Genomic biomarkers to predict resistance to hypomethylating agents in patients with myelodysplastic syndromes using artificial intelligence. JCO Precis. Oncol. 2019, 3, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Zhao, R.; Awada, H.; Kerr, C.M.; Mirzaev, I.; Kongkiatkamon, S.; Nazha, A.; Makishima, H.; Radivoyevitch, T.; Scott, J.G.; et al. Machine learning demonstrates that somatic mutations imprint invariant morphologic features in myelodysplastic syndromes. Blood 2020, 136, 2249–2262. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Gene | Approx. Frequency (%) |
|---|---|---|
| RNA-splicing factors | SF3B1 * | 25–30 |
| SRSF2 | 10–15 | |
| U2AF1 | 5–10 | |
| ZRSR2 | 5 | |
| SF3A1 | 1–2 | |
| SF1 | 1–2 | |
| U2AF65 | 1–2 | |
| PRPF40B | 1–2 | |
| Epigenetic regulators | TET2 | 20–25 |
| DNMT3A | 15 | |
| ASXL1 | 10–15 | |
| EZH2 | 5 | |
| IDH1 | 1–2 | |
| IDH2 | 1–2 | |
| Transcription factors | RUNX1♯ | 10–20 |
| SETBP1 | 1–2 | |
| ETV6♯ | 2 | |
| CEBPA♯ | 1–2 | |
| GATA2♯ | 1–2 | |
| Cell-cycle regulators | TP53 | 5–10 |
| PTEN | 1 | |
| Cohesin complex factors | STAG1 | 1 |
| STAG2 | 6 | |
| RAD21 | 1 | |
| Cell-signaling molecules | NRAS/KRAS | 5–10 |
| NPM1 | 1–2 | |
| JAK2 | 1–2 | |
| FLT3 | 2 | |
| CBL | 1–2 |
|
|
|
|
| If relevant cytopenia in PB is present, consider CCUS (minimal diagnostic criteria for MDS are not fulfilled) or MDS (minimal diagnostic criteria are fulfilled). Annual risk of progression to hematologic neoplasm: 0.5–1% |
| CHIP [20,21,22] | AA [25,26] | CCUS | MDS [10,11] | |
|---|---|---|---|---|
| Frequency | ~2% (40–49 years) ~3% (50–59 years) ~6% (60–69 years) ~10% (70–79 years) ~15% (80–89 years) | 19–47% (median age 44 years) | 35% of ICUS | 70–80% (median age 72 years) |
| Most common mutations | DNMT3A, TET2, ASXL1, JAK2 | Younger patients: PIGA BCOR/BCORL1 Older patients: DNMT3A, ASXL1 | TET2, DNMT3A, ASXL1, TP53 | SF3B1, TET2, ASXL1, DNMT3A, SRSF2, RUNX1 |
| Prognosis | Increased risk for hematological neoplasm, coronary heart disease, ischemic stroke, diabetes mellitus type 2 | Good prognosis: PIGA, BCOR/BCORL1 Poor prognosis: ASXL1, DNMT3A | Increased risk for MDS/AML | Good prognosis: SF3B1 Poor prognosis: TP53, ASXL1, RUNX1, ETV6, EZH2 Neutral: all others |
| Mean VAF (%) | 9% | 20% (10% in 40% of patients) | 30% | 30% |
| Mean number of mutations per patient | 1 (93% of individuals) | 1 (64–90% of patients) | 1 | 3 (range 0–12) |
| Subtype 1 | Number of Dysplastic Lineages | Number of Cytopenic Lineages 2 | % RS of All Erythroid Cells in BM | % Blasts in PB or BM AR: Auer Rods | Conventional Cytogenetics | |||
|---|---|---|---|---|---|---|---|---|
| wtSF3B1 | mSF3B1 | BM | PB | AR | ||||
| MDS-SLD | 1 | 1 or 2 | 15 | 5 | 5 | 1 | - | |
| MDS-MLD | 2 or 3 | 1–3 | 15 | 5 | 5 | 1 | - | |
| MDS-RS-SLD | 1 | 1 or 2 | ≥15 | ≥5 | 5 | 1 | - | |
| MDS-RS-MLD | 2 or 3 | 1–3 | ≥15 | ≥5 | 5 | 1 | - | |
| MDS del(5q) | 1–3 | 1 or 2 | n.a. | n.a. | 5 | 1 | - | Isolated del(5q) with or without one additional cytogenetic aberration without del(7) or −7 |
| MDS-EB-1 | 0–3 | 1–3 | n.a. | n.a. | 5–9 | 2–4 | - | |
| MDS-EB-2 | 0–3 | 1–3 | n.a. | n.a. | 10–19 | 5–19 | + | |
| MDS-U | 15 | 5 | 5 | 1 | - | |||
| (a) 1% blasts in PB | 1–3 | 1–3 | n.a. | n.a. | 5 | 1 3 | - | |
| (b) SLD with pancytopenia | 1 | 3 | n.a. | n.a. | 5 | 1 | - | |
| (c) defining cytogenetic aberration | 0 | 1–3 | 15 4 | n.a. | 5 | 1 | - | MDS defining cytogenetic aberration |
| RCC | 1–3 | 1–3 | 15 | ≤5 | 5 | 1 | - | |
| Peripheral Blood |
|
|
| Bone marrow |
|
|
| Criteria | Diagnostic Test |
|---|---|
| 1. Mandatory criteria (both have to be fulfilled) | |
| Persistent cytopenia(s) for more than 4 months * | PB counts and morphological assessment |
| Exclusion of other disease(s) that may cause cytopenia/dysplasia | BM aspirate and biopsy, cytogenetics, flow cytometry, molecular genetics, other relevant investigations ** |
| 2. MDS-defining criteria (at least one has to be fulfilled) | |
| Morphological criteria of dysplasia 10% in at least one cell lineages investigated in the BM | PB, BM aspirate and biopsy |
| Blasts 5–19% in BM or 2–19% in PB | PB, BM aspirate and biopsy |
| Ring sideroblasts ≥15% or ≥5% with SF3B1 mutation | Iron staining, sequencing |
| MDS-defining cytogenetic alterations *** | Conventional metaphase cytogenetics, interphase fluorescence in situ hybridization, array comparative genomic hybridization |
| 3. Co-criteria (for patients with 1. but not 2., two have to be fulfilled) | |
| Abnormal findings in histologic and/or immunohistochemical studies of supporting the diagnosis of MDS **** | BM biopsy sections with immunohistochemistry |
| Abnormal immunophenotype of BM cells with aberrant immunophenotype indicative for a monoclonal population | Flow cytometry |
| Clonality of myeloid cells revealing MDS-related mutations | Molecular genetics, next generation sequencing |
| Requisite Criteria | Score |
|---|---|
| BM blasts AND/OR CD34+ cells ≥5% | 2 |
| BM blasts AND/OR CD34+ cells 2–4% | 1 |
| Fibrosis grade 2–3 | 1 |
| Dysmegakaryopoiesis | 1 |
| Co-criteria | |
| Ring sideroblasts ≥15% | 2 |
| Ring sideroblasts 5–14% * | 1 |
| Severe dysgranulopoiesis | 1 |
| Karyotype (co-criterion) | |
| Presumptive cytogenetic abnormality * | 2 |
| Somatic mutation (co-criterion) | |
| Specific high-risk mutation pattern ** | 1 |
| Compound | Study Design | Efficacy * | Safety * | NCT |
|---|---|---|---|---|
| Erythropoiesis maturating agents | ||||
| TGFβi luspatercept | Phase 3 (ongoing), open-label, randomized study: efficacy and safety of luspatercept (ACE-536) versus epoetin alpha for the treatment of anemia due to IPSS-R very low, low or intermediate risk according to IPSS-R MDS in ESA naïve subjects requiring red blood transfusions (COMMANDS) | ongoing 38% vs. 13% TI for 8 weeks or longer | AE:
| NCT03682536 [125] |
| Hypoxia-inducible factor prolyl hydroxylase inhibitor (HIF-PHi) | ||||
| roxadustat (FG-4592) | Phase 3, randomized double-blind placebo-controlled study investigating the efficacy and safety of roxadustat (FG-4592) for treatment of anemia in patients with lower risk MDS with low RBC transfusion burden | ongoing | ongoing | NCT03263091 [127] |
| Telomerase inhibitor | ||||
| imetelstat | Phase 2/3 (phase 3 part ongoing), double-blind, randomized study to evaluate imetelstat (GRN163L) versus placebo in transfusion-dependent subjects with IPSS low or intermediate-1 risk MDS that is relapsed/refractory to ESA treatment (IMERGE) | TI in 8- and 24-week: 37%, respectively 23%, median duration of 65 weeks phase 3 ongoing | AE:
| NCT02598661 [133] |
| Spliceosome modulators | ||||
| H3B-8800 | Phase 1 (ongoing), open-label trial to evaluate the safety, pharmacokinetics and pharmacodynamics of splicing modulator H3B-8800 for subjects with MDS, AML and CMML | decreased RBC or TC requirement in 14% | AE:
| NCT02841540 [135,136] |
| DNA-methylation | ||||
| guadecitabine (SGI-110) | Phase 1/2, dose escalation, dose escalation, randomized study of two regimens of SGI-110, in subjects with intermediate or high-risk MDS or AML | ORR 40% with 60 mg/m2 d1-d5 q28d ORR 55% with 90 mg/m2 d1-d5 q28d | AE:
| NCT01261312 [112] |
| guadecitabine (SGI-110) | Phase 3, randomized, open-label study of SGI-110 versus treatment choice in adults with MDS or CMML previously treated with hypomethylating agents | primary endpoint (OS): no statistically significant improvement secondary endpoints: analysis ongoing | comparable to previous studies | NCT02907359 [138] |
| oral AZA/ cedazuridine (CDZ) (ASTX030) | Phase1 (ongoing), multi-phase, dose-escalation followed by an open-label, randomized, crossover study of oral ASTX030 versus subcutaneous azacytidine in subjects with MDS, CMML or AML | parenteral and oral AZA + CDZ similar pharmacokinetic profiles and efficacy against human AML cells | n.a. | NCT04256317 [139] |
| oral DEC/CDZ (ASTX727) | Phase1/2 pharmacokinetic guided dose-escalation and dose-confirmation study of ASTX727 (oral cytidine deaminase inhibitor E7727 with oral decitabine) in subjects with MDS | no difference in pharmakokinetics, pharmacodynamics and efficacy between p.o. and i.v. formulations | similar between p.o. und i.v. | NCT02103478 [112] |
| Immune checkpoint inhibitors | ||||
| CD47-Ab magrolimab | Phase 1b Trial of magrolimab monotherapy versus in combination with azacytidine in patients with hematological malignancies | TI: 58% (MDS), 64% (AML) objective response (CR, marrow CR, HI): 91% (MDS) | AE:
| NCT03248479 [142] |
| TIM3-Ab sabatolimab (MBG453) | Phase 1b, multi-arm, open-label study of PDR001 and/or MBG453 in combination with decitabine in patients with AML or high risk MDS | AML: ORR 41.2% MDS: ORR 62.9% | AE: Grad 3/4 (AML/MDS):
| NCT03066648 [143] |
| Proapoptotic agents | ||||
| BCL-2i venetoclax + AZA | Phase 1b/2 (ongoing), dose escalation study evaluating the safety and pharmacokinetics of venetoclax in combination with azacytidine in subjects with treatment-naïve higher-risk MDS | 18 months: OS 74%, HI 50% | AE, grade 3/4:
| NCT02942290 [147] |
| BCL-2iv enetoclax + AZA | Phase 1b study (ongoing) evaluating the safety and pharmacokinetics of venetoclax as a single-agent and in combination with azacytidine in subjects with relapsed/refractory MDS | median FU 4.7 months:
| Grade 3/4:
| NCT02966782 [148] |
| NEDD8i pevonedistat | Phase 2, randomized, controlled, open-label, study of the efficacy and safety of pevonedistat plus azacytidine versus single-agent azacytidine in patients with higher-risk MDS, CMML and low-blast AML | Combination vs. single-arm
| Grade 3/4 adverse events similar (69% vs. 63% in single-arm)
| NCT02610777 [149] |
| NEDD8i pevonedistat | Phase 3, randomized, controlled, open-label, study of pevonedistat plus azacytidine versus single-agent azacytidine as first-line treatment for patients with Higher-Risk MDS, CMML or low-blast AML (PANTHER) | ongoing | ongoing | NCT03268954 [150] |
| TP53 reconforming agents | ||||
| eprenetapopt (APR-246) | Phase 1b/2 (ongoing) study to evaluate the safety and efficacy of APR-246 in combination with azacytidine for the treatment of TP53 mutant myeloid neoplasms | ongoing | Grade 3/4 (phase 1b):
| NCT03588078 [154] |
| Epigenetic inhibitors | ||||
| IDH1i ivosidenib (AG-120) | Phase 1, open-label, dose-escalation and expansion, safety, pharmacokinetic, pharmacodynamic, and clinical activity study of orally administered AG-120 in subjects with advanced hematologic malignancies with an IDH1 mutation | CR2 plus CRh2: 42.4% CR2 30.3% FU 23.5 months: median OS 12.6 months TI in 9 of 21 TD (42.9%) IDH1 mutation clearance in 9/14 patients | AE:
| NCT02074839 [157] |
| IDH2i enasidenib (AG-221) | Phase 1/2 (ongoing), open-label, dose-escalation and expansion, safety, pharmacokinetic, pharmacodynamic and clinical activity study of orally administered AG-221 in subjects with advanced hematologic malignancies with an IDH2 mutation | phase 1: ORR 53% median duration 9.2 months OS 16.9 months phase 2 ongoing | Grade 3/4 (phase 1):
| NCT01915498 [158] |
| RAS-pathways inhibitors | ||||
| FLT3i quizartinib (AC220) | Phase 1/2 study of the combination of quizartinib (AC220) with 5-azacytidine or low-dose cytarabine for the treatment of patients with AML and MDS | quizartinib/AZA-arm vs quizartinib/LD-AraC-arm:
| AE grade 3/4, quizartinib/AZA-arm vs. quizartinib/LD-AraC-arm:
| NCT01892371 [161] |
| Bispecific antibodies | ||||
| CD3/CD123 Flotetuzumab (MGD006) | Phase 1/2, first in human, dose escalation study of MGD006, a CD123 × CD3 DART® bi-specific antibody based molecule, in patients with relapsed or refractory AML or intermediate-2/high risk MDS | CR/CRh: 26.7% median OS 10.2 months ORR (CR/CRh/CRi): 30% | most frequent AE:
| NCT02152956 [163] |
| CD3/CD33 (AMG 330) | A Phase 1 first-in-human study evaluating the safety, tolerability, pharmacokinetics, pharmacodynamics and efficacy of AMG 330 administered as continuous intravenous infusion in subjects with myeloid malignancies | ongoing | ongoing | NCT02520427 [154] |
| Domain 1 Diagnosis (n = 14) | Domain 2 Therapy (n = 8) | Domain 3 Provider/Infrastructural Characteristics (n = 7) |
|---|---|---|
Diagnostic work-up:
| Supportive care:
| Personnel:
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chanias, I.; Stojkov, K.; Stehle, G.T.; Daskalakis, M.; Simeunovic, H.; Njue, L.M.; Schnegg-Kaufmann, A.S.; Porret, N.A.; Allam, R.; Rao, T.N.; et al. Myelodysplastic Syndromes in the Postgenomic Era and Future Perspectives for Precision Medicine. Cancers 2021, 13, 3296. https://doi.org/10.3390/cancers13133296
Chanias I, Stojkov K, Stehle GT, Daskalakis M, Simeunovic H, Njue LM, Schnegg-Kaufmann AS, Porret NA, Allam R, Rao TN, et al. Myelodysplastic Syndromes in the Postgenomic Era and Future Perspectives for Precision Medicine. Cancers. 2021; 13(13):3296. https://doi.org/10.3390/cancers13133296
Chicago/Turabian StyleChanias, Ioannis, Kristina Stojkov, Gregor Th. Stehle, Michael Daskalakis, Helena Simeunovic, Linet Muthoni Njue, Annatina S. Schnegg-Kaufmann, Naomi A. Porret, Ramanjaneyulu Allam, Tata Nageswara Rao, and et al. 2021. "Myelodysplastic Syndromes in the Postgenomic Era and Future Perspectives for Precision Medicine" Cancers 13, no. 13: 3296. https://doi.org/10.3390/cancers13133296
APA StyleChanias, I., Stojkov, K., Stehle, G. T., Daskalakis, M., Simeunovic, H., Njue, L. M., Schnegg-Kaufmann, A. S., Porret, N. A., Allam, R., Rao, T. N., Benz, R., Ruefer, A., Schmidt, A., Adler, M., Rovo, A., Balabanov, S., Stuessi, G., Bacher, U., & Bonadies, N., on behalf of the Swiss MDS Study Group. (2021). Myelodysplastic Syndromes in the Postgenomic Era and Future Perspectives for Precision Medicine. Cancers, 13(13), 3296. https://doi.org/10.3390/cancers13133296