

Enhancing B-Cell Malignancies—On Repurposing Enhancer Activity towards Cancer


Abstract
:Simple Summary
Abstract
1. Introduction
2. Immunoglobulin Heavy Chain Enhancers in B-Cell Malignancies
2.1. Structure and Function of IGH Enhancers in Normal B Cells
2.1.1. Intronic Eµ Enhancer
2.1.2. The 3′ Regulatory Region
2.1.3. Interplay between Eµ and 3′RR Enhancers
2.2. IGH Translocations in B-Cell Malignancies
2.2.1. Mechanisms of IGH Translocations
2.2.2. Recurrent IGH Translocations in B-Cell Lymphoma and Leukemia
2.3. Role of IGH Enhancers in Regulating Oncogene Expression and Malignant Development
3. Immunoglobulin Light Chain Enhancers in B-Cell Malignancies
3.1. Structure and Function of lGK and IGL Enhancers in Normal B Cells
3.2. IGK and IGL Translocations in B-Cell Malignancies
3.3. Role of IGK and IGL Enhancers in Regulating Oncogene Expression and Malignant Development
4. Enhancer Variants and Mutations in B-Cell Malignancies
4.1. Somatic Mutations
4.2. Germline Sequence Variants
5. Exploiting Enhancers by Deregulated Transcription Factors
6. Enhancer Hijacking by Lymphoma-Associated Viruses
6.1. Epstein–Barr Virus
6.2. Kaposi’s Sarcoma-Associated Herpesvirus
7. Conclusions and Future Perspective
Funding
Acknowledgments
Conflicts of Interest
References
- De Leval, L.; Jaffe, E.S. Lymphoma Classification. Cancer J. 2020, 26, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, E.S.; Barr, P.M.; Smith, S.M. Understanding the New WHO Classification of Lymphoid Malignancies: Why It’s Important and How It Will Affect Practice. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.; Min, Q.; Wang, J.Y. B Cell Lymphoma. In Advances in Experimental Medicine and Biology. Adv. Exp. Med. Biol. 2020. [Google Scholar] [CrossRef]
- Teras, L.R.; DeSantis, C.E.; Cerhan, J.R.; Morton, L.M.; Jemal, A.; Flowers, C.R. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J. Clin. 2016, 66, 443–459. [Google Scholar] [CrossRef]
- Nobili, L.; Ronchetti, D.; Taiana, E.; Neri, A. Long non-coding RNAs in B-cell malignancies: A comprehensive overview. Oncotarget 2017, 8, 60605–60623. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.; Pasqualucci, L.; Dalla-Favera, R. Molecular pathology of lymphoma. In Molecular Oncology; Cambridge University Press (CUP): Cambridge, UK, 2015; pp. 738–750. [Google Scholar]
- Seifert, M.; Scholtysik, R.; Küppers, R. Origin and pathogenesis of B cell lymphomas. Methods Mol. Biol. 2019, 1956, 1–33. [Google Scholar]
- Shaknovich, R.; Melnick, A. Epigenetics and B-cell lymphoma. Curr. Opin. Hematol. 2011, 18, 293. [Google Scholar] [CrossRef] [Green Version]
- Solé, C.; Arnaiz, E.; Lawrie, C.H. MicroRNAs as Biomarkers of B-cell Lymphoma. Biomark. Insights 2018, 13. [Google Scholar] [CrossRef] [Green Version]
- Jin, F.; Li, Y.; Ren, B.; Natarajan, R. Enhancers: Multi-dimensional signal integrators. Transcription 2011, 2, 226–230. [Google Scholar] [CrossRef]
- Merika, M.; Thanos, D. Enhanceosomes. Curr. Opin. Genet. Dev. 2001, 11, 205–208. [Google Scholar] [CrossRef]
- Li, W.; Notani, D.; Rosenfeld, M.G. Enhancers as non-coding RNA transcription units: Recent insights and future perspectives. Nat. Rev. Genet. 2016, 17, 207–223. [Google Scholar] [CrossRef]
- Sakabe, N.J.; Savic, D.; Nobrega, M.A. Transcriptional enhancers in development and disease. Genome Biol. 2012, 13, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sur, I.; Taipale, J. The role of enhancers in cancer. Nat. Rev. Cancer 2016, 16, 483–493. [Google Scholar] [CrossRef]
- Banerji, J.; Olson, L.; Schaffner, W. A lymphocyte-specific cellular enhancer is located downstream of the joining region in immunoglobulin heavy chain genes. Cell 1983, 33, 729–740. [Google Scholar] [CrossRef]
- Choukrallah, M.A.; Matthias, P. The Interplay between Chromatin and Transcription Factor Networks during B Cell Development: Who Pulls the Trigger First? Front. Immunol. 2014, 5, 156. [Google Scholar] [CrossRef] [Green Version]
- Choukrallah, M.-A.; Song, S.; Rolink, A.G.; Burger, L.; Matthias, P. Enhancer repertoires are reshaped independently of early priming and heterochromatin dynamics during B cell differentiation. Nat. Commun. 2015, 6, 8324. [Google Scholar] [CrossRef] [Green Version]
- Mercer, E.M.; Lin, Y.C.; Benner, C.; Jhunjhunwala, S.; Dutkowski, J.; Flores, M.; Sigvardsson, M.; Ideker, T.; Glass, C.K.; Murre, C. Multilineage Priming of Enhancer Repertoires Precedes Commitment to the B and Myeloid Cell Lineages in Hematopoietic Progenitors. Immunity 2011, 35, 413–425. [Google Scholar] [CrossRef] [Green Version]
- Gellert, M. V(D)J Recombination: RAG Proteins, Repair Factors, and Regulation. Annu. Rev. Biochem. 2002, 71, 101–132. [Google Scholar] [CrossRef]
- Maul, R.; Gearhart, P.J. AID and Somatic Hypermutation. Dev. Funct. Myeloid Subsets 2010, 105, 159–191. [Google Scholar] [CrossRef] [Green Version]
- Stavnezer, J.; Guikema, J.E.J.; Schrader, C.E. Mechanism and Regulation of Class Switch Recombination. Annu. Rev. Immunol. 2008, 26, 261–292. [Google Scholar] [CrossRef] [Green Version]
- Gillies, S.D.; Morrison, S.L.; Oi, V.T.; Tonegawa, S. A tissue-specific transcription enhancer element is located in the major intron of a rearranged immunoglobulin heavy chain gene. Cell 1983, 33, 717–728. [Google Scholar] [CrossRef]
- Mercola, M.; Wang, X.F.; Olsen, J.; Calame, K. Transcriptional enhancer elements in the mouse immunoglobulin heavy chain locus. Science 1983, 221, 663–665. [Google Scholar] [CrossRef]
- Neuberger, M. Expression and regulation of immunoglobulin heavy chain gene transfected into lymphoid cells. EMBO J. 1983, 2, 1373–1378. [Google Scholar] [CrossRef] [PubMed]
- Ernst, P.; Smale, S.T. Combinatorial regulation of transcription II: The immunoglobulin μ heavy chain gene. Immunity 1995, 2, 427–438. [Google Scholar] [CrossRef] [Green Version]
- Calame, K.; Sen, R. Transcription of Immunoglobulin Genes. In Molecular Biology of B Cells; Elsevier BV: Amsterdam, The Netherlands, 2004; pp. 83–100. [Google Scholar]
- Herrscher, R.F.; Kaplan, M.H.; Lelsz, D.L.; Das, C.; Scheuermann, R.; Tucker, P.W. The immunoglobulin heavy-chain matrix-associating regions are bound by Bright: A B cell-specific trans-activator that describes a new DNA-binding protein family. Genes Dev. 1995, 9, 3067–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, C.; Zong, R.-T.; Lin, D.; Wang, Z.; Kaplan, M.; Paulin, Y.; Smith, E.; Probst, L.; Bryant, J.; Goldstein, A.; et al. Differential Regulation of Immunoglobulin Gene Transcription via Nuclear Matrix-associated Regions. Cold Spring Harb. Symp. Quant. Biol. 1999, 64, 109–118. [Google Scholar] [CrossRef]
- Wang, Z.; Goldstein, A.; Zong, R.; Lin, D.; Neufeld, E.J.; Scheuermann, R.H.; Tucker, P.W. Cux/CDP Homeoprotein Is a Component of NF-μNR and Represses the Immunoglobulin Heavy Chain Intronic Enhancer by Antagonizing the Bright Transcription Activator. Mol. Cell Biol. 1999, 19, 284–295. [Google Scholar] [CrossRef] [Green Version]
- Nelsen, B.; Tian, G.; Erman, B.; Gregoire, J.; Mäki, R.; Graves, B.; Sen, R. Regulation of lymphoid-specific immunoglobulin mu heavy chain gene enhancer by ETS-domain proteins. Science 1993, 261, 82–86. [Google Scholar] [CrossRef]
- Nikolajczyk, B.S.; Cortes, M.; Feinman, R.; Sen, R. Combinatorial determinants of tissue-specific transcription in B cells and macrophages. Mol. Cell. Biol. 1997, 17, 3527–3535. [Google Scholar] [CrossRef] [Green Version]
- Annweiler, A.; Muller, U.; Wirth, T. Functional analysis of defined mutations in the immunoglobulin heavy-chain enhancer in transgenic mice. Nucleic Acids Res. 1992, 20, 1503–1509. [Google Scholar] [CrossRef] [Green Version]
- Forrester, W.; van Genderen, C.; Jenuwein, T.; Grosschedl, R. Dependence of enhancer-mediated transcription of the immunoglobulin mu gene on nuclear matrix attachment regions. Science 1994, 265, 1221–1225. [Google Scholar] [CrossRef]
- Jenuwein, T.; Grosschedl, R. Complex pattern of immunoglobulin mu gene expression in normal and transgenic mice: Nonoverlapping regulatory sequences govern distinct tissue specificities. Genes Dev. 1991, 5, 932–943. [Google Scholar] [CrossRef] [Green Version]
- Afshar, R.; Pierce, S.; Bolland, D.J.; Corcoran, A.; Oltz, E.M. Regulation of IgH Gene Assembly: Role of the Intronic Enhancer and 5′D Q52 Region in Targeting D H J H Recombination. J. Immunol. 2006, 176, 2439–2447. [Google Scholar] [CrossRef] [Green Version]
- Ferrier, P.; Krippl, B.; Blackwell, T.; Furley, A.; Suh, H.; Winoto, A.; Cook, W.; Hood, L.; Costantini, F.; Alt, F. Separate elements control DJ and VDJ rearrangement in a transgenic recombination substrate. EMBO J. 1990, 9, 117–125. [Google Scholar] [CrossRef]
- Perlot, T.; Alt, F.W.; Bassing, C.H.; Suh, H.; Pinaud, E. Elucidation of IgH intronic enhancer functions via germ-line deletion. Proc. Natl. Acad. Sci. USA 2005, 102, 14362–14367. [Google Scholar] [CrossRef] [Green Version]
- Sakai, E.; Bottaro, A.; Alt, F.W. The Ig heavy chain intronic enhancer core region is necessary and sufficient to promote efficient class switch recombination. Int. Immunol. 1999, 11, 1709–1713. [Google Scholar] [CrossRef] [Green Version]
- Lennon, G.G.; Perry, R.P. Cμ-containing transcripts initiate heterogeneously within the IgH enhancer region and contain a novel 5′-nontranslatable exon. Nat. Cell Biol. 1985, 318, 475–478. [Google Scholar] [CrossRef]
- Bolland, D.J.; Wood, A.L.; Afshar, R.; Featherstone, K.; Oltz, E.M.; Corcoran, A.E. Antisense Intergenic Transcription Precedes Igh D-to-J Recombination and Is Controlled by the Intronic Enhancer Eμ. Mol. Cell. Biol. 2007, 27, 5523–5533. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, T.; Perlot, T.; Subrahmanyam, R.; Jani, A.; Goff, P.H.; Zhang, Y.; Ivanova, I.; Alt, F.W.; Sen, R. A 220-nucleotide deletion of the intronic enhancer reveals an epigenetic hierarchy in immunoglobulin heavy chain locus activation. J. Exp. Med. 2009, 206, 1019–1027. [Google Scholar] [CrossRef]
- Inlay, M.A.; Lin, T.; Gao, H.H.; Xu, Y. Critical roles of the immunoglobulin intronic enhancers in maintaining the sequential rearrangement of IgH and Igk loci. J. Exp. Med. 2006, 203, 1721–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Gerasimova, T.; Hao, H.; Ivanova, I.; Chakraborty, T.; Selimyan, R.; Oltz, E.M.; Sen, R. Two Forms of Loops Generate the Chromatin Conformation of the Immunoglobulin Heavy-Chain Gene Locus. Cell 2011, 147, 332–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, X.; Kumari, G.; Gerasimova, T.; Du, H.; Labaran, L.; Singh, A.; De, S.; Wood, W.H.; Becker, K.G.; Zhou, W.; et al. Sequential Enhancer Sequestration Dysregulates Recombination Center Formation at the IgH Locus. Mol. Cell 2018, 70, 21–33.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Zhang, Y.; Zhao, L.; Frock, R.L.; Du, Z.; Meyers, R.; Meng, F.-L.; Schatz, D.G.; Alt, F.W. Chromosomal Loop Domains Direct the Recombination of Antigen Receptor Genes. Cell 2015, 163, 947–959. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Eckhardt, L.A. A role for the IgH intronic enhancer Eμ in enforcing allelic exclusion. J. Exp. Med. 2009, 206, 153–167. [Google Scholar] [CrossRef] [Green Version]
- Peng, C.; Eckhardt, L.A. Role of the IgH Intronic Enhancer Eμ in Clonal Selection at the Pre-B to Immature B Cell Transition1. J. Immunol. 2013, 191, 4399–4411. [Google Scholar] [CrossRef] [Green Version]
- Perlot, T.; Alt, F.W. Cis-Regulatory Elements and Epigenetic Changes Control Genomic Reaarangements of the IgH Locus. Adv. Immunol. 2008, 99, 1–32. [Google Scholar]
- Li, F.; Yan, Y.; Pieretti, J.; Feldman, D.A.; Eckhardt, L.A. Comparison of Identical and Functional Igh Alleles Reveals a Nonessential Role for Eμ in Somatic Hypermutation and Class-Switch Recombination. J. Immunol. 2010, 185, 6049–6057. [Google Scholar] [CrossRef] [Green Version]
- Birshtein, B.K. Epigenetic Regulation of Individual Modules of the immunoglobulin heavy chain locus 3’ Regulatory Region. Front. Immunol. 2014, 5, 163. [Google Scholar] [CrossRef] [Green Version]
- D’Addabbo, P.; Scascitelli, M.; Giambra, V.; Rocchi, M.; Frezza, D. Position and sequence conservation in Amniota of polymorphic enhancer HS1.2 within the palindrome of IgH 3’Regulatory Region. BMC Evol. Biol. 2011, 11, 71. [Google Scholar] [CrossRef] [Green Version]
- Garot, A.; Marquet, M.; Saintamand, A.; Bender, S.; Le Noir, S.; Rouaud-Tinguely, P.; Carrion, C.; Oruc, Z.; Bébin, A.-G.; Moreau, J.; et al. Sequential activation and distinct functions for distal and proximal modules within the IgH 3′ regulatory region. Proc. Natl. Acad. Sci. USA 2016, 113, 1618–1623. [Google Scholar] [CrossRef] [Green Version]
- Khamlichi, A.A.; Pinaud, E.; Decourt, C.; Chauveau, C.; Cogne, M. The 3′ IgH Regulatory Region: A Complex. Structure in a Search for a Function; Elsevier BV: Amsterdam, The Netherlands, 2000; Volume 75, pp. 317–345. [Google Scholar]
- Sepúlveda, M.; Garrett, F.; Price-Whelan, A.; Birshtein, B. Comparative analysis of human and mouse 3′ Igh regulatory regions identifies distinctive structural features. Mol. Immunol. 2005, 42, 605–615. [Google Scholar] [CrossRef]
- Chauveau, C.; Cogné, M. Palindromic structure of the IgH 3′locus control region. Nat. Genet. 1996, 14, 15–16. [Google Scholar] [CrossRef]
- Chauveau, C.; Decourt, C.; Cogné, M. Insertion of the IgH locus 3’ regulatory palindrome in expression vectors warrants sure and efficient expression in stable B cell transfectants. Gene 1998, 222, 279–285. [Google Scholar] [CrossRef]
- Saintamand, A.; Vincent-Fabert, C.; Garot, A.; Rouaud, P.; Oruc, Z.; Magnone, V.; Cogne, M.; Denizot, Y. Deciphering the importance of the palindromic architecture of the immunoglobulin heavy-chain 3’ regulatory region. Nat. Commun. 2016, 7, 10730. [Google Scholar] [CrossRef] [Green Version]
- Le Noir, S.; Boyer, F.; Lecardeur, S.; Brousse, M.; Oruc, Z.; Cook-Moreau, J.; Denizot, Y.; Cogné, M. Functional anatomy of the immunoglobulin heavy chain 3’ super-enhancer needs not only core enhancer elements but also their unique DNA context. Nucleic Acids Res. 2017, 45, 5829–5837. [Google Scholar] [CrossRef]
- Giambra, V.; Fruscalzo, A.; Giufre, M.; Martinez-Labarga, C.; Favaro, M.; Rocchi, M.; Frezza, D. Evolution of human IgH3′EC duplicated structures: Both enhancers HS1,2 are polymorphic with variation of transcription factor’s consensus sites. Gene 2005, 346, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Pinaud, E.; Marquet, M.; Fiancette, R.; Péron, S.; Vincent-Fabert, C.; Denizot, Y.; Cogné, M. The IgH locus 3’ regulatory region: Pulling the strings from behind. Adv. Immunol. 2011, 110, 27–70. [Google Scholar]
- Cianci, R.; D’Addabbo, P.; Gambassi, G.; Lolli, S.; Serone, E.; Rizzi, A.; Pitocco, D.; Pandolfi, F.; Frezza, D. Association between IgH enhancer hs1.2 and type 1 diabetes. Acta Diabetol. 2018, 55, 443–448. [Google Scholar] [CrossRef]
- Giambra, V.; Martinez-Labarga, C.; Giufre, M.; Modiano, D.; Simpore, J.; Gisladottir, B.K.; Francavilla, R.; Zhelezova, G.; Kilic, S.S.; Crawford, M.; et al. Immunoglobulin Enhancer HS1,2 polymorphism: A new powerful anthropogenetic marker. Ann. Hum. Genet. 2006, 70, 946–950. [Google Scholar] [CrossRef]
- Giambra, V.; Cianci, R.; Lolli, S.; Mattioli, C.; Tampella, G.; Cattalini, M.; Kilic, S.S.; Pandolfi, F.; Plebani, A.; Frezza, D. Allele *1 of HS1.2 Enhancer Associates with Selective IgA Deficiency and IgM Concentration. J. Immunol. 2009, 183, 8280–8285. [Google Scholar] [CrossRef] [Green Version]
- Rouaud-Tinguely, P.; Vincent-Fabert, C.; Fiancette, R.; Cogné, M.; Pinaud, E.; Denizot, Y. Enhancers Located in Heavy Chain Regulatory Region (hs3a, hs1,2, hs3b, and hs4) Are Dispensable for Diversity of VDJ Recombination. J. Biol. Chem. 2012, 287, 8356–8360. [Google Scholar] [CrossRef] [Green Version]
- Vincent-Fabert, C.; Fiancette, R.; Cogne, M.; Pinaud, E.; Denizot, Y. The IgH 3′ regulatory region and its implication in lymphomagenesis. Eur. J. Immunol. 2010, 40, 3306–3311. [Google Scholar] [CrossRef]
- Braikia, F.-Z.; Conte, C.; Moutahir, M.; Denizot, Y.; Cogné, M.; Khamlichi, A.A. Developmental Switch in the Transcriptional Activity of a Long-Range Regulatory Element. Mol. Cell. Biol. 2015, 35, 3370–3380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cogné, M.; Lansford, R.; Bottaro, A.; Zhang, J.; Gorman, J.; Young, F.; Cheng, H.-L.; Alt, F.W. A class switch control region at the 3′ end of the immunoglobulin heavy chain locus. Cell 1994, 77, 737–747. [Google Scholar] [CrossRef]
- Hussein, I.; Nour, G.; François, B.; Yves, D.; Alexis, S. Deletion of the immunoglobulin heavy chain 3′ regulatory region super-enhancer affects somatic hypermutation in B1 B cells. Cell. Mol. Immunol. 2018, 16, 195–197. [Google Scholar] [CrossRef]
- Rouaud-Tinguely, P.; Vincent-Fabert, C.; Saintamand, A.; Fiancette, R.; Marquet, M.; Robert, I.; Reina-San-Martin, B.; Pinaud, E.; Cogné, M.; Denizot, Y. The IgH 3′ regulatory region controls somatic hypermutation in germinal center B cells. J. Exp. Med. 2013, 210, 1501–1507. [Google Scholar] [CrossRef] [Green Version]
- Le Morvan, C.; Pinaud, E.; Decourt, C.; Cuvillier, A.; Cogneé, M.; Morvan, C.L. The immunoglobulin heavy-chain locus hs3b and hs4 3′ enhancers are dispensable for VDJ assembly and somatic hypermutation. Blood 2003, 102, 1421–1427. [Google Scholar] [CrossRef] [Green Version]
- Manis, J.P.; Van Der Stoep, N.; Tian, M.; Ferrini, R.; Davidson, L.; Bottaro, A.; Alt, F.W. Class Switching in B Cells Lacking 3′ Immunoglobulin Heavy Chain Enhancers. J. Exp. Med. 1998, 188, 1421–1431. [Google Scholar] [CrossRef] [Green Version]
- Pinaud, E.; Khamlichi, A.A.; Le Morvan, C.; Drouet, M.; Nalesso, V.; Le Bert, M.; Cogné, M. Localization of the 3′ IgH Locus Elements that Effect Long-Distance Regulation of Class Switch Recombination. Immunity 2001, 15, 187–199. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Pieretti, J.; Ju, Z.; Wei, S.; Christin, J.R.; Bah, F.; Birshtein, B.K.; Eckhardt, L.A. Homologous elements hs3a and hs3b in the 3′ regulatory region of the murine immunoglobulin heavy chain (Igh) locus are both dispensable for class-switch recombination. J. Biol. Chem. 2011, 286, 27123–27131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birshtein, B.K. The role of CTCF binding sites in the 3′ immunoglobulin heavy chain regulatory region. Front. Genet. 2012, 3, 251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bébin, A.-G.; Carrion, C.; Marquet, M.; Cogné, N.; Lecardeur, S.; Cogné, M.; Pinaud, E. In Vivo Redundant Function of the 3′ IgH Regulatory Element HS3b in the Mouse. J. Immunol. 2010, 184, 3710–3717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saintamand, A.; Rouaud, P.; Garot, A.; Saad, F.; Carrion, C.; Oblet, C.; Cogné, M.; Pinaud, E.; Denizot, Y. The IgH 3′ regulatory region governs μ chain transcription in mature B lymphocytes and the B cell fate. Oncotarget 2015, 6, 4845–4852. [Google Scholar] [CrossRef] [Green Version]
- Issaoui, H.; Ghazzaui, N.; Saintamand, A.; Denizot, Y.; Boyer, F. IgD class switch recombination is not controlled through the immunoglobulin heavy chain 3′ regulatory region super-enhancer. Cell. Mol. Immunol. 2017, 14, 871–874. [Google Scholar] [CrossRef]
- Rouaud-Tinguely, P.; Saintamand, A.; Saad, F.; Carrion, C.; Lecardeur, S.; Cogné, M.; Denizot, Y. Elucidation of the enigmatic IgD class-switch recombination via germline deletion of the IgH 3′ regulatory region. J. Exp. Med. 2014, 211, 975–985. [Google Scholar] [CrossRef]
- Rothschild, G.; Zhang, W.; Lim, J.; Giri, P.K.; Laffleur, B.; Chen, Y.; Fang, M.; Chen, Y.; Nair, L.; Liu, Z.-P.; et al. Noncoding RNA transcription alters chromosomal topology to promote isotype-specific class switch recombination. Sci. Immunol. 2020, 5, eaay5864. [Google Scholar] [CrossRef]
- Chatterjee, S.; Ju, Z.; Hassan, R.; Volpi, S.A.; Emelyanov, A.V.; Birshtein, B.K. Dynamic Changes in Binding of Immunoglobulin Heavy Chain 3′ Regulatory Region to Protein Factors during Class Switching. J. Biol. Chem. 2011, 286, 29303–29312. [Google Scholar] [CrossRef] [Green Version]
- Ju, Z.; Chatterjee, S.; Birshtein, B.K. Interaction between the immunoglobulin heavy chain 3′ regulatory region and the IgH transcription unit during B cell differentiation. Mol. Immunol. 2011, 49, 297–303. [Google Scholar] [CrossRef] [Green Version]
- Mocikat, R.; Harloff, C.; Kütemeier, G. The effect of the rat immunoglobulin heavy-chain 3′ enhancer is position dependent. Gene 1993, 136, 349–353. [Google Scholar] [CrossRef]
- Mocikat, R.; Kardinal, C.; Klobeck, H.-G. Differential interactions between the immunoglobulin heavy chain μ intron and 3′ enhancer. Eur. J. Immunol. 1995, 25, 3195–3198. [Google Scholar] [CrossRef]
- Ju, Z.; Volpi, S.A.; Hassan, R.; Martinez, N.; Giannini, S.L.; Gold, T.; Birshtein, B.K. Evidence for physical interaction between the immunoglobulin heavy chain variable region and the 3′ regulatory region. J. Biol. Chem. 2007, 282, 35169–35178. [Google Scholar] [CrossRef] [Green Version]
- Sette, M.; D’Addabbo, P.; Kelly, G.; Cicconi, A.; Micheli, E.; Cacchione, S.; Poma, A.; Gargioli, C.; Giambra, V.; Frezza, D. Evidence for a quadruplex structure in the polymorphic hs1.2 enhancer of the immunoglobulin heavy chain 3’ regulatory regions and its conservation in mammals. Biopolymers 2016, 105, 768–778. [Google Scholar] [CrossRef] [Green Version]
- Kenter, A.L.; Feldman, S.; Wuerffel, R.; Achour, I.; Wang, L.; Kumar, S. Three-dimensional architecture of the IgH locus facilitates class switch recombination. Ann. N. Y. Acad. Sci. 2012, 1267, 86–94. [Google Scholar] [CrossRef] [Green Version]
- Wuerffel, R.; Wang, L.; Grigera, F.; Manis, J.; Selsing, E.; Perlot, T.; Alt, F.W.; Cogné, M.; Pinaud, E.; Kenter, A.L. S-S Synapsis during Class Switch Recombination Is Promoted by Distantly Located Transcriptional Elements and Activation-Induced Deaminase. Immunity 2007, 27, 711–722. [Google Scholar] [CrossRef] [Green Version]
- Saintamand, A.; Vincent-Fabert, C.; Marquet, M.; Ghazzaui, N.; Magnone, V.; Pinaud, E.; Cogné, M.; Denizot, Y. Eμ and 3′RR IgH enhancers show hierarchic unilateral dependence in mature B-cells. Sci. Rep. 2017, 7, 442. [Google Scholar] [CrossRef] [Green Version]
- Lecluse, Y.; LeBailly, P.; Roulland, S.; Gac, A.-C.; Nadel, B.; Gauduchon, P. t(11;14)-positive clones can persist over a long period of time in the peripheral blood of healthy individuals. Leukemia 2009, 23, 1190–1193. [Google Scholar] [CrossRef] [Green Version]
- Limpens, J.; De Jong, D.; Van Krieken, J.H.; Price, C.G.; Young, B.D.; Van Ommen, G.J.; Kluin, P.M. Bcl-2/JH rearrangements in benign lymphoid tissues with follicular hyperplasia. Oncogene 1991, 6, 2271–2276. [Google Scholar]
- Schmitt, C.; Balogh, B.; Grundt, A.; Buchholtz, C.; Leo, A.; Benner, A.; Hensel, M.; Ho, A.D.; Leo, E. The bcl-2/IgH rearrangement in a population of 204 healthy individuals: Occurrence, age and gender distribution, breakpoints, and detection method validity. Leuk. Res. 2006, 30, 745–750. [Google Scholar] [CrossRef]
- Brassesco, M. ReviewLeukemia/lymphoma-associated gene fusions in normal individuals. Genet. Mol. Res. 2008, 7, 782–790. [Google Scholar] [CrossRef]
- Ghazzaui, N.; Issaoui, H.; Boyer, F.; Martin, O.A.; Saintamand, A.; Denizot, Y. 3′RR and 5′E μ immunoglobulin heavy chain enhancers are independent engines of locus remodeling. Cell. Mol. Immunol. 2019, 16, 198–200. [Google Scholar] [CrossRef] [Green Version]
- Baens, M.; Fevery, S.; Sagaert, X.; Noels, H.; Hagens, S.; Broeckx, V.; Billiau, A.D.; De Wolf-Peeters, C.; Marynen, P. Selective Expansion of Marginal Zone B Cells in Eμ-API2-MALT1 Mice Is Linked to Enhanced IκB Kinase γ Polyubiquitination. Cancer Res. 2006, 66, 5270–5277. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Wang, H.; Xue, L.; Shin, D.M.; Roopenian, D.; Xu, W.; Qi, C.F.; Sangster, M.Y.; Orihuela, C.J.; Tuomanen, E.; et al. Eμ-BCL10 mice exhibit constitutive activation of both canonical and noncanonical NF-κB pathways generating marginal zone (MZ) B-cell expansion as a precursor to splenic MZ lymphoma. Blood 2009, 114, 4158–4168. [Google Scholar] [CrossRef] [Green Version]
- Küppers, R.; Dalla-Favera, R. Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene 2001, 20, 5580–5594. [Google Scholar] [CrossRef] [Green Version]
- Merelli, I.; Guffanti, A.; Fabbri, M.; Cocito, A.; Furia, L.; Grazini, U.; Bonnal, R.; Milanesi, L.; McBlane, F. RSSsite: A reference database and prediction tool for the identification of cryptic Recombination Signal Sequences in human and murine genomes. Nucleic Acids Res. 2010, 38, W262–W267. [Google Scholar] [CrossRef]
- Tsai, A.; Lu, H.; Raghavan, S.C.; Muschen, M.; Hsieh, C.-L.; Lieber, M.R. Human Chromosomal Translocations at CpG Sites and a Theoretical Basis for Their Lineage and Stage Specificity. Cell 2008, 135, 1130–1142. [Google Scholar] [CrossRef] [Green Version]
- Maman, Y.; Teng, G.; Seth, R.; Kleinstein, S.H.; Schatz, D.G. RAG1 targeting in the genome is dominated by chromatin interactions mediated by the non-core regions of RAG1 and RAG2. Nucleic Acids Res. 2016, 44, 9624–9637. [Google Scholar] [CrossRef] [Green Version]
- Muramatsu, M.; Kinoshita, K.; Fagarasan, S.; Yamada, S.; Shinkai, Y.; Honjo, T. Class Switch Recombination and Hypermutation Require Activation-Induced Cytidine Deaminase (AID), a Potential RNA Editing Enzyme. Cell 2000, 102, 553–563. [Google Scholar] [CrossRef] [Green Version]
- Revy, P.; Muto, T.; Levy, Y.; Geissmann, F.; Plebani, A.; Sanal, O.; Catalan, N.; Forveille, M.; Dufourcq-Lagelouse, R.; Gennery, A.; et al. Activation-Induced Cytidine Deaminase (AID) Deficiency Causes the Autosomal Recessive Form of the Hyper-IgM Syndrome (HIGM2). Cell 2000, 102, 565–575. [Google Scholar] [CrossRef] [Green Version]
- Pham, P.; Bransteitter, R.; Petruska, J.; Goodman, M.F. Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature 2003, 424, 103–107. [Google Scholar] [CrossRef]
- Di Noia, J.M.; Neuberger, M.S. Molecular Mechanisms of Antibody Somatic Hypermutation. Annu. Rev. Biochem. 2007, 76, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Zarrin, A.A.; Alt, F.W.; Chaudhuri, J.; Stokes, N.; Kaushal, D.; Du Pasquier, L.; Tian, M. An evolutionarily conserved target motif for immunoglobulin class-switch recombination. Nat. Immunol. 2004, 5, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Yeap, L.-S.; Hwang, J.K.; Du, Z.; Meyers, R.; Meng, F.-L.; Jakubauskaitė, A.; Liu, M.; Mani, V.; Neuberg, D.; Kepler, T.B.; et al. Sequence-Intrinsic Mechanisms that Target AID Mutational Outcomes on Antibody Genes. Cell 2015, 163, 1124–1137. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Masani, S.; Yu, K. Overlapping activation-induced cytidine deaminase hotspot motifs in Ig class-switch recombination. Proc. Natl. Acad. Sci. USA 2011, 108, 11584–11589. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.M.; Peters, A.; Baron, B.; Zhu, X.; Storb, U. Mutation of BCL-6 Gene in Normal B Cells by the Process of Somatic Hypermutation of Ig Genes. Science 1998, 280, 1750–1752. [Google Scholar] [CrossRef]
- Pasqualucci, L.; Migliazza, A.; Fracchiolla, N.; William, C.; Neri, A.; Baldini, L.; Chaganti, R.S.K.; Klein, U.; Küppers, R.; Rajewsky, K.; et al. BCL-6 mutations in normal germinal center B cells: Evidence of somatic hypermutation acting outside Ig loci. Proc. Natl. Acad. Sci. USA 1998, 95, 11816–11821. [Google Scholar] [CrossRef] [Green Version]
- Müschen, M.; Re, D.; Jungnickel, B.; Diehl, V.; Rajewsky, K.; Küppers, R. Somatic Mutation of the Cd95 Gene in Human B Cells as a Side-Effect of the Germinal Center Reaction. J. Exp. Med. 2000, 192, 1833–1840. [Google Scholar] [CrossRef]
- Gordon, M.S.; Kanegai, C.M.; Doerr, J.R.; Wall, R. Somatic hypermutation of the B cell receptor genes B29 (Ig, CD79b) and mb1 (Ig, CD79a). Proc. Natl. Acad. Sci. USA 2003, 100, 4126–4131. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Duke, J.L.; Richter, D.; Vinuesa, C.; Goodnow, C.; Kleinstein, S.H.; Schatz, D.G. Two levels of protection for the B cell genome during somatic hypermutation. Nature 2008, 451, 841–845. [Google Scholar] [CrossRef]
- Alt, F.W.; Zhang, Y.; Meng, F.-L.; Guo, C.; Schwer, B. Mechanisms of Programmed DNA Lesions and Genomic Instability in the Immune System. Cell 2013, 152, 417–429. [Google Scholar] [CrossRef] [Green Version]
- Storb, U. Why Does Somatic Hypermutation by AID Require Transcription of Its Target Genes? Dev. Funct. Myeloid Subsets 2014, 122, 253–277. [Google Scholar] [CrossRef]
- Qian, J.; Wang, Q.; Dose, M.; Pruett, N.; Kieffer-Kwon, K.-R.; Resch, W.; Liang, G.; Tang, Z.; Mathé, E.; Benner, C.; et al. B Cell Super-Enhancers and Regulatory Clusters Recruit AID Tumorigenic Activity. Cell 2014, 159, 1524–1537. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.-L.; Du, Z.; Federation, A.; Hu, J.; Wang, Q.; Kieffer-Kwon, K.-R.; Meyers, R.; Amor, C.; Wasserman, C.R.; Neuberg, D.; et al. Convergent Transcription at Intragenic Super-Enhancers Targets AID-Initiated Genomic Instability. Cell 2014, 159, 1538–1548. [Google Scholar] [CrossRef] [Green Version]
- Basu, U.; Meng, F.-L.; Keim, C.; Grinstein, V.; Pefanis, E.; Eccleston, J.; Zhang, T.; Myers, D.; Wasserman, C.R.; Wesemann, D.R.; et al. The RNA Exosome Targets the AID Cytidine Deaminase to Both Strands of Transcribed Duplex DNA Substrates. Cell 2011, 144, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Pavri, R.; Gazumyan, A.; Jankovic, M.; Di Virgilio, M.; Klein, I.; Ansarah-Sobrinho, C.; Resch, W.; Yamane, A.; Reina-San-Martin, B.; Barreto, V.M.; et al. Activation-Induced Cytidine Deaminase Targets DNA at Sites of RNA Polymerase II Stalling by Interaction with Spt5. Cell 2010, 143, 122–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.; Pannunzio, N.R.; Greisman, H.A.; Casero, D.; Parekh, C.; Lieber, M.R. Convergent BCL6 and lncRNA promoters demarcate the major breakpoint region for BCL6 translocations. Blood 2015, 126, 1730–1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinäniemi, M.; Vuorenmaa, T.; Teppo, S.; Kaikkonen, M.U.; Bouvy-Liivrand, M.; Mehtonen, J.; Niskanen, H.; Zachariadis, V.; Laukkanen, S.; Liuksiala, T.; et al. Transcription-coupled genetic instability marks acute lymphoblastic leukemia structural variation hotspots. eLife 2016, 5, 13087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roix, J.J.; McQueen, P.G.; Munson, P.J.; Parada, L.A.; Misteli, T. Spatial proximity of translocation-prone gene loci in human lymphomas. Nat. Genet. 2003, 34, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; McCord, R.; Ho, Y.-J.; Lajoie, B.R.; Hildebrand, D.G.; Simon, A.C.; Becker, M.S.; Alt, F.W.; Dekker, J. Spatial Organization of the Mouse Genome and Its Role in Recurrent Chromosomal Translocations. Cell 2012, 148, 908–921. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Benner, C.; Mansson, R.; Heinz, S.; Miyazaki, K.; Miyazaki, M.; Chandra, V.; Bossen, C.; Glass, C.K.; Murre, C. Global changes in the nuclear positioning of genes and intra- and interdomain genomic interactions that orchestrate B cell fate. Nat. Immunol. 2012, 13, 1196–1204. [Google Scholar] [CrossRef] [Green Version]
- Lieberman-Aiden, E.; Van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Engreitz, J.M.; Agarwala, V.; Mirny, L.A. Three-Dimensional Genome Architecture Influences Partner Selection for Chromosomal Translocations in Human Disease. PLoS ONE 2012, 7, e44196. [Google Scholar] [CrossRef] [Green Version]
- Strongin, D.E.; Groudine, M.; Politz, J.C.R. Nucleolar tethering mediates pairing between theIgHandMycloci. Nucleus 2014, 5, 474–481. [Google Scholar] [CrossRef] [Green Version]
- Sklyar, I.V.; Pichugin, A.M.; Razin, S.V.; Vassetzky, E.S.; Iarovaia, O.V. Nuclear localization of translocation partners in differentiating B-cells. Dokl. Biochem. Biophys. 2015, 464, 312–314. [Google Scholar] [CrossRef]
- Gerasimova, T.I.; Guo, C.; Ghosh, A.; Qiu, X.; Montefiori, L.; Verma-Gaur, J.; Choi, N.M.; Feeney, A.J.; Sen, R. A structural hierarchy mediated by multiple nuclear factors establishesIgHlocus conformation. Genes Dev. 2015, 29, 1683–1695. [Google Scholar] [CrossRef] [Green Version]
- Lieber, M.R. Mechanisms of human lymphoid chromosomal translocations. Nat. Rev. Cancer 2016, 16, 387–398. [Google Scholar] [CrossRef] [Green Version]
- Burotto, M.; Berkovits, A.; Dunleavy, K. Double hit lymphoma: From biology to therapeutic implications. Expert Rev. Hematol. 2016, 9, 669–678. [Google Scholar] [CrossRef]
- Campbell, L.J. Cytogenetics of lymphomas. Pathology 2005, 37, 493–507. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Gorham, J.; Cossman, J.; Jaffe, E.; Croce, C.M. The t(14;18) chromosome translocations involved in B-cell neoplasms result from mistakes in VDJ joining. Science 1985, 229, 1390–1393. [Google Scholar] [CrossRef]
- Weiss, L.M.; Warnke, R.A.; Sklar, J.; Cleary, M.L. Molecular Analysis of the T(14;18) Chromosomal Translocation in Malignant Lymphomas. N. Engl. J. Med. 1987, 317, 1185–1189. [Google Scholar] [CrossRef]
- Jaäger, U.; Boöcskoör, S.; Le, T.; Mitterbauer, G.; Bolz, I.; Chott, A.; Kneba, M.; Mannhalter, C.; Nadel, B. Follicular lymphomas’ BCL-2/IgH junctions contain templated nucleotide insertions: Novel insights into the mechanism of t(14;18) translocation. Blood 2000, 95, 3520–3529. [Google Scholar] [CrossRef]
- Willis, T.; Dyer, M. The role of immunoglobulin translocations in the pathogenesis of B-cell malignancies. Blood 2000, 96, 808–822. [Google Scholar] [CrossRef]
- Attygalle, A.D. t(14;19)(q32;q13)-associated B-cell neoplasms—A review. J. Hematop. 2011, 5, 159–163. [Google Scholar] [CrossRef]
- Huh, Y.O.; Schweighofer, C.D.; Ketterling, R.P.; Knudson, R.A.; Vega, F.; Kim, J.E.; Luthra, R.; Keating, M.J.; Medeiros, L.J.; Abruzzo, L.V. Chronic Lymphocytic Leukemia With t(14;19)(q32;q13) Is Characterized by Atypical Morphologic and Immunophenotypic Features and Distinctive Genetic Features. Am. J. Clin. Pathol. 2011, 135, 686–696. [Google Scholar] [CrossRef] [Green Version]
- Martin-Subero, J.I.; Ibbotson, R.; Klapper, W.; Michaux, L.; Callet-Bauchu, E.; Berger, F.; Calasanz, M.J.; De Wolf-Peeters, C.; Dyer, M.; Felman, P.; et al. A comprehensive genetic and histopathologic analysis identifies two subgroups of B-cell malignancies carrying a t(14;19)(q32;q13) or variant BCL3-translocation. Leukemia 2007, 21, 1532–1544. [Google Scholar] [CrossRef] [Green Version]
- Michaux, L.; Dierlamm, J.; Wlodarska, I.; Bours, V.; Van Den Berghe, H.; Hagemeijer, A. t(14;19)/BCL3 rearrangements in lymphoproliferative disorders: A review of 23 cases. Cancer Genet. Cytogenet. 1997, 94, 36–43. [Google Scholar] [CrossRef]
- Bastard, C.; Tilly, H.; Lenormand, B.; Bigorgne, C.; Boulet, D.; Kunlin, A.; Monconduit, M.; Piguet, H. Translocations involving band 3q27 and Ig gene regions in non-Hodgkin’s lymphoma [see comments]. Blood 1992, 79, 2527–2531. [Google Scholar] [CrossRef] [Green Version]
- Bastard, C.; Deweindt, C.; Kerckaert, J.P.; Lenormand, B.; Rossi, A.; Pezzella, F.; Fruchart, C.; Duval, C.; Monconduit, M.; Tilly, H. LAZ3 rearrangements in non-Hodgkin’s lymphoma: Correlation with histology, immunophenotype, karyotype, and clinical outcome in 217 patients. Blood 1994, 83, 2423–2427. [Google Scholar] [CrossRef] [Green Version]
- Kerckaert, J.-P.; Deweindt, C.; Tilly, H.; Quief, S.; Lecocq, G.; Bastard, C. LAZ3, a novel zinc—finger encoding gene, is disrupted by recurring chromosome 3q27 translocations in human lymphomas. Nat. Genet. 1993, 5, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Offit, K.; Coco, F.L.; Louie, D.C.; Parsa, N.Z.; Leung, D.H.Y.; Portlock, C.; Ye, B.H.; Lista, F.; Filippa, D.A.; Rosenbaum, A.; et al. Rearrangement of the bcl-6 Gene as a Prognostic Marker in Diffuse Large-Cell Lymphoma. N. Engl. J. Med. 1994, 331, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Ohno, H.; Fukuhara, S. Significance of Rearrangement of the BCL6 Gene in B-Cell Lymphoid Neoplasms. Leuk. Lymphoma 1997, 27, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Adachi, M.; Tefferi, A.; Greipp, P.R.; Kipps, T.J.; Tsujimoto, Y.; Adachi, M.; Tefferi, A.; Greipp, P.R.; Kipps, T.J.; Tsujimoto, Y. Preferential linkage of bcl-2 to immunoglobulin light chain gene in chronic lymphocytic leukemia. J. Exp. Med. 1990, 171, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Akasaka, T.; Akasaka, H.; Ohno, H. Polymerase chain reaction amplification of long DNA targets: Application to analysis of chromosomal translocations in human B-cell tumors (review). Int. J. Oncol. 1998, 12, 113–134. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Nishida, K.; Miki, T.; Horiike, S.; Kaneko, H.; Yokota, S.; Misawa, S.; Abe, T.; Kashima, K.; Taniwaki, M. Interphase Detection of BCL6/IgH Fusion Gene in Non-Hodgkin Lymphoma by Fluorescence In Situ Hybridization. Cancer Genet. Cytogenet. 1997, 99, 102–107. [Google Scholar] [CrossRef]
- Du, M.; Peng, H.; Liu, H.; Hamoudi, R.A.; Diss, T.C.; Willis, T.G.; Ye, H.; Dogan, A.; Wotherspoon, A.C.; Dyer, M.J.S.; et al. BCL10 gene mutation in lymphoma. Blood 2000, 95, 3885–3890. [Google Scholar] [CrossRef]
- Zhang, Q.; Siebert, R.; Yan, M.; Hinzmann, B.; Cui, X.; Xue, L.; Rakestraw, K.M.; Naeve, C.W.; Beckmann, G.; Weisenburger, D.D.; et al. Inactivating mutations and overexpression of BCL10, a caspase recruitment domain-containing gene, in MALT lymphoma with t(1;14)(p22;q32). Nat. Genet. 1999, 22, 63–68. [Google Scholar] [CrossRef]
- Willis, T.G.; Jadayel, D.M.; Du, M.-Q.; Peng, H.; Perry, A.R.; Abdul-Rauf, M.; Price, H.; Karran, L.; Majekodunmi, O.; Wlodarska, I.; et al. Bcl10 Is Involved in t(1;14)(p22;q32) of MALT B Cell Lymphoma and Mutated in Multiple Tumor Types. Cell 1999, 96, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Avet-Loiseau, H.; Brigaudeau, C.; Morineau, N.; Talmant, P.; La, J.; Daviet, A.; Li, J.; Praloran, V.; Rapp, M.; Harousseau, J.; et al. High incidence of cryptic translocations involving the Ig heavy chain gene in multiple myeloma, as shown by fluorescence in situ hybridization. Genes Chromosom. Cancer 1999, 24, 9–15. [Google Scholar] [CrossRef]
- Resnitzky, P.; Matutes, E.; Hedges, M.; Morilla, R.; Brito-Babapulle, V.; Khokhar, T.; Catovsky, D. The ultrastructure of mantle cell lymphoma and other B-cell disorders with translocation t(11;14)(q13;q32). Br. J. Haematol. 1996, 94, 352–361. [Google Scholar] [CrossRef]
- Vaandrager, J.; Schuuring, E.; Zwikstra, E.; De Boer, C.; Kleiverda, K.; Van Krieken, J.; Kluin-Nelemans, H.; Van Ommen, G.; Raap, A.; Kluin, P. Direct visualization of dispersed 11q13 chromosomal translocations in mantle cell lymphoma by multicolor DNA fiber fluorescence in situ hybridization. Blood 1996, 88, 1177–1182. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, V.R.; Valdes, R.F.; Vose, J.M. Mantle Cell Lymphoma. Hematology 2018, 88, 1298–1308.e2. [Google Scholar] [CrossRef]
- Greisman, H.A.; Lu, Z.; Tsai, A.; Greiner, T.C.; Yi, H.S.; Lieber, M.R. IgH partner breakpoint sequences provide evidence that AID initiates t(11;14) and t(8;14) chromosomal breaks in mantle cell and Burkitt lymphomas. Blood 2012, 120, 2864–2867. [Google Scholar] [CrossRef] [Green Version]
- Hasanali, Z.; Sharma, K.; Epner, E. Flipping the cyclin D1 switch in mantle cell lymphoma. Best Pract. Res. Clin. Haematol. 2012, 25, 143–152. [Google Scholar] [CrossRef]
- Akasaka, T.; Balasas, T.; Russell, L.J.; Sugimoto, K.-J.; Majid, A.; Walewska, R.; Karran, E.L.; Brown, D.G.; Cain, K.; Harder, L.; et al. Five members of the CEBP transcription factor family are targeted by recurrent IGH translocations in B-cell precursor acute lymphoblastic leukemia (BCP-ALL). Blood 2006, 109, 3451–3461. [Google Scholar] [CrossRef]
- Hayashi, Y.; Pui, C.H.; Behm, F.G.; Fuchs, A.H.; Raimondi, S.C.; Kitchingman, G.R.; Mirro, J.J.; Williams, D.L. 14q32 translocations are associated with mixed-lineage expression in childhood acute leukemia. Blood 1990, 76, 150–156. [Google Scholar] [CrossRef]
- Messinger, Y.H.; Higgins, R.R.; Devidas, M.; Hunger, S.P.; Carroll, A.J.; Heerema, N.A. Pediatric acute lymphoblastic leukemia with a t(8;14)(q11.2;q32): B-cell disease with a high proportion of Down syndrome: A Children’s Oncology Group study. Cancer Genet. 2012, 205, 453–458. [Google Scholar] [CrossRef] [Green Version]
- Moore, S.; Suttle, J.; Bain, S.; Story, C.; Rice, M. Acute lymphoblastic leukemia characterized by t(8;14)(q11.2;q32). Cancer Genet. Cytogenet. 2003, 141, 1–4. [Google Scholar] [CrossRef]
- Dring, A.M.; Davies, F.; Fenton, J.A.L.; Roddam, P.L.; Scott, K.; Gonzalez, D.; Rollinson, S.; Rawstron, A.C.; Rees-Unwin, K.S.; Li, C.; et al. A Global Expression-based Analysis of the Consequences of the t(4;14) Translocation in Myeloma. Clin. Cancer Res. 2004, 10, 5692–5701. [Google Scholar] [CrossRef] [Green Version]
- Chesi, M.; Nardini, E.; Brents, L.A.; Schröck, E.; Ried, T.; Kuehl, W.M.; Bergsagel, P.L. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat. Genet. 1997, 16, 260–264. [Google Scholar] [CrossRef]
- Kalff, A.; Spencer, A. The t(4;14) translocation and FGFR3 overexpression in multiple myeloma: Prognostic implications and current clinical strategies. Blood Cancer J. 2012, 2, e89. [Google Scholar] [CrossRef] [Green Version]
- Richelda, R.; Ronchetti, D.; Baldini, L.; Cro, L.; Viggiano, L.; Marzella, R.; Rocchi, M.; Otsuki, T.; Lombardi, L.; Maiolo, A.T.; et al. A Novel Chromosomal Translocation t(4; 14)(p16.3; q32) in Multiple Myeloma Involves the Fibroblast Growth-Factor Receptor 3 Gene. Blood 1997, 90, 4062–4070. [Google Scholar] [CrossRef]
- Santra, M.; Zhan, F.; Tian, E.; Barlogie, B.; Shaughnessy, J. A subset of multiple myeloma harboring the t(4;14)(p16;q32) translocation lacks FGFR3 expression but maintains anIGH/MMSET fusion transcript. Blood 2003, 101, 2374–2376. [Google Scholar] [CrossRef]
- Hudlebusch, H.R.; Theilgaard-Mönch, K.; Lodahl, M.; Johnsen, H.E.; Rasmussen, T. Identification of ID-1 as a potential target gene of MMSET in multiple myeloma. Br. J. Haematol. 2005, 130, 700–708. [Google Scholar] [CrossRef]
- Farinha, P.; Gascoyne, R.D. Molecular Pathogenesis of Mucosa-Associated Lymphoid Tissue Lymphoma. J. Clin. Oncol. 2005, 23, 6370–6378. [Google Scholar] [CrossRef]
- Sasaki, Y.; Shiozawa, E.; Watanabe, N.; Homma, M.; Noh, J.Y.; Ito, K.; Takimoto, M.; Yamochi-Onizuka, T. t(3;14)(p14.1;q32)/FOXP1-IGH translocation in thyroid extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma). Leuk. Res. 2020, 95, 106399. [Google Scholar] [CrossRef]
- Streubel, B.; Vinatzer, U.; Lamprecht, A.; Raderer, M.; Chott, A. T(3;14)(p14.1;q32) involving IGH and FOXP1 is a novel recurrent chromosomal aberration in MALT lymphoma. Leukemia 2005, 19, 652–658. [Google Scholar] [CrossRef]
- Fournier, B.; Balducci, E.; Duployez, N.; Clappier, E.; Cuccuini, W.; Arfeuille, C.; Caye-Eude, A.; Delabesse, E.; Colomb, E.B.-L.; Nebral, K.; et al. B-ALL With t(5;14)(q31;q32); IGH-IL3 Rearrangement and Eosinophilia: A Comprehensive Analysis of a Peculiar IGH-Rearranged B-ALL. Front. Oncol. 2019, 9, 9. [Google Scholar] [CrossRef]
- Grimaldi, J.C.; Meeker, T.C. The t(5;14) chromosomal translocation in a case of acute lymphocytic leukemia joins the interleukin-3 gene to the immunoglobulin heavy chain gene. Blood 1989, 73, 2081–2085. [Google Scholar] [CrossRef] [Green Version]
- Chesi, M.; Bergsagel, P.L.; Shonukan, O.O.; Martelli, M.L.; Brents, L.A.; Chen, T.; Schroöck, E.; Ried, T.; Kuehl, W.M. Frequent Dysregulation of the c-maf Proto-Oncogene at 16q23 by Translocation to an Ig Locus in Multiple Myeloma. Blood 1998, 91, 4457–4463. [Google Scholar] [CrossRef]
- Hurt, E.M.; Wiestner, A.; Rosenwald, A.; Shaffer, A.; Campo, E.; Grogan, T.; Bergsagel, P.; Kuehl, W.; Staudt, L.M. Overexpression of c-maf is a frequent oncogenic event in multiple myeloma that promotes proliferation and pathological interactions with bone marrow stroma. Cancer Cell 2004, 5, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Bergsagel, P.L.; Kuehl, W.M. Chromosome translocations in multiple myeloma. Oncogene 2001, 20, 5611–5622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, B.; Wardell, C.P.; Johnson, D.C.; Kaiser, M.F.; Begum, D.B.; Dahir, N.B.; Ross, F.M.; Davies, F.E.; Gonzalez, D.; Morgan, G. Characterization of IGH locus breakpoints in multiple myeloma indicates a subset of translocations appear to occur in pregerminal center B cells. Blood 2013, 121, 3413–3419. [Google Scholar] [CrossRef] [PubMed]
- Streubel, B.; Lamprecht, A.; Dierlamm, J.; Cerroni, L.; Stolte, M.; Ott, G.; Raderer, M.; Chott, A. T(14;18)(q32;q21) involving IGH andMALT1 is a frequent chromosomal aberration in MALT lymphoma. Blood 2003, 101, 2335–2339. [Google Scholar] [CrossRef] [PubMed]
- Penas, E.M.M.; Callet-Bauchu, E.; Ye, H.; Gazzo, S.; Berger, F.; Schilling, G.; Albert-Konetzny, N.; Vettorazzi, E.; Salles, G.; Wlodarska, I.; et al. The t(14;18)(q32;q21)/IGH-MALT1 translocation in MALT lymphomas contains templated nucleotide insertions and a major breakpoint region similar to follicular and mantle cell lymphoma. Blood 2010, 115, 2214–2219. [Google Scholar] [CrossRef] [Green Version]
- Dalla-Favera, R.; Martinotti, S.; Gallo, R.C.; Erikson, J.; Croce, C.M. Translocation and rearrangements of the c-myc oncogene locus in human undifferentiated B-cell lymphomas. Science 1983, 219, 963–967. [Google Scholar] [CrossRef]
- Kornblau, S.M.; Goodacre, A.; Cabanillas, F. Chromosomal abnormalities in adult non-endemic burkitt’s lymphoma and leukemia: 22 new reports and a review of 148 cases from the literature. Hematol. Oncol. 2006, 9, 63–78. [Google Scholar] [CrossRef]
- Taub, R.; Kirsch, I.; Morton, C.; Lenoir, G.; Swan, D.; Tronick, S.; Aaronson, S.; Leder, P. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7837–7841. [Google Scholar] [CrossRef] [Green Version]
- Ladanyi, M.; Offit, K.; Jhanwar, S.C.; Filippa, D.A.; Chaganti, R.S. MYC rearrangement and translocations involving band 8q24 in diffuse large cell lymphomas. Blood 1991, 77, 1057–1063. [Google Scholar] [CrossRef] [Green Version]
- Quintanilla-Martinez, L. The 2016 updated WHO classification of lymphoid neoplasias. Hematol. Oncol. 2017, 35, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.-J.; Dyer, P.N.; Tremethick, D.J.; Luger, K. A New Fluorescence Resonance Energy Transfer Approach Demonstrates That the Histone Variant H2AZ Stabilizes the Histone Octamer within the Nucleosome. J. Biol. Chem. 2004, 279, 24274–24282. [Google Scholar] [CrossRef] [Green Version]
- Neri, A.; Chang, C.-C.; Lombardi, L.; Salina, M.; Corradini, P.; Maiolo, A.T.; Chaganti, R.; Dalla-Favera, R. B cell lymphoma-associated chromosomal translocation involves candidate oncogene lyt-10, homologous to NF-κB p50. Cell 1991, 67, 1075–1087. [Google Scholar] [CrossRef]
- Migliazza, A.; Lombardi, L.; Rocchi, M.; Trecca, D.; Chang, C.; Antonacci, R.; Fracchiolla, N.; Ciana, P.; Maiolo, A.; Neri, A. Heterogeneous chromosomal aberrations generate 3’ truncations of the NFKB2/lyt-10 gene in lymphoid malignancies. Blood 1994, 84, 3850–3860. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Zhang, J.; Lombardi, L.; Neri, A.; Dalla-Favera, R. Rearranged NFKB-2 genes in lymphoid neoplasms code for constitutively active nuclear transactivators. Mol. Cell. Biol. 1995, 15, 5180–5187. [Google Scholar] [CrossRef] [Green Version]
- Busslinger, M.; Klix, N.; Pfeffer, P.; Graninger, P.G.; Kozmik, Z. Deregulation of PAX-5 by translocation of the Emu enhancer of the IgH locus adjacent to two alternative PAX-5 promoters in a diffuse large-cell lymphoma. Proc. Natl. Acad. Sci. USA 1996, 93, 6129–6134. [Google Scholar] [CrossRef] [Green Version]
- Iida, S.; Rao, P.H.; Nallasivam, P.; Hibshoosh, H.; Butler, M.; Louie, D.C.; Dyomin, V.; Ohno, H.; Chaganti, R.S.; Dalla-Favera, R. The t(9;14)(p13;q32) chromosomal translocation associated with lymphoplasmacytoid lymphoma involves the PAX-5 gene. Blood 1996, 88, 4110–4117. [Google Scholar] [CrossRef] [Green Version]
- Ferrad, M.; Ghazzaui, N.; Issaoui, H.; Cook-Moreau, J.; Denizot, Y. Mouse Models of c-myc Deregulation Driven by IgH Locus Enhancers as Models of B-Cell Lymphomagenesis. Front. Immunol. 2020, 11, 1564. [Google Scholar] [CrossRef]
- Ghazzaui, N.; Saintamand, A.; Issaoui, H.; Vincent-Fabert, C.; Denizot, Y. The IgH 3’ regulatory region and c-myc-induced B-cell lymphomagenesis. Oncotarget 2016, 8, 7059–7067. [Google Scholar] [CrossRef] [Green Version]
- Ghazzaui, N.; Issaoui, H.; Ferrad, M.; Carrion, C.; Cook-Moreau, J.; Denizot, Y.; Boyer, F. Eμ and 3′RR transcriptional enhancers of the IgH locus cooperate to promote c-myc–induced mature B-cell lymphomas. Blood Adv. 2020, 4, 28–39. [Google Scholar] [CrossRef] [Green Version]
- Ghazzaui, N.; Saintamand, A.; Issaoui, H.; Saad, F.; Denizot, Y. Efficient role of IgH 3′ regulatory region deficient B-cells in the development of oil granulomas. Oncotarget 2016, 7, 38741–38749. [Google Scholar] [CrossRef] [Green Version]
- Donnou, S.; Galand, C.; Touitou, V.; Sautès-Fridman, C.; Fabry, Z.; Fisson, S. Murine Models of B-Cell Lymphomas: Promising Tools for Designing Cancer Therapies. Adv. Hematol. 2012, 2012, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Xiang, H.; Noonan, E.J.; Wang, J.; Duan, H.; Ma, L.; Michie, S.; Boxer, L.M. The immunoglobulin heavy chain gene 3′ enhancers induce Bcl2 deregulation and lymphomagenesis in murine B cells. Leukemia 2011, 25, 1484–1493. [Google Scholar] [CrossRef] [Green Version]
- Baron, B.W.; Anastasi, J.; Montag, A.; Huo, D.; Baron, R.M.; Karrison, T.; Thirman, M.J.; Subudhi, S.K.; Chin, R.K.; Felsher, D.W.; et al. The human BCL6 transgene promotes the development of lymphomas in the mouse. Proc. Natl. Acad. Sci. USA 2004, 101, 14198–14203. [Google Scholar] [CrossRef] [Green Version]
- Fiancette, R.; Amin, R.; Truffinet, V.; Vincent-Fabert, C.; Cogné, N.; Cogné, M.; Denizot, Y. A myeloma translocation-like model associating CCND1 with the immunoglobulin heavy-chain locus 3′ enhancers does not promote by itself B-cell malignancies. Leuk. Res. 2010, 34, 1043–1051. [Google Scholar] [CrossRef]
- Lovec, H.; Grzeschiczek, A.; Kowalski, M.; Möröy, T. Cyclin D1/bcl-1 cooperates with myc genes in the generation of B-cell lymphoma in transgenic mice. EMBO J. 1994, 13, 3487–3495. [Google Scholar] [CrossRef]
- Morito, N.; Yoh, K.; Maeda, A.; Nakano, T.; Fujita, A.; Kusakabe, M.; Hamada, M.; Kudo, T.; Yamagata, K.; Takahashi, S. A novel transgenic mouse model of the human multiple myeloma chromosomal translocation t(14;16)(q32;q23). Cancer Res. 2011, 71, 339–348. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.; Harris, A.W.; Pinkert, C.A.; Corcoran, L.M.; Alexander, W.S.; Cory, S.; Palmiter, R.D.; Brinster, R.L. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nat. Cell Biol. 1985, 318, 533–538. [Google Scholar] [CrossRef]
- Han, S.S.; Shaffer, A.L.; Peng, L.; Chung, S.T.; Lim, J.H.; Maeng, S.; Kim, J.S.; McNeil, N.; Ried, T.; Staudt, L.M.; et al. Molecular and cytological features of the mouse B-cell lymphoma line iMyc-Eμ-1. Mol. Cancer 2005, 4, 1–10. [Google Scholar]
- Han, S.S.; Yun, H.; Son, D.J.; Tompkins, V.S.; Peng, L.; Chung, S.T.; Kim, J.S.; Park, E.S.; Janz, S. NF-κB/STAT3/PI3K signaling crosstalk in iMycEμB lymphoma. Mol. Cancer 2010, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Boxer, L.M. Regulatory Elements in the Immunoglobulin Heavy Chain Gene 3′-Enhancers Induce c-myc Deregulation and Lymphomagenesis in Murine B Cells. J. Biol. Chem. 2005, 280, 12766–12773. [Google Scholar] [CrossRef] [Green Version]
- Truffinet, V.; Pinaud, E.; Cogné, N.; Petit, B.; Guglielmi, L.; Cogné, M.; Denizot, Y. The 3′ IgH Locus Control Region Is Sufficient to Deregulate a c-myc Transgene and Promote Mature B Cell Malignancies with a Predominant Burkitt-Like Phenotype. J. Immunol. 2007, 179, 6033–6042. [Google Scholar] [CrossRef] [Green Version]
- Kovalchuk, A.L.; Sakai, T.; Qi, C.-F.; Du Bois, W.; Dunnick, W.A.; Cogné, M.; Morse, H.C. 3’Igh enhancers hs3b/hs4 are dispensable for Myc deregulation in mouse plasmacytomas with T(12;15) translocations. Oncotarget 2018, 9, 34528–34542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gostissa, M.; Yan, C.T.; Bianco, J.M.; Cogne, M.; Pinaud, E.; Alt, F.W. Long-range oncogenic activation of Igh–c-myc translocations by the Igh 3′ regulatory region. Nature 2009, 462, 803–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.S.; Shaffer, A.L.; Kim, J.S.; Dubois, W.; Potter, M.; Staudt, L.M.; Janz, S. Insertion of Myc into Igh Accelerates Peritoneal Plasmacytomas in Mice. Cancer Res. 2005, 65, 7644–7652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, W.C.; Kim, J.S.; Linden, M.; Peng, L.; Van Ness, B.; Polakiewicz, R.D.; Janz, S. Novel targeted deregulation of c-Myc cooperates with Bcl-XL to cause plasma cell neoplasms in mice. J. Clin. Investig. 2004, 113, 1763–1773. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.; Xiang, H.; Ma, L.; Boxer, L.M. Functional long-range interactions of the IgH 3′ enhancers with the bcl-2 promoter region in t(14;18) lymphoma cells. Oncogene 2008, 27, 6720–6728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Nikolaidis, N.; Nei, M. Genomic organization and evolution of immunoglobulin kappa gene enhancers and kappa deleting element in mammals. Mol. Immunol. 2009, 46, 3171–3177. [Google Scholar] [CrossRef] [Green Version]
- McDevit, D.C.; Perkins, L.; Atchison, M.L.; Nikolajczyk, B.S. The Igκ3′ Enhancer Is Activated by Gradients of Chromatin Accessibility and Protein Association. J. Immunol. 2005, 174, 2834–2842. [Google Scholar] [CrossRef] [Green Version]
- Grange, S.; Boyes, J. Chromatin opening is tightly linked to enhancer activation at the κ light chain locus. Biochem. Biophys. Res. Commun. 2007, 363, 223–228. [Google Scholar] [CrossRef]
- Nikolajczyk, B.S.; Sardi, S.H.; Tumang, J.R.; Ganley-Leal, L.M. Immunoglobulin kappa enhancers are differentially regulated at the level of chromatin structure. Mol. Immunol. 2007, 44, 3407–3415. [Google Scholar] [CrossRef] [Green Version]
- Stadhouders, R.; De Bruijn, M.J.W.; Rother, M.B.; Yuvaraj, S.; De Almeida, C.R.; Kolovos, P.; Van Zelm, M.C.; Van Ijcken, W.; Grosveld, F.; Soler, E.; et al. Pre-B Cell Receptor Signaling Induces Immunoglobulin κ Locus Accessibility by Functional Redistribution of Enhancer-Mediated Chromatin Interactions. PLoS ONE 2014, 12, e1001791. [Google Scholar] [CrossRef] [Green Version]
- Hewitt, S.L.; Farmer, D.; Marszalek, K.; Cadera, E.; Liang, H.-E.; Xu, Y.; Schlissel, M.S.; Skok, J.A. Association between the Igk and Igh immunoglobulin loci mediated by the 3′ Igk enhancer induces ’decontraction’ of the Igh locus in pre–B cells. Nat. Immunol. 2008, 9, 396–404. [Google Scholar] [CrossRef]
- Fulton, R.; Van Ness, B. Selective synergy of immunoglobulin enhancer elements in B-cell development: A characteristic of kappa light chain enhancers, but not heavy chain enhancers. Nucleic Acids Res. 1994, 22, 4216–4223. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Garrard, W.T. Long-Range Interactions between Three Transcriptional Enhancers, Active Vκ Gene Promoters, and a 3′ Boundary Sequence Spanning 46 Kilobases. Mol. Cell. Biol. 2005, 25, 3220–3231. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Xiang, Y.; Garrard, W.T. The Igκ Gene Enhancers, E3′ and Ed, Are Essential for Triggering Transcription. J. Immunol. 2010, 185, 7544–7552. [Google Scholar] [CrossRef] [Green Version]
- Asenbauer, H.; Combriato, G.; Klobeck, H. The immunoglobulin lambda light chain enhancer consists of three modules which synergize in activation of transcription. Eur. J. Immunol. 1999, 29, 713–724. [Google Scholar] [CrossRef]
- Haque, S.F.; Bevington, S.L.; Boyes, J. The Eλ3–1 enhancer is essential for V(D)J recombination of the murine immunoglobulin lambda light chain locus. Biochem. Biophys. Res. Commun. 2013, 441, 482–487. [Google Scholar] [CrossRef] [Green Version]
- Hagman, J.; Rudin, C.M.; Haasch, D.; Chaplin, D.; Storb, U. A novel enhancer in the immunoglobulin lambda locus is duplicated and functionally independent of NF kappa B. Genes Dev. 1990, 4, 978–992. [Google Scholar] [CrossRef] [Green Version]
- Blomberg, B.B.; Rudin, C.M.; Storb, U. Identification and localization of an enhancer for the human lambda L chain Ig gene complex. J. Immunol. 1991, 147, 2354–2358. [Google Scholar]
- Combriato, G.; Klobeck, H.-G. Regulation of Human Igλ Light Chain Gene Expression by NF-κB. J. Immunol. 2002, 168, 1259–1266. [Google Scholar] [CrossRef] [Green Version]
- Chong, L.C.; Ben-Neriah, S.; Slack, G.W.; Freeman, C.; Ennishi, D.; Mottok, A.; Collinge, B.; Abrisqueta, P.; Farinha, P.; Boyle, M.; et al. High-resolution architecture and partner genes of MYC rearrangements in lymphoma with DLBCL morphology. Blood Adv. 2018, 2, 2755–2765. [Google Scholar] [CrossRef]
- Einerson, R.R.; Law, M.E.; Blair, H.E.; Kurtin, P.J.; McClure, R.F.; Ketterling, R.P.; Flynn, H.C.; Dogan, A.; Remstein, E.D. Novel FISH probes designed to detect IGK-MYC and IGL-MYC rearrangements in B-cell lineage malignancy identify a new breakpoint cluster region designated BVR2. Leukemia 2006, 20, 1790–1799. [Google Scholar] [CrossRef]
- Kroenlein, H.; Schwartz, S.; Reinhardt, R.; Rieder, H.; Molkentin, M.; Gökbuget, N.; Hoelzer, D.; Thiel, E.; Burmeister, T. Molecular analysis of the t(2;8)/MYC-IGK translocation in high-grade lymphoma/leukemia by long-distance inverse PCR. Genes Chromosomes Cancer 2012, 51, 290–299. [Google Scholar] [CrossRef]
- Angi, M.; Kamath, V.; Yuvarani, S.; Meena, J.; Sitaram, U.; Manipadam, M.T.; Nair, S.; Ganapule, A.; Fouzia, N.; Abraham, A.; et al. The t(8;14)(q24.1;q32) and its variant translocations: A study of 34 cases. Hematol. Stem Cell Ther. 2017, 10, 126–134. [Google Scholar] [CrossRef]
- Tomita, N.; Tokunaka, M.; Nakamura, N.; Takeuchi, K.; Koike, J.; Motomura, S.; Miyamoto, K.; Kikuchi, A.; Hyo, R.; Yakushijin, Y.; et al. Clinicopathological features of lymphoma/leukemia patients carrying both BCL2 and MYC translocations. Haematologica 2009, 94, 935–943. [Google Scholar] [CrossRef]
- Rätsch, A.; Joos, S.; Kioschis, P.; Lichter, P. Topological Organization of the MYC/IGK Locus in Burkitt’s Lymphoma Cells Assessed by Nuclear Halo Preparations. Exp. Cell Res. 2002, 273, 12–20. [Google Scholar] [CrossRef]
- Barwick, B.G.; Neri, P.; Bahlis, N.J.; Nooka, A.K.; Dhodapkar, M.V.; Jaye, D.L.; Hofmeister, C.C.; Kaufman, J.L.; Gupta, V.; Auclair, D.; et al. Multiple myeloma immunoglobulin lambda translocations portend poor prognosis. Nat. Commun. 2019, 10, 1911. [Google Scholar] [CrossRef] [Green Version]
- Aventin, A.; Mecucci, C.; Guanyabens, C.; Brunet, S.; Soler, J.; Bordes, R.; Berghe, H.V.D. Variant T(2;18) Translocation in a Burkitt Conversion of Follicular Lymphoma. Br. J. Haematol. 1990, 74, 367–369. [Google Scholar] [CrossRef]
- Bentley, G.; Palutke, M.; Mohamed, A.N. Variant t(14;18) in malignant lymphoma: A report of seven cases. Cancer Genet. Cytogenet. 2005, 157, 12–17. [Google Scholar] [CrossRef]
- Bertheas, M.-F.; Rimokh, R.; Berger, F.; Gaucherand, M.; Machado, P.; Vasselon, C.; Calmard-Oriol, P.; Jaubert, J.; Guyotat, D.; Magaud, J.-P. Molecular Study of a Variant Translocation T(2;18)(P11;Q21) in a Follicular Lymphoma. Br. J. Haematol. 1991, 78, 132–134. [Google Scholar] [CrossRef] [PubMed]
- Hillion, J.; Mecucci, C.; Aventin, A.; Leroux, D.; Wlodarska, I.; Berghe, H.V.D.; Larsen, C.J. A variant translocation t(2;18) in follicular lymphoma involves the 5’ end of bcl-2 and Ig kappa light chain gene. Oncogene 1991, 6, 169–172. [Google Scholar] [PubMed]
- Larsen, C.J.; Mecucci, C.; Leroux, D. t(2;18) and t(18;22) variant chromosomal translocations and bcl-2 gene rearrangements in human malignant lymphomas. Nouv. Rev. Fr. D’hematologie 1990, 32, 401–403. [Google Scholar]
- Leroux, D.; Monteil, M.; Sotto, J.J.; Jacob, M.C.; Le Marc’Hadour, F.; Bonnefoi, H.; Jalbert, P. Variant t(2;18) translocation in a follicular lymphoma. Br. J. Haematol. 1990, 75, 290–292. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Jetly, R.; Lennon, P.A.; Abruzzo, L.V.; Prajapati, S.; Medeiros, L.J. Translocation (18;22)(q21;q11) in B-cell lymphomas: A report of 4 cases and review of the literature. Hum. Pathol. 2008, 39, 1664–1672. [Google Scholar] [CrossRef]
- Tashiro, S.; Takechi, M.; Asou, H.; Takauchi, K.; Kyo, T.; Dohy, H.; Kikuchi, M.; Kamada, N.; Tsujimoto, Y. Cytogenetic 2; 18 and 18; 22 translocation in chronic lymphocytic leukemia with juxtaposition of bcl-2 and immunoglobulin light chain genes. Oncogene 1992, 7, 573–577. [Google Scholar]
- Wlodarska, I.; Meeus, P.; Stul, M.; Thienpont, L.; Wouters, E.; Marcelis, L.; Demuynck, H.; Rummens, J.; Madoe, V.; Hagemeijer, A. Variant t(2;11)(p11;q13) associated with the IgK-CCND1 rearrangement is a recurrent translocation in leukemic small-cell B-non-Hodgkin lymphoma. Leukemia 2004, 18, 1705–1710. [Google Scholar] [CrossRef]
- Gesk, S.; Klapper, W.; Martin-Subero, J.I.; Nagel, I.; Harder, L.; Fu, K.; Bernd, H.-W.; Weisenburger, D.D.; Parwaresch, R.; Siebert, R. A chromosomal translocation in cyclin D1–negative/cyclin D2–positive mantle cell lymphoma fuses the CCND2 gene to the IGK locus. Blood 2006, 108, 1109–1110. [Google Scholar] [CrossRef]
- Rocha, C.K.; Praulich, I.; Gehrke, I.; Hallek, M.; Kreuzer, K.-A. A rare case of t(11;22) in a mantle cell lymphoma like B-cell neoplasia resulting in a fusion of IGL and CCND1: Case report. Mol. Cytogenet. 2011, 4, 8. [Google Scholar] [CrossRef] [Green Version]
- Shiller, S.M.; Zieske, A.; Holmes, H.; Feldman, A.L.; Law, M.E.; Saad, R. CD5-positive, cyclinD1-negative mantle cell lymphoma with a translocation involving the CCND2 gene and the IGL locus. Cancer Genet. 2011, 204, 162–164. [Google Scholar] [CrossRef]
- Fuster, C.; Martín-Garcia, D.; Balagué, O.; Navarro, A.; Nadeu, F.; Costa, D.; Prieto, M.; Salaverria, I.; Espinet, B.; Rivas-Delgado, A.; et al. Cryptic insertions of the immunoglobulin light chain enhancer region near CCND1 in t(11;14)-negative mantle cell lymphoma. Haematology 2019, 105, e408–e411. [Google Scholar] [CrossRef] [Green Version]
- Marrero, W.D.; Cruz-Chacón, A.; Cabanillas, F. Mantle Cell Lymphoma with t(11;22) (q13;q11.2) an indolent clinical variant? Leuk. Lymphoma 2018, 59, 2509–2511. [Google Scholar] [CrossRef]
- Martín-Garcia, D.; Navarro, A.; Valdés-Mas, R.; Clot, G.; Gutiérrez-Abril, J.; Prieto, M.; Ribera-Cortada, I.; Woroniecka, R.; Rymkiewicz, G.; Bens, S.; et al. CCND2 and CCND3 hijack immunoglobulin light-chain enhancers in cyclin D1—mantle cell lymphoma. Blood 2019, 133, 940–951. [Google Scholar] [CrossRef]
- Martin-Subero, J.I.; Klapper, W.; Sotnikova, A.; Callet-Bauchu, E.; Harder, L.; Bastard, C.; Schmitz, R.; Grohmann, S.; Höppner, J.; Riemke, J.; et al. Chromosomal Breakpoints Affecting Immunoglobulin Loci Are Recurrent in Hodgkin and Reed-Sternberg Cells of Classical Hodgkin Lymphoma. Cancer Res. 2006, 66, 10332–10338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, Y.; Nomura, K.; Fukada, S.; Shimizu, D.; Shimura, K.; Matsumoto, Y.; Horiike, S.; Nishida, K.; Shimazaki, C.; Abe, M.; et al. Immunoglobulin light chain gene translocations in non-Hodgkin’s lymphoma as assessed by fluorescence in situ hybridisation. Eur. J. Haematol. 2008, 80, 143–150. [Google Scholar] [CrossRef]
- Suzuki, K.; Miki, T.; Kawamata, N.; Hirosawa, S.; Yoshizawa, K.; Kiyosawa, K.; Aoki, N. Variant translocation of the BCL6 gene to immunoglobulin kappa light chain gene in B-cell lymphoma. Japanese journal of cancer research. Gann 1994, 85, 911–917. [Google Scholar]
- Achuthan, R.; Bell, S.; Leek, J.; Roberts, P.; Horgan, K.; Markham, A.; Selby, P.; MacLennan, K. Novel translocation of theBCL10 gene in a case of mucosa associated lymphoid tissue lymphoma. Genes Chromosom. Cancer 2000, 29, 347–349. [Google Scholar] [CrossRef]
- Chuang, S.-S.; Liu, H.; Ye, H.; Martin-Subero, J.I.; Siebert, R.; Huang, W.-T. Pulmonary mucosa-associated lymphoid tissue lymphoma with strong nuclear B-cell CLL/lymphoma 10 (BCL10) expression and novel translocation t(1;2)(p22;p12)/immunoglobulin chain-BCL10. J. Clin. Pathol. 2007, 60, 727–728. [Google Scholar] [CrossRef] [Green Version]
- Hebert, J.; Romana, S.P.; Hillion, J.; Kerkaert, J.P.; Bastard, C.; Berger, R. Translocation t(3;22)(q27;q11) in non-Hodgkin’s malignant lymphoma: Chromosome painting and molecular studies. Leukemia 1993, 7, 1971–1974. [Google Scholar]
- Hörtnagel, K.; Mautner, J.; Strobl, L.J.; Wolf, D.; Christoph, B.; Geltinger, C.; Polack, A. The role of immunoglobulin kappa elements in c-myc activation. Oncogene 1995, 10, 1393–1401. [Google Scholar]
- Wittekindt, N.E.; Hörtnagel, K.; Geltinger, C.; Polack, A. Activation of c-myc promoter P1 by immunoglobulin kappa gene enhancers in Burkitt lymphoma: Functional characterization of the intron enhancer motifs kappaB, E box 1 and E box 2, and of the 3’ enhancer motif PU. Nucleic Acids Res. 2000, 28, 800–808. [Google Scholar] [CrossRef] [Green Version]
- Kovalchuk, A.L.; Qi, C.-F.; Torrey, T.A.; Taddesse-Heath, L.; Feigenbaum, L.; Park, S.S.; Gerbitz, A.; Klobeck, G.; Hoertnagel, K.; Polack, A.; et al. Burkitt Lymphoma in the Mouse. J. Exp. Med. 2000, 192, 1183–1190. [Google Scholar] [CrossRef]
- Axelson, H.; Panda, C.K.; Silva, S.; Sugiyama, H.; Wiener, F.; Klein, G.; Sumegi, J. A new variant 15; 16 translocation in mouse plasmacytoma leads to the juxtaposition of c-myc and immunoglobulin lambda. Oncogene 1991, 6, 2263–2270. [Google Scholar] [PubMed]
- Axelson, H.; Wang, Y.; Silva, S.; Mattei, M.-G.; Klein, G. Juxtaposition ofN-myc andIgk through a reciprocal t(6;12) translocation in a mouse plasmacytoma. Genes Chromosom. Cancer 1994, 11, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, M.; Wiener, F.; Spira, J.; Babonits, M.; Nilsson, M.; Sumegi, J.; Klein, G. Mapping of the c-myc, pvt-1 and immunoglobulin kappa genes in relation to the mouse plasmacytoma-associated variant (6;15) translocation breakpoint. EMBO J. 1985, 4, 3183–3188. [Google Scholar] [CrossRef] [PubMed]
- Gadó, K.; Silva, S.; Pálóczi, K.; Domján, G.; Falus, A. Mouse plasmacytoma: An experimental model of human multiple myeloma. Haematology 2001, 86, 227–236. [Google Scholar]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef] [Green Version]
- Rheinbay, E.; PCAWG Drivers and Functional Interpretation Working Group; Nielsen, M.M.; Abascal, F.; Wala, J.A.; Shapira, O.; Tiao, G.; Hornshøj, H.; Hess, J.M.; Juul, R.I.; et al. Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nat. Cell Biol. 2020, 578, 102–111. [Google Scholar] [CrossRef] [Green Version]
- Puente, X.S.; Beà, S.; Valdés-Mas, R.; Villamor, N.; Gutiérrez-Abril, J.; Martín-Subero, J.I.; Munar, M.; Rubio-Pérez, C.; Jares, P.; Aymerich, M.; et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015, 526, 519–524. [Google Scholar] [CrossRef]
- Arthur, S.E.; Jiang, A.; Grande, B.M.; Alcaide, M.; Cojocaru, R.; Rushton, C.K.; Mottok, A.; Hilton, L.K.; Lat, P.K.; Zhao, E.Y.; et al. Genome-wide discovery of somatic regulatory variants in diffuse large B-cell lymphoma. Nat. Commun. 2018, 9, 4001. [Google Scholar] [CrossRef] [Green Version]
- Grande, B.M.; Gerhard, D.S.; Jiang, A.; Griner, N.B.; Abramson, J.S.; Alexander, T.; Allen, H.; Ayers, L.W.; Bethony, J.M.; Bhatia, K.; et al. Genome-wide discovery of somatic coding and noncoding mutations in pediatric endemic and sporadic Burkitt lymphoma. Blood 2019, 133, 1313–1324. [Google Scholar] [CrossRef]
- Cobaleda, C.; Schebesta, A.; Delogu, A.; Busslinger, M. Pax5: The guardian of B cell identity and function. Nat. Immunol. 2007, 8, 463–470. [Google Scholar] [CrossRef]
- Medvedovic, J.; Ebert, A.; Tagoh, H.; Busslinger, M. Pax5: A master regulator of B cell development and leukemogenesis. Adv. Immunol. 2011, 111, 179–206. [Google Scholar] [CrossRef]
- Mathelier, A.; Lefebvre, C.; Zhang, A.W.; Arenillas, D.J.; Ding, J.; Wasserman, W.W.; Shah, S.P. Cis-regulatory somatic mutations and gene-expression alteration in B-cell lymphomas. Genome Biol. 2015, 16, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Cornish, A.J.; Hoang, P.H.; Dobbins, S.E.; Law, P.J.; Chubb, D.; Orlando, G.; Houlston, R.S. Identification of recurrent noncoding mutations in B-cell lymphoma using capture Hi-C. Blood Adv. 2019, 3, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Klanova, M.; Klener, P. BCL-2 Proteins in Pathogenesis and Therapy of B-Cell Non-Hodgkin Lymphomas. Cancers 2020, 12, 938. [Google Scholar] [CrossRef] [Green Version]
- Wagner, S.D.; Ahearne, M.; Ferrigno, P.K. The role of BCL6 in lymphomas and routes to therapy. Br. J. Haematol. 2010, 152, 3–12. [Google Scholar] [CrossRef]
- Garnham, R.; Scott, E.; Livermore, K.E.; Munkley, J. ST6GAL1: A key player in cancer (Review). Oncol. Lett. 2019, 18, 983–989. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.-C.; Kamath-Loeb, A.S.; Kohrn, B.F.; Loeb, K.R.; Preston, B.D.; Loeb, L.A. A high-resolution landscape of mutations in the BCL6 super-enhancer in normal human B cells. Proc. Natl. Acad. Sci. USA 2019, 116, 24779–24785. [Google Scholar] [CrossRef]
- Speedy, H.E.; Beekman, R.; Chapaprieta, V.; Orlando, G.; Law, P.J.; Martín-García, D.; Gutiérrez-Abril, J.; Catovsky, D.; Beà, S.; Clot, G.; et al. Insight into genetic predisposition to chronic lymphocytic leukemia from integrative epigenomics. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Yan, H.; Tian, S.; Kleinstern, G.; Wang, Z.; Lee, J.-H.; Boddicker, N.J.; Cerhan, J.R.; Kay, N.E.; Braggio, E.; Slager, S.L. Chronic lymphocytic leukemia (CLL) risk is mediated by multiple enhancer variants within CLL risk loci. Hum. Mol. Genet. 2020, 29, 2761–2774. [Google Scholar] [CrossRef]
- Kandaswamy, R.; Sava, G.P.; Speedy, H.E.; Beà, S.; Martín-Subero, J.I.; Studd, J.B.; Migliorini, G.; Law, P.; Suarez-Puente, X.; Martín-García, D.; et al. Genetic Predisposition to Chronic Lymphocytic Leukemia Is Mediated by a BMF Super-Enhancer Polymorphism. Cell Rep. 2016, 16, 2061–2067. [Google Scholar] [CrossRef] [Green Version]
- De Smith, A.J.; Walsh, K.M.; Francis, S.S.; Zhang, C.; Hansen, H.M.; Smirnov, I.; Morimoto, L.; Whitehead, T.P.; Kang, A.; Shao, X.; et al. BMI1enhancer polymorphism underlies chromosome 10p12.31 association with childhood acute lymphoblastic leukemia. Int. J. Cancer 2018, 143, 2647–2658. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Zhang, H.; Luan, Y.; Liu, T.; Roberts, K.G.; Qian, M.; Zhang, B.; Yang, W.; Perez-Andreu, V.; Xu, J.; et al. Non-coding germline GATA3 variants alter chromatin topology and contribute to pathogenesis of acute lymphoblastic leukemia. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Koues, O.I.; Kowalewski, R.A.; Chang, L.-W.; Pyfrom, S.C.; Schmidt, J.A.; Luo, H.; Sandoval, L.E.; Hughes, T.B.; Bednarski, J.J.; Cashen, A.F.; et al. Enhancer Sequence Variants and Transcription-Factor Deregulation Synergize to Construct Pathogenic Regulatory Circuits in B-Cell Lymphoma. Immunity 2015, 42, 186–198. [Google Scholar] [CrossRef] [Green Version]
- Spitz, F.; Furlong, E. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef]
- Kron, K.J.; Bailey, S.D.; Lupien, M. Enhancer alterations in cancer: A source for a cell identity crisis. Genome Med. 2014, 6, 77. [Google Scholar] [CrossRef] [Green Version]
- Richart, L.; Bidard, F.-C.; Margueron, R. Enhancer rewiring in tumors: An opportunity for therapeutic intervention. Oncogene 2021, 40, 3475–3491. [Google Scholar] [CrossRef]
- Decker, T.; di Magliano, M.P.; McManus, S.; Sun, Q.; Bonifer, C.; Tagoh, H.; Busslinger, M. Stepwise Activation of Enhancer and Promoter Regions of the B Cell Commitment Gene Pax5 in Early Lymphopoiesis. Immunity 2009, 30, 508–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McManus, S.; Ebert, A.; Salvagiotto, G.; Medvedovic, J.; Sun, Q.; Tamir, I.; Jaritz, M.; Tagoh, H.; Busslinger, M. The transcription factor Pax5 regulates its target genes by recruiting chromatin-modifying proteins in committed B cells. EMBO J. 2011, 30, 2388–2404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bullerwell, C.E.; Robichaud, P.P.; Deprez, P.M.L.; Joy, A.P.; Wajnberg, G.; D’Souza, D.; Chacko, S.; Fournier, S.; Crapoulet, N.; Barnett, D.A.; et al. EBF1 drives hallmark B cell gene expression by enabling the interaction of PAX5 with the MLL H3K4 methyltransferase complex. Sci. Rep. 2021, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, R.; Jensen, C.T.; Tingvall-Gustafsson, J.; Åhsberg, J.; Okuyama, K.; Prasad, M.; Hagman, J.R.; Wang, X.; Soneji, S.; Strid, T.; et al. EBF1 and PAX5 control pro-B cell expansion via opposing regulation of the Myc gene. Blood 2021, 137, 3037–3049. [Google Scholar] [CrossRef]
- Ramamoorthy, S.; Kometani, K.; Herman, J.S.; Bayer, M.; Boller, S.; Edwards-Hicks, J.; Ramachandran, H.; Li, R.; Klein-Geltink, R.; Pearce, E.L.; et al. EBF1 and Pax5 safeguard leukemic transformation by limiting IL-7 signaling, Myc expression, and folate metabolism. Genes Dev. 2020, 34, 1503–1519. [Google Scholar] [CrossRef]
- Sood, R.; Kamikubo, Y.; Liu, P. Role of RUNX1 in hematological malignancies. Blood 2017, 129, 2070–2082. [Google Scholar] [CrossRef] [Green Version]
- Debaize, L.; Jakobczyk, H.; Avner, S.; Gaudichon, J.; Rio, A.; Sérandour, A.A.; Dorsheimer, L.; Chalmel, F.; Carroll, J.S.; Zörnig, M.; et al. Interplay between transcription regulators RUNX1 and FUBP1 activates an enhancer of the oncogene c-KIT and amplifies cell proliferation. Nucleic Acids Res. 2018, 46, 11214–11228. [Google Scholar] [CrossRef] [Green Version]
- Jakobczyk, H.; Debaize, L.; Soubise, B.; Avner, S.; Rouger-Gaudichon, J.; Commet, S.; Jiang, Y.; Sérandour, A.A.; Rio, A.-G.; Carroll, J.S.; et al. Reduction of RUNX1 transcription factor activity by a CBFA2T3-mimicking peptide: Application to B cell precursor acute lymphoblastic leukemia. J. Hematol. Oncol. 2021, 14, 1–17. [Google Scholar] [CrossRef]
- Gandemer, V.; Rio, A.-G.; De Tayrac, M.; Sibut, V.; Mottier, S.; Sunnaram, B.L.; Henry, C.; Monnier, A.; Berthou, C.; Le Gall, E.; et al. Five distinct biological processes and 14 differentially expressed genes characterize TEL/AML1-positive leukemia. BMC Genom. 2007, 8, 385. [Google Scholar] [CrossRef] [Green Version]
- Hunger, S. Chromosomal translocations involving the E2A gene in acute lymphoblastic leukemia: Clinical features and molecular pathogenesis. Blood 1996, 87, 1211–1224. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Mouttet, B.; Warnatz, H.-J.; Risch, T.; Rietmann, F.; Frommelt, F.; Ngo, Q.A.; Dobay, M.P.; Marovca, B.; Jenni, S.; et al. The Leukemogenic TCF3-HLF Complex Rewires Enhancers Driving Cellular Identity and Self-Renewal Conferring EP300 Vulnerability. Cancer Cell 2019, 36, 630–644.e9. [Google Scholar] [CrossRef]
- Ying, C.Y.; Dominguez-Sola, D.; Fabi, M.; Lorenz, I.C.; Hussein, S.; Bansal, M.; Califano, A.; Pasqualucci, L.; Basso, K.; Dalla-Favera, R. MEF2B mutations lead to deregulated expression of the oncogene BCL6 in diffuse large B cell lymphoma. Nat. Immunol. 2013, 14, 1084–1092. [Google Scholar] [CrossRef] [Green Version]
- Ryan, R.J.; Drier, Y.; Whitton, H.; Cotton, M.J.; Kaur, J.; Issner, R.; Gillespie, S.M.; Epstein, C.B.; Nardi, V.; Sohani, A.R.; et al. Detection of Enhancer-Associated Rearrangements Reveals Mechanisms of Oncogene Dysregulation in B-cell Lymphoma. Cancer Discov. 2015, 5, 1058–1071. [Google Scholar] [CrossRef] [Green Version]
- Benito, J.M.; Godfrey, L.; Kojima, K.; Hogdal, L.; Wunderlich, M.; Geng, H.; Marzo, I.; Harutyunyan, K.G.; Golfman, L.; North, P.; et al. MLL-Rearranged Acute Lymphoblastic Leukemias Activate BCL-2 through H3K79 Methylation and Are Sensitive to the BCL-2-Specific Antagonist ABT-199. Cell Rep. 2015, 13, 2715–2727. [Google Scholar] [CrossRef] [Green Version]
- Godfrey, L.; Kerry, J.; Thorne, R.; Repapi, E.; Davies, J.; Tapia, M.; Ballabio, E.; Hughes, J.R.; Geng, H.; Konopleva, M.; et al. MLL-AF4 binds directly to a BCL-2 specific enhancer and modulates H3K27 acetylation. Exp. Hematol. 2017, 47, 64–75. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Zhang, L.; Ke, L.; Ding, W.; Jiang, S.; Li, D.; Narita, Y.; Hou, I.; Liang, J.; Li, S.; et al. Primary effusion lymphoma enhancer connectome links super-enhancers to dependency factors. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Saha, A.; Robertson, E.S. Epstein-Barr Virus–Associated B-cell Lymphomas: Pathogenesis and Clinical Outcomes. Clin. Cancer Res. 2011, 17, 3056–3063. [Google Scholar] [CrossRef] [Green Version]
- Shannon-Lowe, C.; Rickinson, A.B.; Bell, A.I. Epstein-Barr virus-associated lymphomas. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2017, 372, 20160271. [Google Scholar] [CrossRef]
- Zhou, H.; Schmidt, S.C.; Jiang, S.; Willox, B.; Bernhardt, K.; Liang, J.; Johannsen, E.C.; Kharchenko, P.; Gewurz, B.E.; Kieff, E.; et al. Epstein-Barr Virus Oncoprotein Super-enhancers Control B Cell Growth. Cell Host Microbe 2015, 17, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Zou, J.; Wang, H.; Johannsen, E.; Peng, C.-W.; Quackenbush, J.; Mar, J.; Morton, C.C.; Freedman, M.L.; Blacklow, S.C.; et al. Epstein-Barr virus exploits intrinsic B-lymphocyte transcription programs to achieve immortal cell growth. Proc. Natl. Acad. Sci. USA 2011, 108, 14902–14907. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Zhou, H.; Gerdt, C.; Tan, M.; Colson, T.; Kaye, K.M.; Kieff, E.; Zhao, B. Epstein–Barr virus super-enhancer eRNAs are essential for MYC oncogene expression and lymphoblast proliferation. Proc. Natl. Acad. Sci. USA 2016, 113, 14121–14126. [Google Scholar] [CrossRef] [Green Version]
- Wood, C.D.; Veenstra, H.; Khasnis, S.; Gunnell, A.; Webb, H.M.; Shannon-Lowe, C.; Andrews, S.; Osborne, C.; West, M.J. MYC activation and BCL2L11 silencing by a tumour virus through the large-scale reconfiguration of enhancer-promoter hubs. eLife 2016, 5, e18270. [Google Scholar] [CrossRef]
- Jiang, S.; Zhou, H.; Liang, J.; Gerdt, C.; Wang, C.; Ke, L.; Schmidt, S.C.; Narita, Y.; Ma, Y.; Wang, S.; et al. The Epstein-Barr Virus Regulome in Lymphoblastoid Cells. Cell Host Microbe 2017, 22, 561–573.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClellan, M.J.; Wood, C.D.; Ojeniyi, O.; Cooper, T.J.; Kanhere, A.; Arvey, A.; Webb, H.M.; Palermo, R.D.; Harth-Hertle, M.L.; Kempkes, B.; et al. Modulation of Enhancer Looping and Differential Gene Targeting by Epstein-Barr Virus Transcription Factors Directs Cellular Reprogramming. PLoS Pathog. 2013, 9, e1003636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunnell, A.; Webb, H.M.; Wood, C.D.; McClellan, M.J.; Wichaidit, B.; Kempkes, B.; Jenner, R.G.; Osborne, C.; Farrell, P.J.; West, M.J. RUNX super-enhancer control through the Notch pathway by Epstein-Barr virus transcription factors regulates B cell growth. Nucleic Acids Res. 2016, 44, 4636–4650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spender, L.C.; Whiteman, H.; Karstegl, C.E.; Farrell, P. Transcriptional cross-regulation of RUNX1 by RUNX3 in human B cells. Oncogene 2005, 24, 1873–1881. [Google Scholar] [CrossRef] [PubMed]
- Kalchschmidt, J.; Bashford-Rogers, R.; Paschos, K.; Gillman, A.; Styles, C.; Kellam, P.; Allday, M.J. Epstein-Barr virus nuclear protein EBNA3C directly induces expression of AID and somatic mutations in B cells. J. Exp. Med. 2016, 213, 921–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazot, Q.; Paschos, K.; Skalska, L.; Kalchschmidt, J.; Parker, G.A.; Allday, M.J. Epstein-Barr Virus Proteins EBNA3A and EBNA3C Together Induce Expression of the Oncogenic MicroRNA Cluster miR-221/miR-222 and Ablate Expression of Its Target p57KIP2. PLoS Pathog. 2015, 11, e1005031. [Google Scholar] [CrossRef] [Green Version]
- Wood, C.D.; Carvell, T.; Gunnell, A.; Ojeniyi, O.O.; Osborne, C.; West, M.J. Enhancer Control of MicroRNA miR-155 Expression in Epstein-Barr Virus-Infected B Cells. J. Virol. 2018, 92, e00716-18. [Google Scholar] [CrossRef] [Green Version]
- Narkhede, M.; Arora, S.; Ujjani, C. Primary effusion lymphoma: Current perspectives. Oncotarget. Ther. 2018, 11, 3747–3754. [Google Scholar] [CrossRef] [Green Version]
- Carbone, A.; Gloghini, A.; Cozzi, M.R.; Capello, D.; Steffan, A.; Monini, P.; De Marco, L.; Gaidano, G. Expression of MUM1/IRF4 selectively clusters with primary effusion lymphoma among lymphomatous effusions: Implications for disease histogenesis and pathogenesis. Br. J. Haematol. 2000, 111, 247–257. [Google Scholar] [CrossRef]
- Manzano, M.; Günther, T.; Ju, H.; Nicholas, J.; Bartom, E.T.; Grundhoff, A.; Gottwein, E. Kaposi’s Sarcoma-Associated Herpesvirus Drives a Super-Enhancer-Mediated Survival Gene Expression Program in Primary Effusion Lymphoma. mBio 2020, 11. [Google Scholar] [CrossRef]
- Li, X.; Chen, S.; Feng, J.; Deng, H.; Sun, R. Myc Is Required for the Maintenance of Kaposi’s Sarcoma-Associated Herpesvirus Latency. J. Virol. 2010, 84, 8945–8948. [Google Scholar] [CrossRef] [Green Version]
- Park, A.; Oh, S.; Jung, K.L.; Choi, U.Y.; Lee, H.-R.; Rosenfeld, M.G.; Jung, J.U. Global epigenomic analysis of KSHV-infected primary effusion lymphoma identifies functionalMYCsuperenhancers and enhancer RNAs. Proc. Natl. Acad. Sci. USA 2020, 117, 21618–21627. [Google Scholar] [CrossRef]
- White, M.E.; Fenger, J.M.; Carson, W.E., 3rd. Emerging roles of and therapeutic strategies targeting BRD4 in cancer. Cell Immunol. 2019, 337, 48–53. [Google Scholar] [CrossRef]
- Verza, F.A.; Das, U.; Fachin, A.L.; Dimmock, J.R.; Marins, M. Roles of Histone Deacetylases and Inhibitors in Anticancer Therapy. Cancers 2020, 12, 1664. [Google Scholar] [CrossRef]
- Saad, F.; Saintamand, A.; Rouaud, P.; Denizot, Y. Targeting the oncogene B lymphoma deregulator IgH 3’ regulatory region does not impede the in vivo inflammatory response in mice. Oncoscience 2014, 1, 591–598. [Google Scholar] [CrossRef]
- Lu, Z.-P.; Ju, Z.-L.; Shi, G.-Y.; Zhang, J.-W.; Sun, J. Histone deacetylase inhibitor Trichostatin A reduces anti-DNA autoantibody production and represses IgH gene transcription. Biochem. Biophys. Res. Commun. 2005, 330, 204–209. [Google Scholar] [CrossRef]
- Dolloff, N.G. Discovery platform for inhibitors of IgH gene enhancer activity. Cancer Biol. Ther. 2019, 20, 571–581. [Google Scholar] [CrossRef]
- Wourms, M.J.; Sulentic, C.E. The aryl hydrocarbon receptor regulates an essential transcriptional element in the immunoglobulin heavy chain gene. Cell. Immunol. 2015, 295, 60–66. [Google Scholar] [CrossRef] [Green Version]

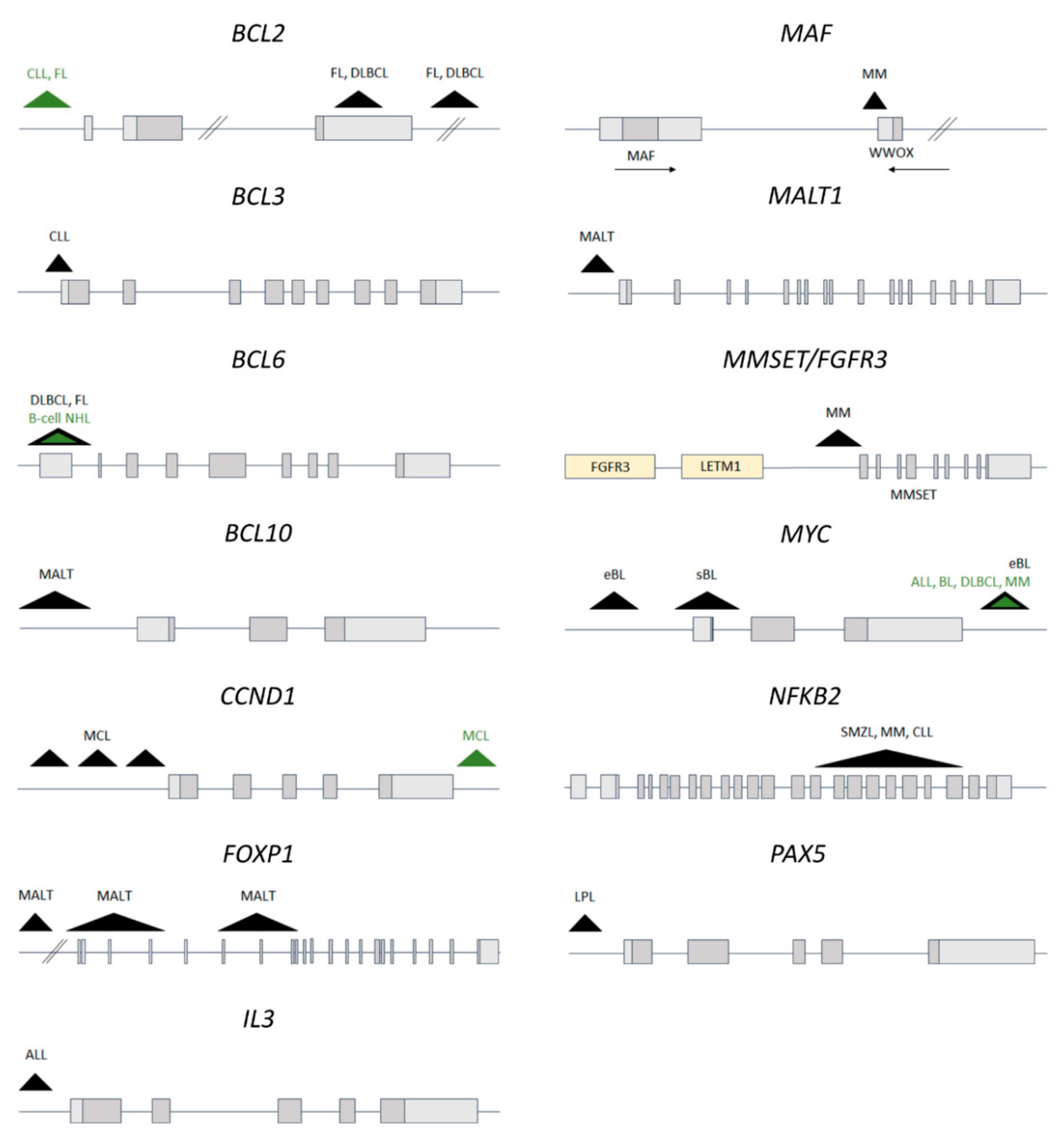
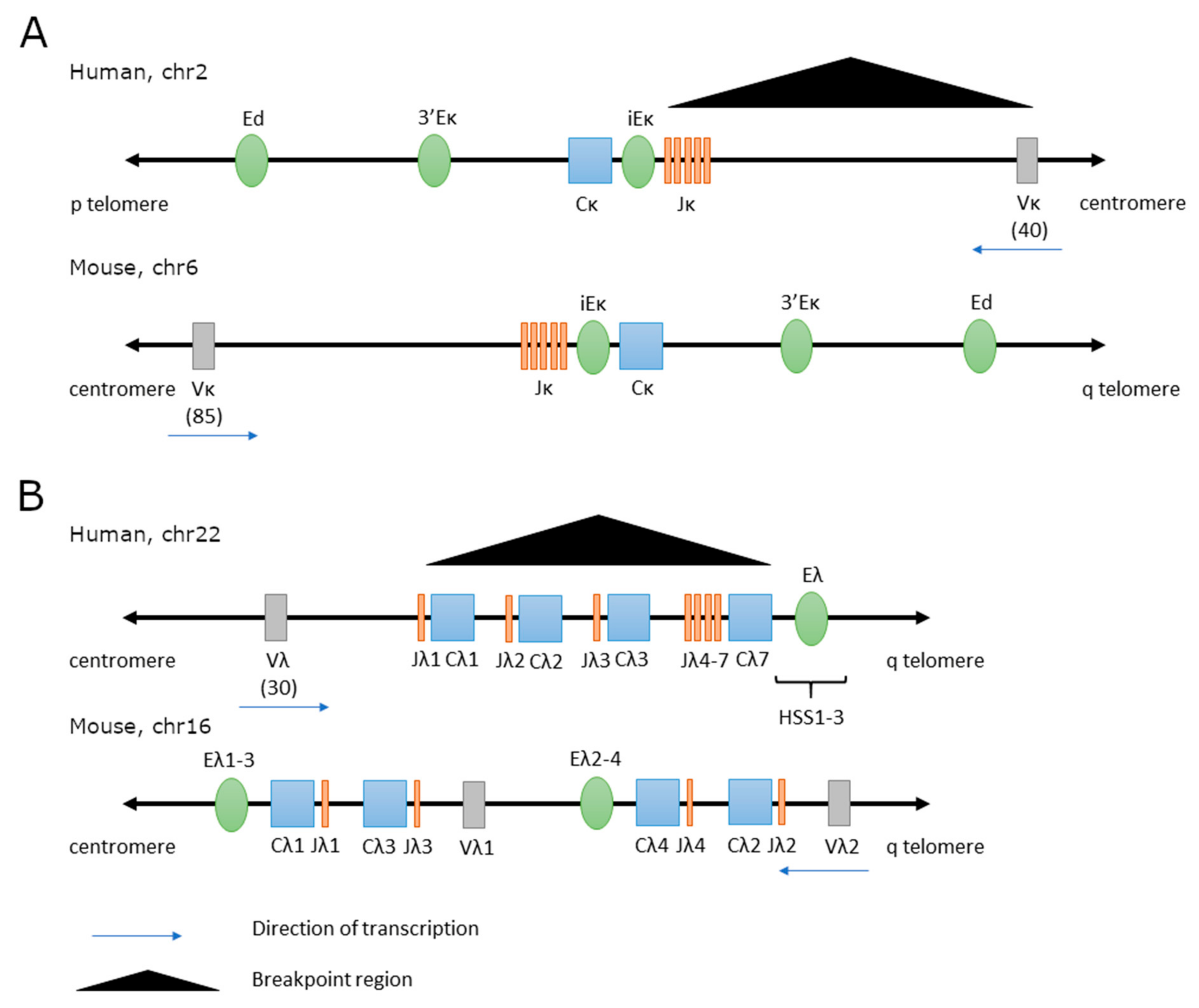
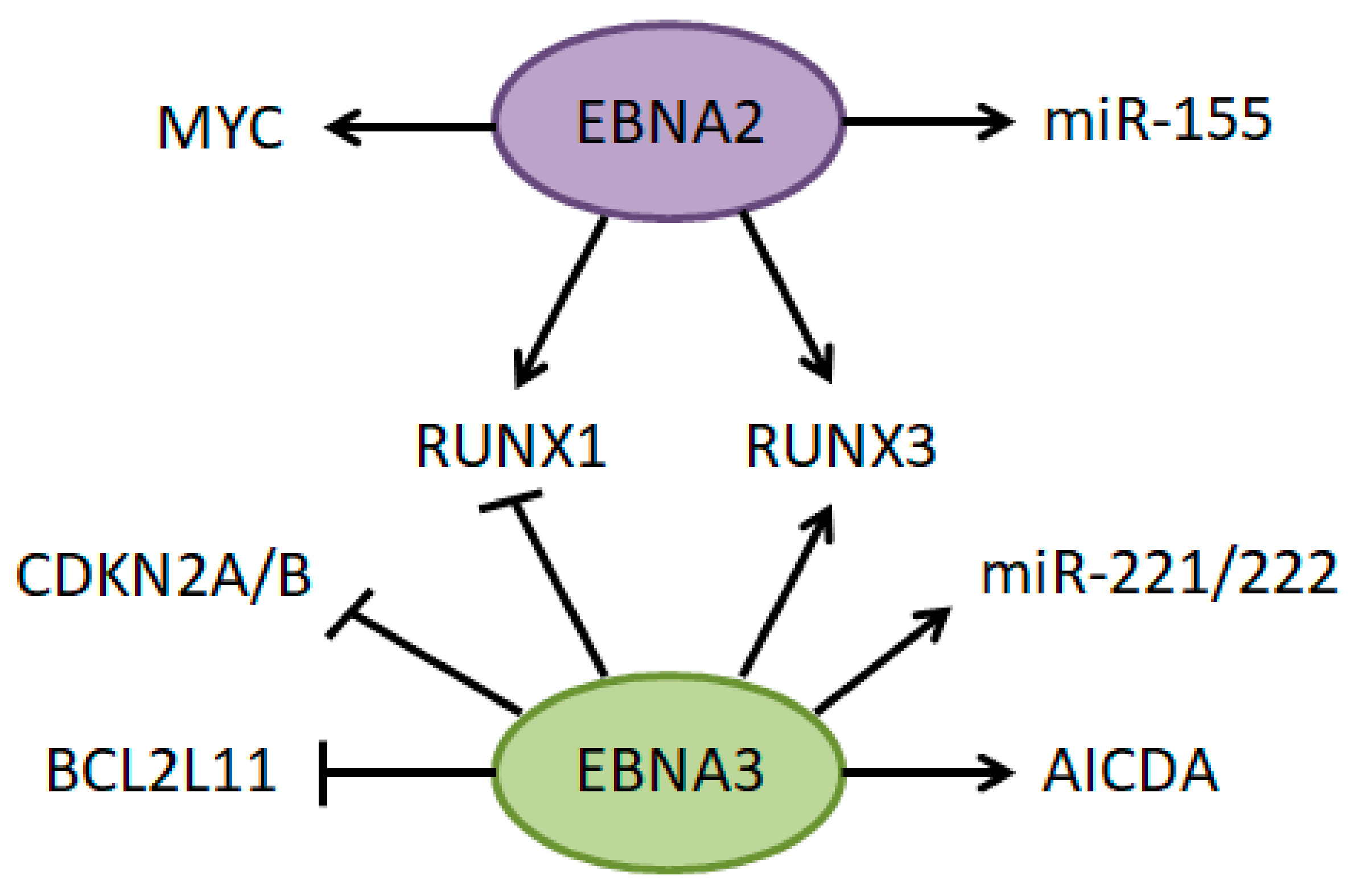

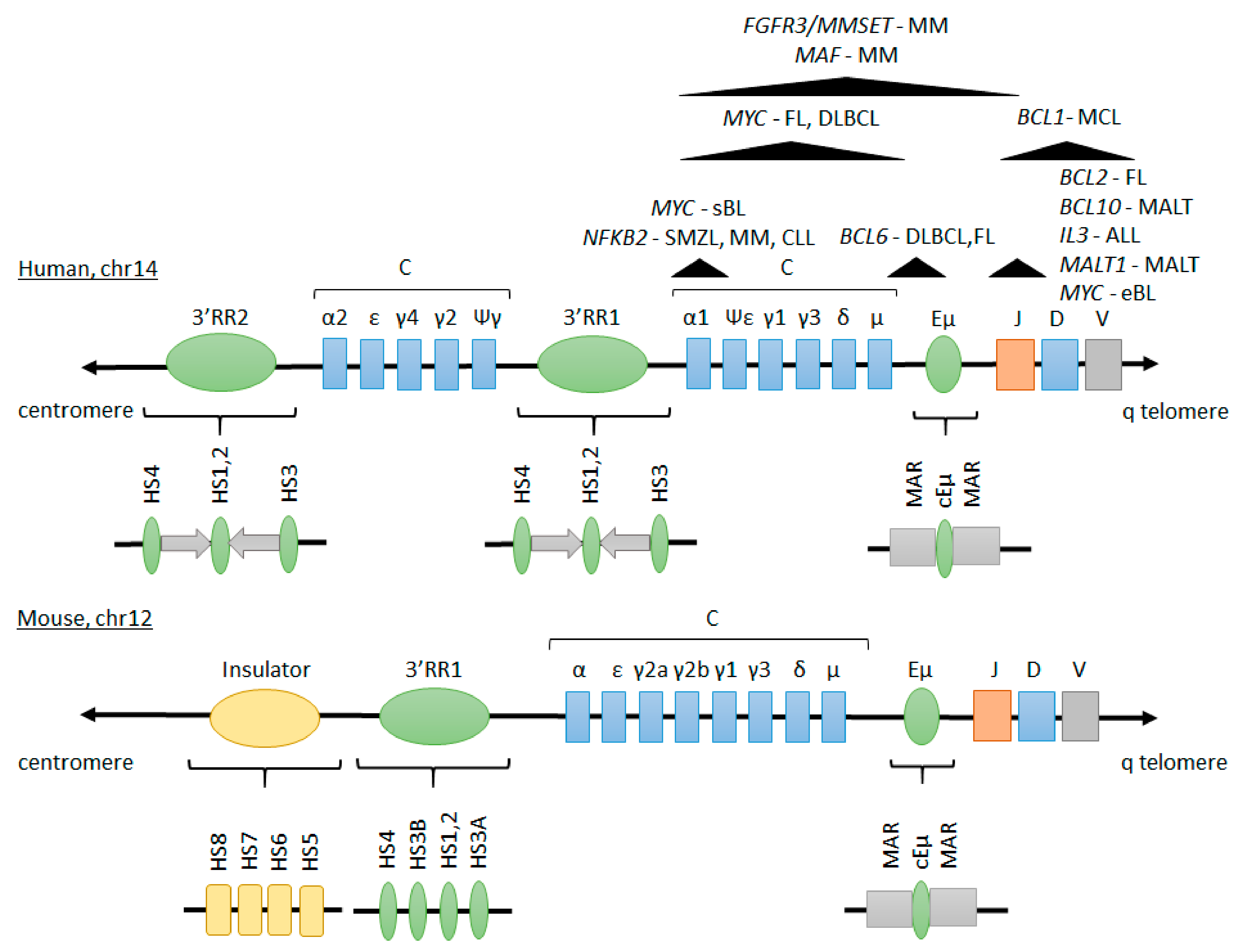{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes Involved | Translocation | Disease | Consequences | References |
|---|---|---|---|---|
| BCL2 | t(14;18)(q32;q2) | 90% FL | Delayed apoptosis and accumulation of aberrant cells | [132,133,134,135] |
| 15–30% DLBCL | ||||
| BCL3 | t(14;19)(q32;q13) | CLL, NHL | Modulation of the NF-kB pathway | [136,137,138,139] |
| BCL6 | t(3;14)(q27;q32) | 30% DLBCL | Increased cell proliferation, block of terminal differentiation | [140,141,142,143,144,145,146,147] |
| 4-14% FL | ||||
| BCL10 | t(1;14)(p21;q32) | 5% MALT | Activation of the NF-kB pathway (translocation involves a mutant BCL10 which lost pro-apoptotic functions) | [148,149,150] |
| CCND1 (BCL1) | t(11;14)(q13;q32) | 95% MCL | Accelerated passage through the G1 phase | [151,152,153,154,155,156] |
| 15–20% MM | ||||
| B-PLL, PCL, SLVL | ||||
| CEBPA | t(14;19)(q32;q13) | ALL | Deregulated cellular proliferation and differentiation | [157,158,159,160] |
| CEBPB | t(14;20)(q32;q13) | |||
| CEBPD | t(8;14)(q11;q32) | |||
| CEBPE | t(14;14)(q11;q32) | |||
| CEBPG | t(14;19)(q32;q13) | |||
| FGFR3/MMSET | t(4;14)(p16;q32) | 10% MM | Increased cell proliferation and survival | [161,162,163,164,165,166] |
| FOXP1 | t(3;14)(p14;q32) | 10% MALT | Enhanced tumor cell survival | [167,168,169] |
| DLBCL | ||||
| IL3 | t(5;14)(q31;q32) | ALL | Increased cell proliferation and survival | [170,171] |
| MAF | t(14;16)(q32;q23) | MM | Increased cell proliferation | [172,173,174,175] |
| MALT1 | t(14;18)(q32;q21) | 15–20% MALT | Activation of the NF-kB pathway | [167,176,177] |
| MYC | t(8;14)(q24;q32) | 70% BL | Increased cell proliferation | [135,178,179,180,181,182,183] |
| ALL | ||||
| DLBCL | ||||
| NFKB2 | t(10;14)(q24;q32) | SMZL | Constitutional activation of the non-canonical NF-kB pathway | [184,185,186] |
| MM, CLL | ||||
| PAX5 | t(9;14)(p13;q32) | 50% LPL | Dysregulation of PAX5 target genes | [187,188] |
| Gene | Translocation | Enhacer Involved | Model Name | Disease | References |
|---|---|---|---|---|---|
| BCL2 | t(14;18)(q32;q21) | 3′RR | Igh-3′E-bcl2 | FL | [194] |
| BCL6 | t(3;14)(q27;q32) | Eμ | Eμ-tTA-BCL6 | DLBCL, TL | [195] |
| BCL10 | t(1;14)(p22;q32) | Eμ | Eμ-BCL10 | MZL | [96] |
| CCND1 | t(11;14)(q13;q32) | Eμ | Eμ-CCND1 | no * | [196,197] |
| 3′RR | CCND1-3′RR | no | |||
| MAF | t(14;16)(q32;q23) | Eμ | Eμ-c-MAF | MM | [198] |
| MYC | t(8;14)(q24;q32) | Eµ | Eμ-myc iMycEμ | BL | [189,190,191,199,200,201,202,203,204,205] |
| Eµ + 3′RR | iMycCα | ||||
| 3′RR | IgH-3′E-myc, minimal 3′RR, iMycCµ |
| Gene | IG Light Chain | Translocation | Disease | References |
|---|---|---|---|---|
| BCL2 | lambda | t(18;22)(q21;q11) | CLL, FL | [145,230,231,232,233,234,235,236,237] |
| kappa | t(2;18)(p11;q21) | |||
| BCL3 | lambda | t(19;22)(q13;q11) | FL, DLBCL | [138] |
| kappa | t(2;19)(p12;q13) | HL, B-cell NHL | ||
| BCL6 | lambda | t(3;22)(q27;q11) | B-cell NHL | [247,250] |
| kappa | t(2;3)(p11;q27) | |||
| BCL10 | kappa | t(1; 2)(p22; p12) | MALT | [248,249] |
| CCND1 | lambda | t(11;22)(q13;q11) | MCL | [238,240,242,243] |
| kappa | t(2;11)(p11;q13) | |||
| CCND2 | lambda | t(12;22)(p13;q11) | MCL | [239,241,244] |
| kappa | t(2;12)(p11;p13) | |||
| CCND3 | lambda | t(6;22)(p21;q11) | MCL | [244] |
| kappa | t(2;6)(p11;p21) | |||
| MYC | lambda | t(8;22)(q24;q11) | ALL, BL, DLBCL, MM | [223,224,225,226,227,229] |
| kappa | t(2;8)(p11;q24) | |||
| REL | lambda | t(2;22)(p16;q11) | HL | [245] |
| Gene | Disease | Effect on Gene Expression | Reference |
|---|---|---|---|
| BCL2 | DLBCL | ND | [261,265] |
| BCL6 | BL, DLBCL | ND | [261,262,265] |
| PAX5 | BL, CLL, DLBCL, FL, MCL | Decreased | [260,261,262] |
| ST6GAL1 | BL | ND | [262,265] |
| TPRG1 | DLBCL | Increased | [266] |
| Gene | SNP ID | Disease | Gene Expression | TF Binding | Reference |
|---|---|---|---|---|---|
| BMF | rs539846 | CLL | Decreased | RELA (disrupted) | [273] |
| BMI1 | rs11591377 | ALL | ND | MYBL2, p300 (enhanced) | [274] |
| GATA3 | rs3824662 | ALL | Increased | NFIC (enhanced) | [275] |
| PIP4K2A | rs4748812 | ALL | ND | RUNX1 (enhanced) | [274] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasprzyk, M.E.; Sura, W.; Dzikiewicz-Krawczyk, A. Enhancing B-Cell Malignancies—On Repurposing Enhancer Activity towards Cancer. Cancers 2021, 13, 3270. https://doi.org/10.3390/cancers13133270
Kasprzyk ME, Sura W, Dzikiewicz-Krawczyk A. Enhancing B-Cell Malignancies—On Repurposing Enhancer Activity towards Cancer. Cancers. 2021; 13(13):3270. https://doi.org/10.3390/cancers13133270
Chicago/Turabian StyleKasprzyk, Marta Elżbieta, Weronika Sura, and Agnieszka Dzikiewicz-Krawczyk. 2021. "Enhancing B-Cell Malignancies—On Repurposing Enhancer Activity towards Cancer" Cancers 13, no. 13: 3270. https://doi.org/10.3390/cancers13133270
APA StyleKasprzyk, M. E., Sura, W., & Dzikiewicz-Krawczyk, A. (2021). Enhancing B-Cell Malignancies—On Repurposing Enhancer Activity towards Cancer. Cancers, 13(13), 3270. https://doi.org/10.3390/cancers13133270