
AML-Related NPM Mutations Drive p53 Delocalization into the Cytoplasm with Possible Impact on p53-Dependent Stress Response

 ,
,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultivation
2.2. Plasmid Construction and Cell Transfection
2.3. Cell Lysis
2.4. Western Blotting
2.5. Co-Immunoprecipitation Trap Assay
2.6. Live Cell Imaging
2.7. FLIM—Data Acquisition and Analysis
3. Results
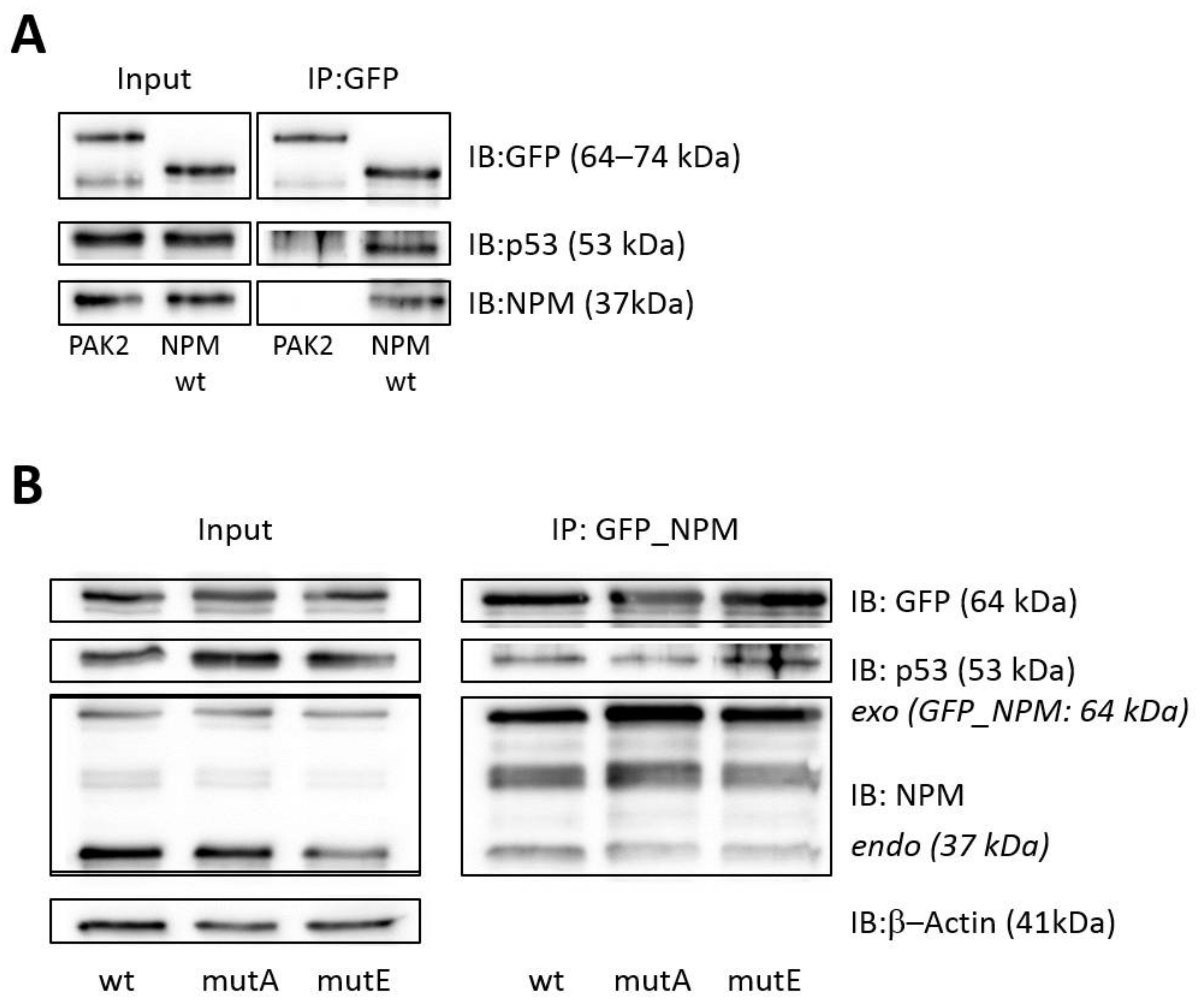
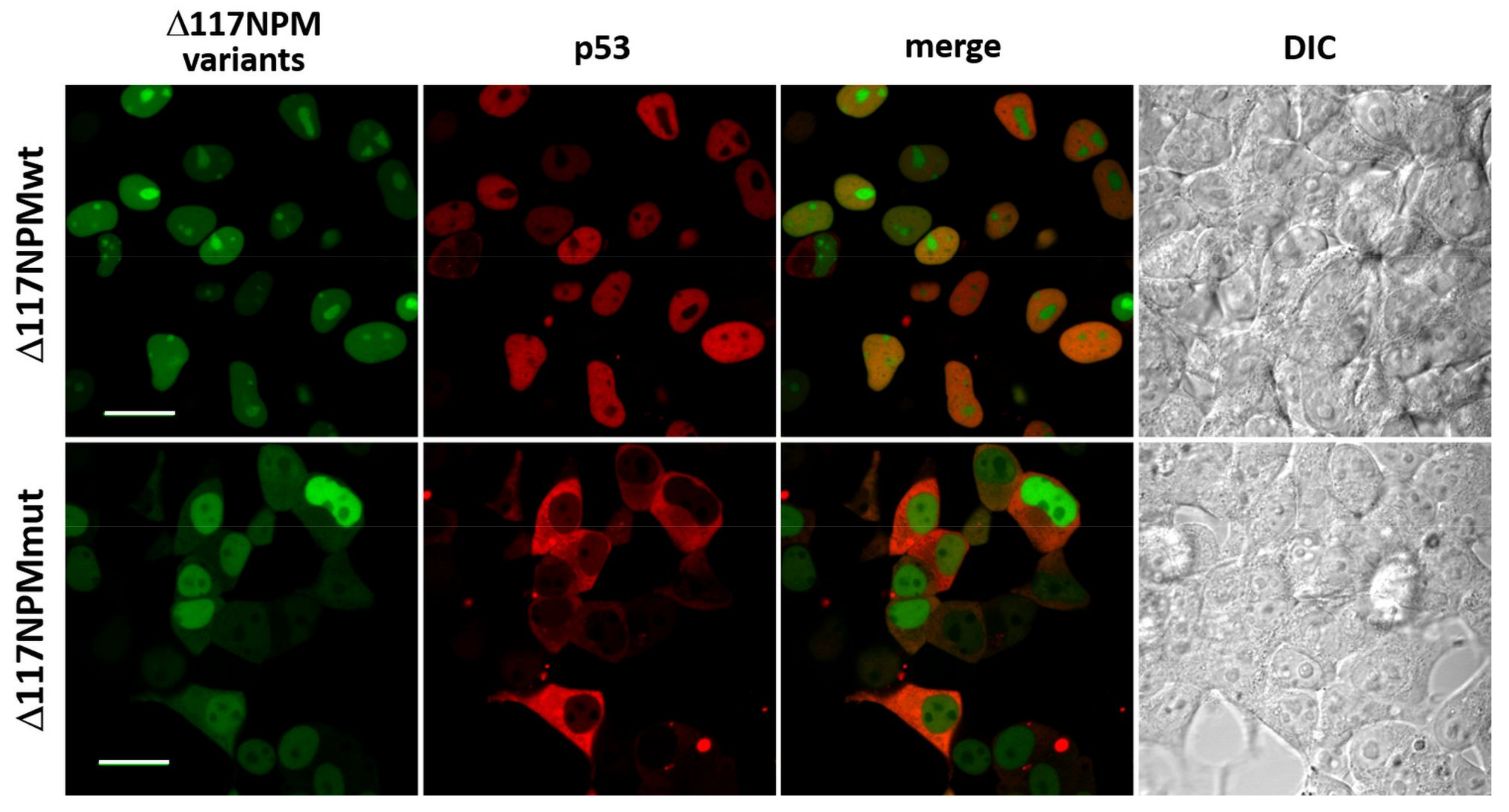
3.1. Interaction of p53 with NPMwt and NPMmut
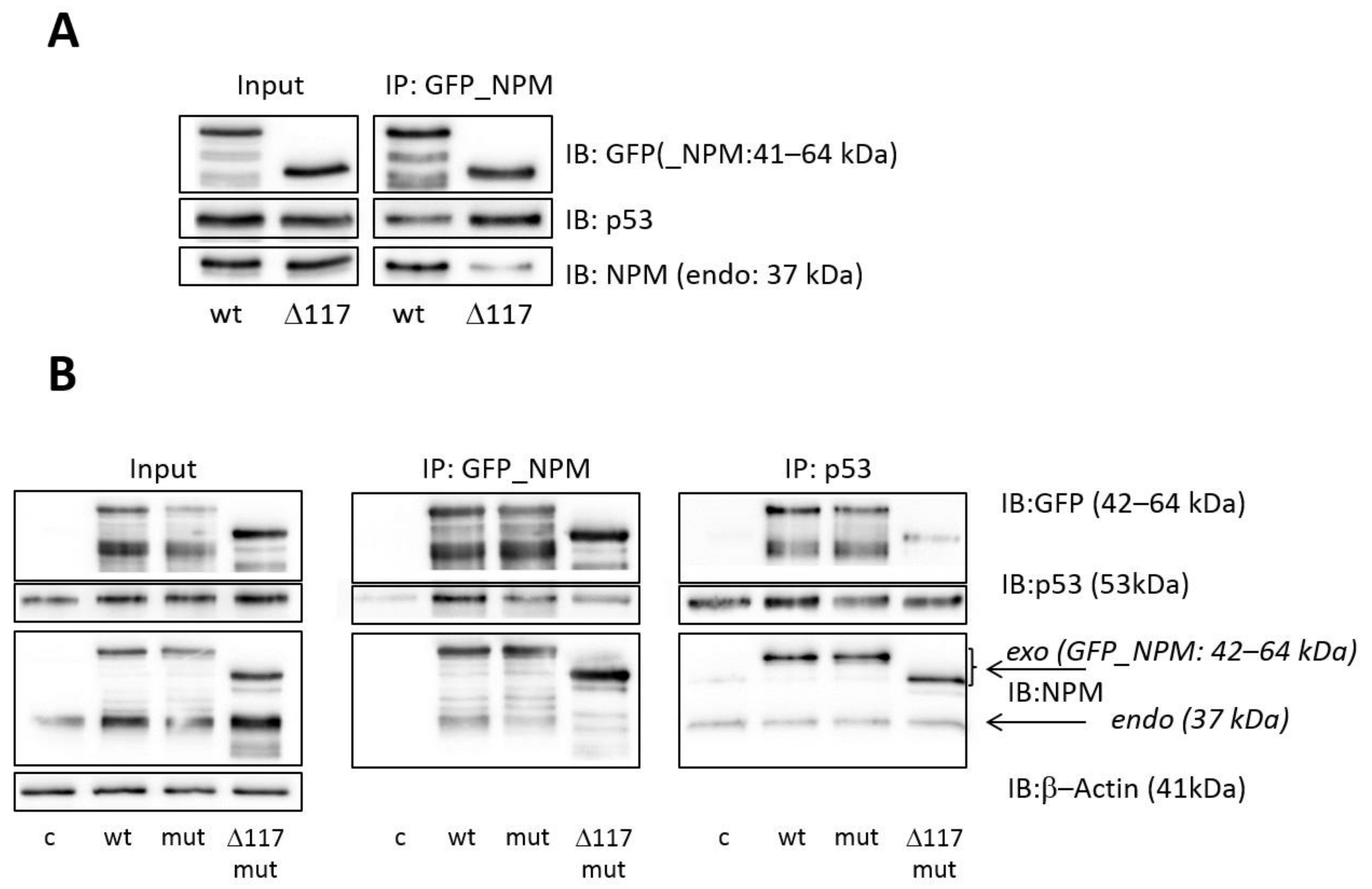
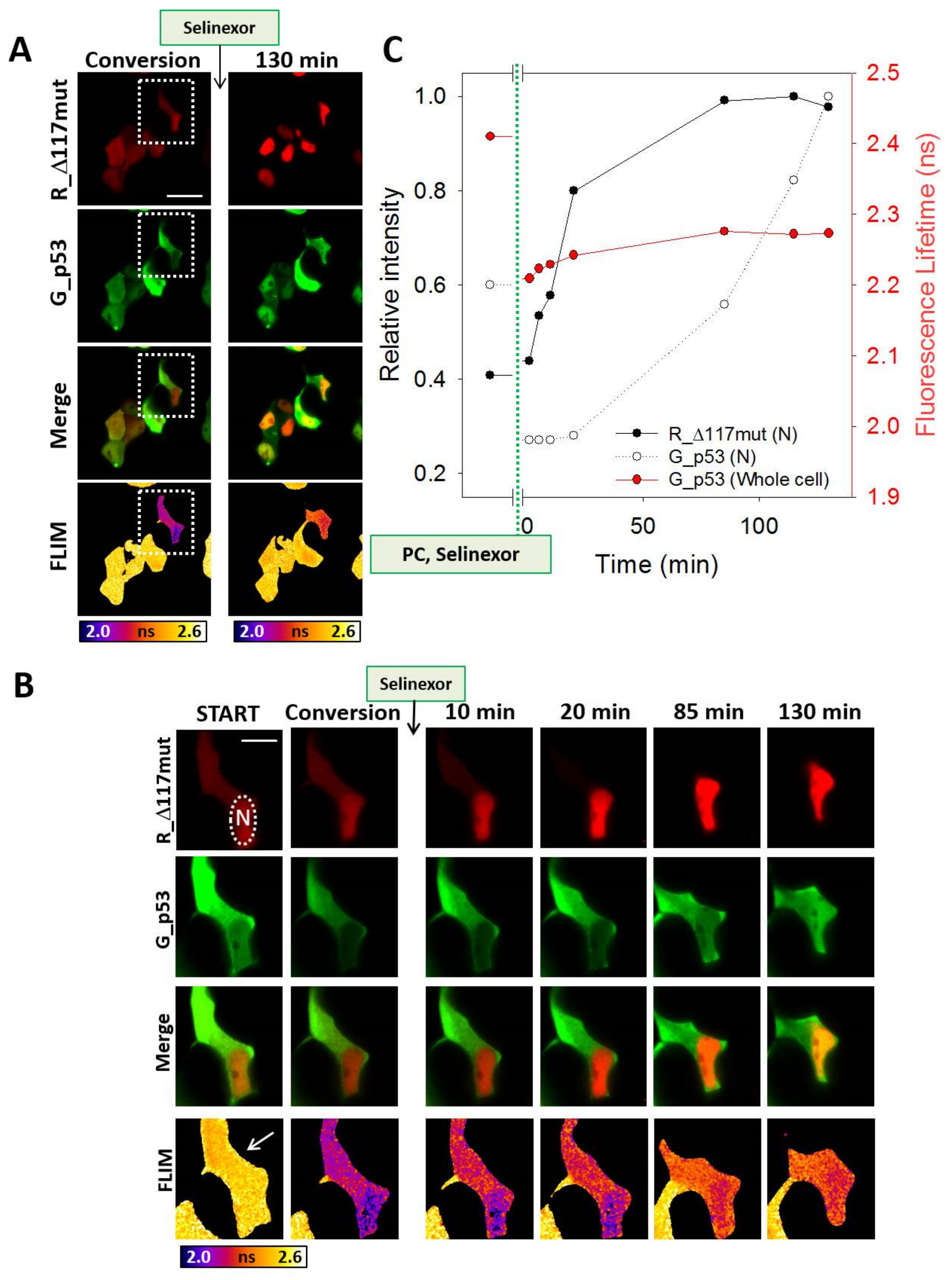
3.2. Effect of the NPM Oligomerization Domain
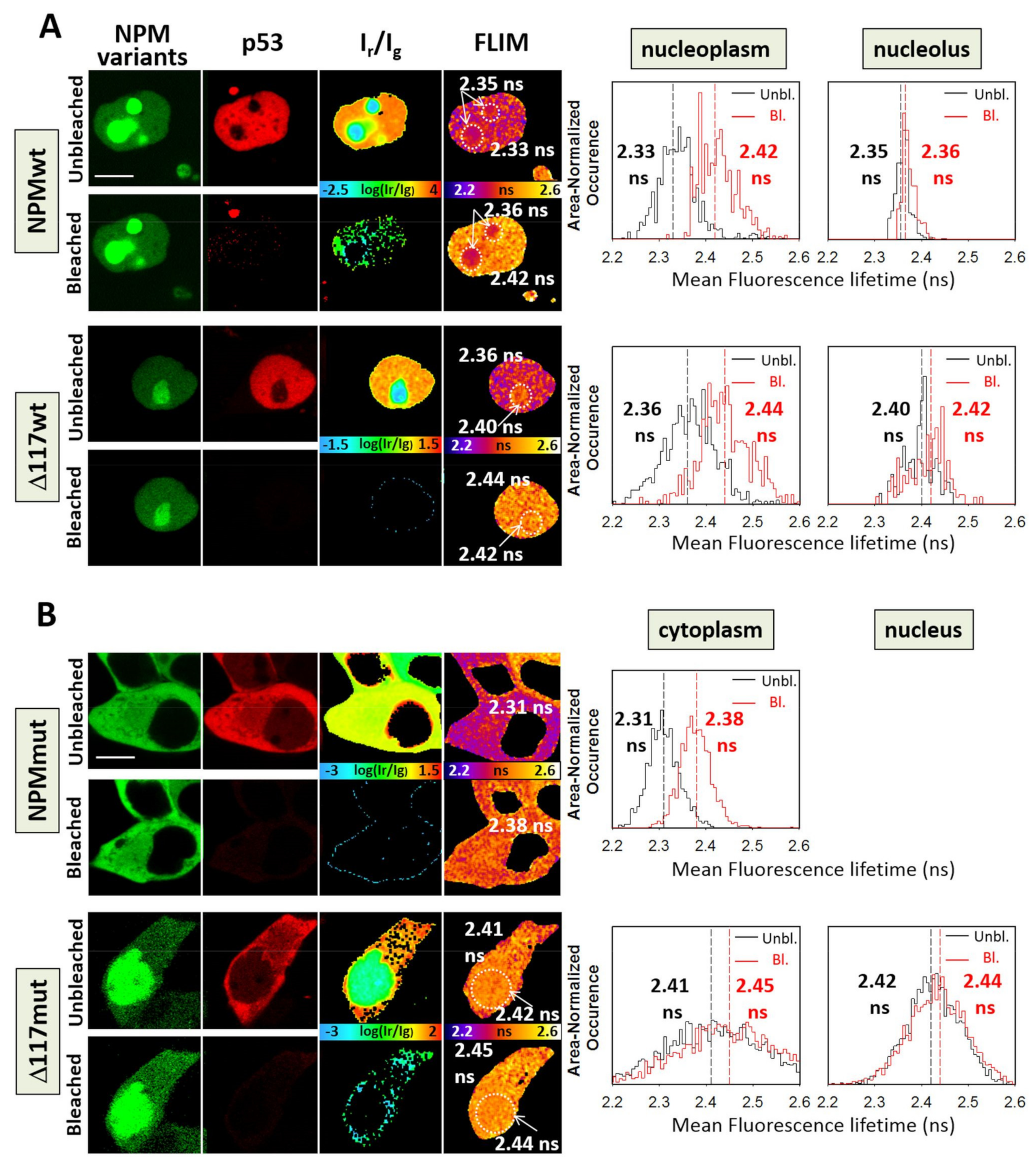
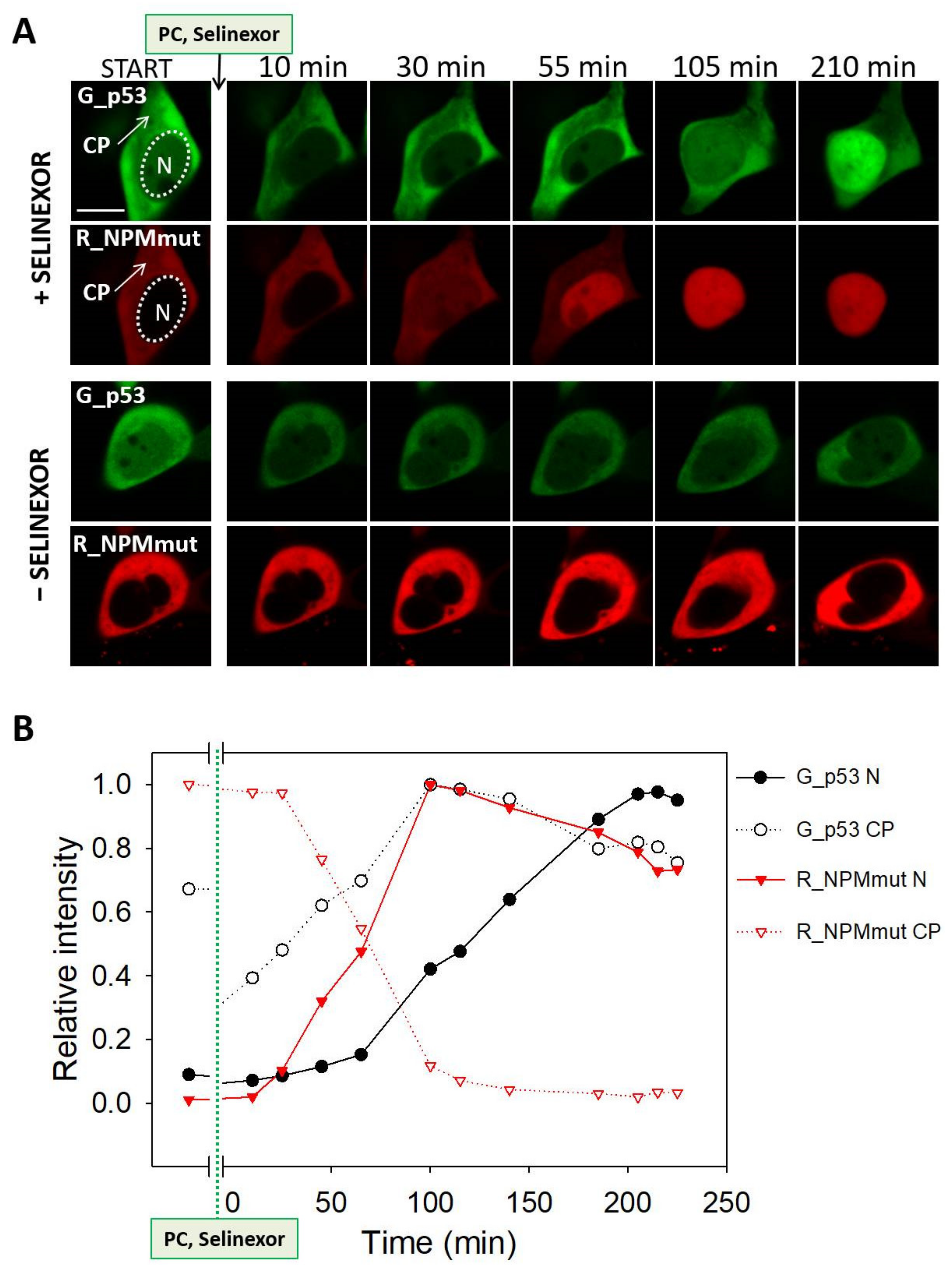
3.3. Interaction of p53 and NPM in Live Cells
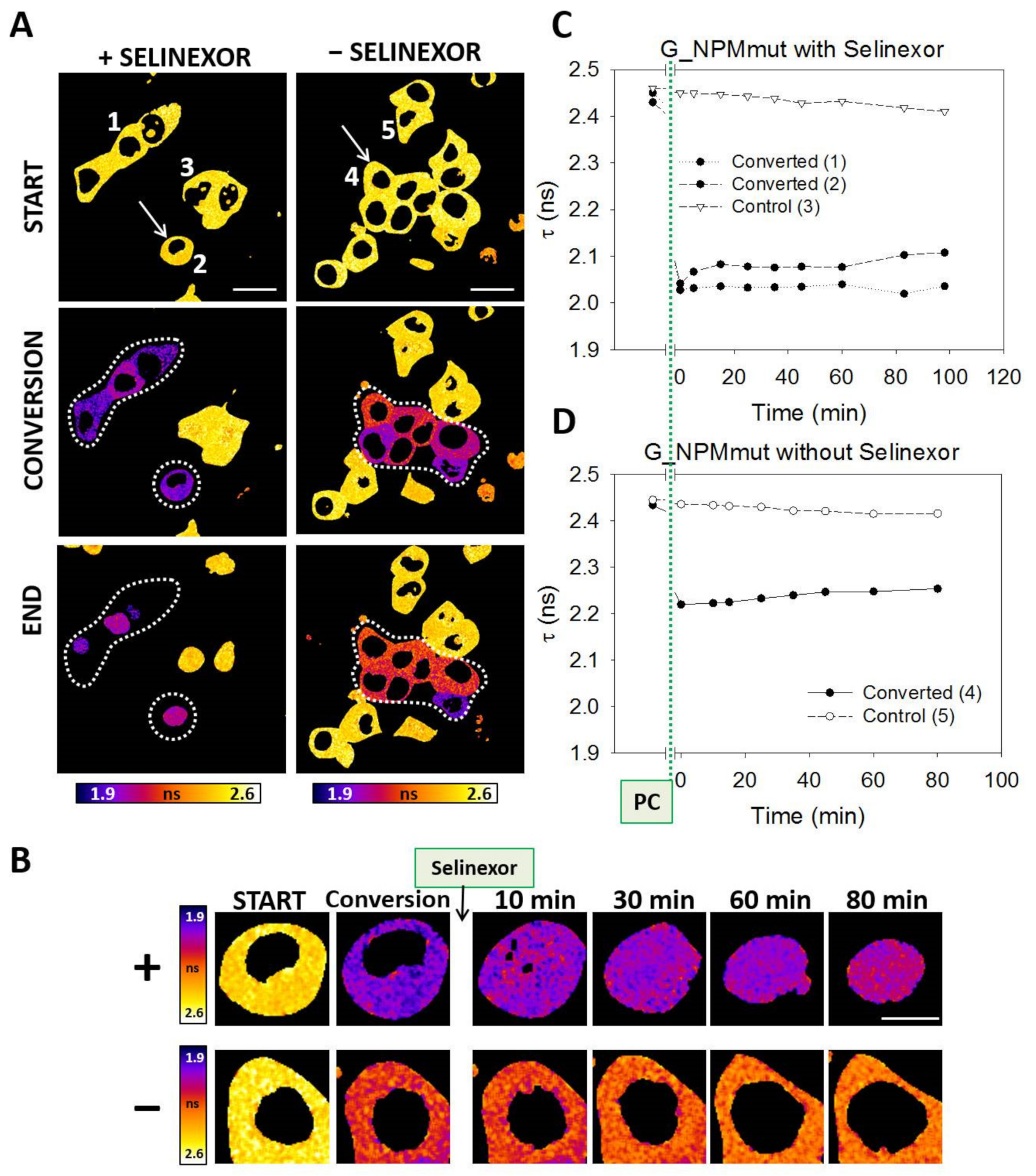
3.4. Selinexor-Induced p53 and NPM Relocalization
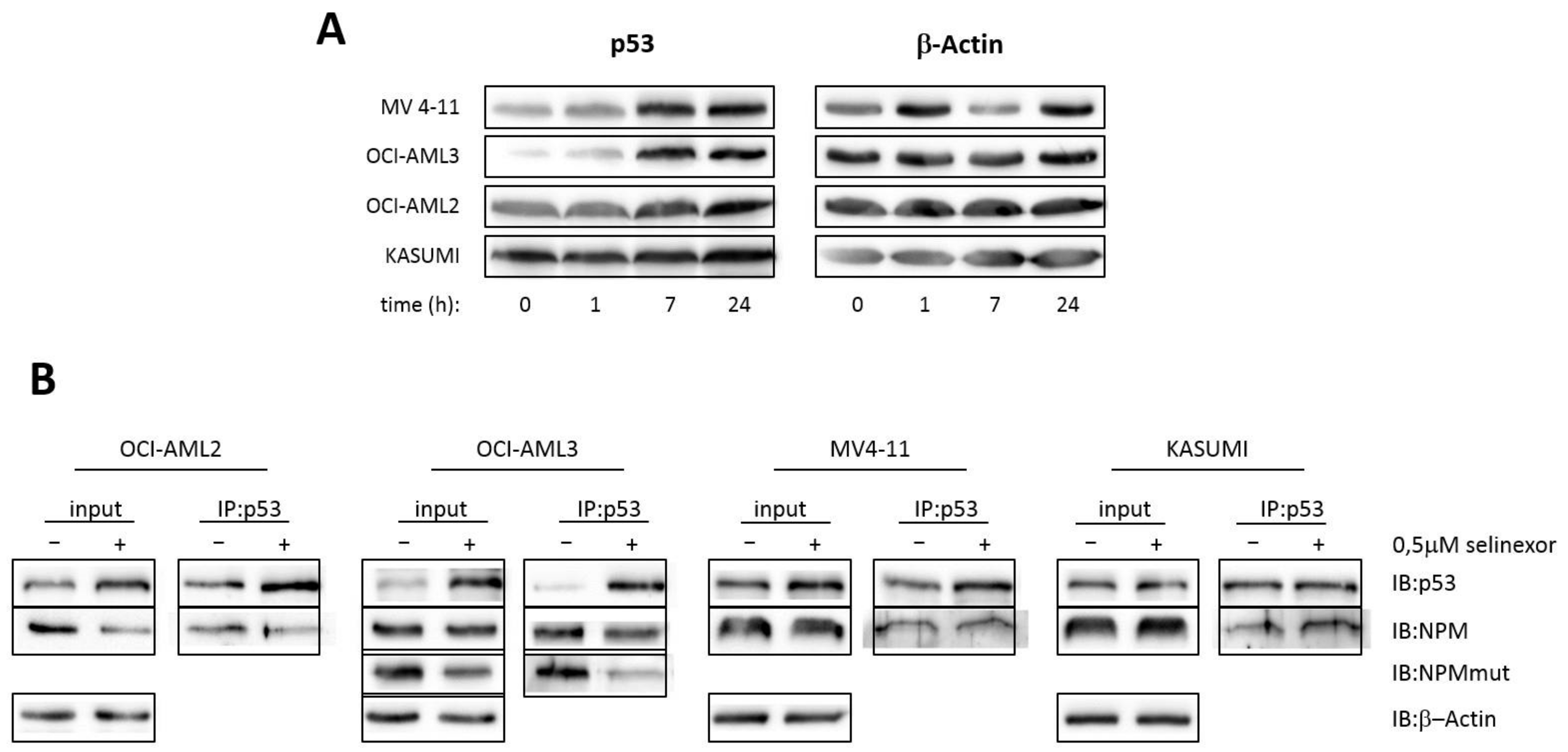
3.5. Interaction of p53 and NPM in AML Cell Lines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, K.; Yang, J.; Yi, J. Nucleolar Stress: Hallmarks, sensing mechanism and diseases. Cell Stress 2018, 2, 125–140. [Google Scholar] [CrossRef]
- Colombo, E.; Alcalay, M.; Pelicci, P.G. Nucleophosmin and its complex network: A possible therapeutic target in hematological diseases. Oncogene 2011, 30, 2595–2609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, E.; Marine, J.C.; Danovi, D.; Falini, B.; Pelicci, P.G. Nucleophosmin regulates the stability and transcriptional activity of p53. Nat. Cell Biol. 2002, 4, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Lambert, B.; Buckle, M. Characterisation of the interface between nucleophosmin (NPM) and p53: Potential role in p53 stabilisation. FEBS Lett. 2006, 580, 345–350. [Google Scholar] [CrossRef]
- Daniely, Y.; Dimitrova, D.D.; Borowiec, J.A. Stress-dependent nucleolin mobilization mediated by p53-nucleolin complex formation. Mol. Cell. Biol. 2002, 22, 6014–6022. [Google Scholar] [CrossRef] [Green Version]
- Dhar, S.K.; St Clair, D.K. Nucleophosmin blocks mitochondrial localization of p53 and apoptosis. J. Biol. Chem. 2009, 284, 16409–16418. [Google Scholar] [CrossRef] [Green Version]
- Saxena, A.; Rorie, C.J.; Dimitrova, D.; Daniely, Y.; Borowiec, J.A. Nucleolin inhibits Hdm2 by multiple pathways leading to p53 stabilization. Oncogene 2006, 25, 7274–7288. [Google Scholar] [CrossRef] [Green Version]
- Matt, S.; Hofmann, T.G. The DNA damage-induced cell death response: A roadmap to kill cancer cells. Cell Mol. Life Sci. 2016, 73, 2829–2850. [Google Scholar] [CrossRef] [PubMed]
- Lindstrom, M.S. NPM1/B23: A Multifunctional Chaperone in Ribosome Biogenesis and Chromatin Remodeling. Biochem. Res. Int. 2011, 2011, 195209. [Google Scholar] [CrossRef] [Green Version]
- Brodska, B.; Sasinkova, M.; Kuzelova, K. Nucleophosmin in leukemia: Consequences of anchor loss. Int. J. Biochem. Cell Biol. 2019, 111, 52–62. [Google Scholar] [CrossRef]
- Meani, N.; Alcalay, M. Role of nucleophosmin in acute myeloid leukemia. Expert Rev. Anticancer Ther. 2009, 9, 1283–1294. [Google Scholar] [CrossRef]
- Federici, L.; Falini, B. Nucleophosmin mutations in acute myeloid leukemia: A tale of protein unfolding and mislocalization. Protein Sci. 2013, 22, 545–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michael, D.; Oren, M. The p53-Mdm2 module and the ubiquitin system. Semin. Cancer Biol. 2003, 13, 49–58. [Google Scholar] [CrossRef]
- Brooks, C.L.; Gu, W. P53 Regulation by Ubiquitin. FEBS Lett. 2011, 585, 2803–2809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marine, J.C.; Lozano, G. Mdm2-mediated ubiquitylation: p53 and beyond. Cell Death Differ. 2010, 17, 93–102. [Google Scholar] [CrossRef] [Green Version]
- Gjerset, R.A. DNA damage, p14ARF, nucleophosmin (NPM/B23), and cancer. J. Mol. Histol. 2006, 37, 239–251. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, S.J.; Kefford, R.F.; Rizos, H. The ARF tumour suppressor. Int. J. Biochem. Cell Biol. 2006, 38, 1637–1641. [Google Scholar] [CrossRef]
- Korgaonkar, C.; Hagen, J.; Tompkins, V.; Frazier, A.A.; Allamargot, C.; Quelle, F.W.; Quelle, D.E. Nucleophosmin (B23) targets ARF to nucleoli and inhibits its function. Mol. Cell. Biol. 2005, 25, 1258–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalabothula, N.; Indig, F.E.; Carrier, F. The Nucleolus Takes Control of Protein Trafficking Under Cellular Stress. Mol. Cell. Pharmacol. 2010, 2, 203–212. [Google Scholar]
- Li, Y.P.; Busch, R.K.; Valdez, B.C.; Busch, H. C23 interacts with B23, a putative nucleolar-localization-signal-binding protein. Eur. J. Biochem. 1996, 237, 153–158. [Google Scholar] [CrossRef]
- Mitrea, D.M.; Cika, J.A.; Guy, C.S.; Ban, D.; Banerjee, P.R.; Stanley, C.B.; Nourse, A.; Deniz, A.A.; Kriwacki, R.W. Nucleophosmin integrates within the nucleolus via multi-modal interactions with proteins displaying R-rich linear motifs and rRNA. Elife 2016, 5. [Google Scholar] [CrossRef]
- Chen, D.; Huang, S. Nucleolar components involved in ribosome biogenesis cycle between the nucleolus and nucleoplasm in interphase cells. J. Cell Biol. 2001, 153, 169–176. [Google Scholar] [CrossRef] [Green Version]
- Olson, M.O.; Dundr, M. The moving parts of the nucleolus. Histochem. Cell Biol. 2005, 123, 203–216. [Google Scholar] [CrossRef]
- Phair, R.D.; Misteli, T. High mobility of proteins in the mammalian cell nucleus. Nature 2000, 404, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Holoubek, A.; Heřman, P.; Sýkora, J.; Brodská, B.; Humpolickova, J.; Kráčmarová, M.; Gášková, D.; Hof, M.; Kuzelová, K. Monitoring of nucleophosmin oligomerization in live cells. Methods Appl. Fluoresc. 2018, 6, 035016. [Google Scholar] [CrossRef] [PubMed]
- Sasinkova, M.; Herman, P.; Holoubek, A.; Strachotova, D.; Otevrelova, P.; Grebenova, D.; Kuzelova, K.; Brodska, B. NSC348884 cytotoxicity is not mediated by inhibition of nucleophosmin oligomerization. Sci. Rep. 2021, 11, 1084. [Google Scholar] [CrossRef] [PubMed]
- Sasinkova, M.; Holoubek, A.; Otevrelova, P.; Kuzelova, K.; Brodska, B. AML-associated mutation of nucleophosmin compromises its interaction with nucleolin. Int. J. Biochem. Cell Biol. 2018, 103, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hu, J. Nucleophosmin1 (NPM1) abnormality in hematologic malignancies, and therapeutic targeting of mutant NPM1 in acute myeloid leukemia. Ther. Adv. Hematol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Mecucci, C.; Tiacci, E.; Alcalay, M.; Rosati, R.; Pasqualucci, L.; La Starza, R.; Diverio, D.; Colombo, E.; Santucci, A.; et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl. J. Med. 2005, 352, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Bullinger, L.; Dohner, K.; Dohner, H. Genomics of Acute Myeloid Leukemia Diagnosis and Pathways. J. Clin. Oncol. 2017, 35, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Forghieri, F.; Comoli, P.; Marasca, R.; Potenza, L.; Luppi, M. Minimal/Measurable Residual Disease Monitoring in NPM1-Mutated Acute Myeloid Leukemia: A Clinical Viewpoint and Perspectives. Int. J. Mol. Sci. 2018, 19, 3492. [Google Scholar] [CrossRef] [Green Version]
- Falini, B.; Brunetti, L.; Martelli, M.P. How I diagnose and treat NPM1-mutated AML. Blood 2021, 137, 589–599. [Google Scholar] [CrossRef]
- Bolli, N.; De Marco, M.F.; Martelli, M.P.; Bigerna, B.; Pucciarini, A.; Rossi, R.; Mannucci, R.; Manes, N.; Pettirossi, V.; Pileri, S.A.; et al. A dose-dependent tug of war involving the NPM1 leukaemic mutant, nucleophosmin, and ARF. Leukemia 2009, 23, 501–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodska, B.; Kracmarova, M.; Holoubek, A.; Kuzelova, K. Localization of AML-related nucleophosmin mutant depends on its subtype and is highly affected by its interaction with wild-type NPM. PLoS ONE 2017, 12, e0175175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falini, B.; Bolli, N.; Shan, J.; Martelli, M.P.; Liso, A.; Pucciarini, A.; Bigerna, B.; Pasqualucci, L.; Mannucci, R.; Rosati, R.; et al. Both carboxy-terminus NES motif and mutated tryptophan(s) are crucial for aberrant nuclear export of nucleophosmin leukemic mutants in NPMc+ AML. Blood 2006, 107, 4514–4523. [Google Scholar] [CrossRef]
- Falini, B.; Albiero, E.; Bolli, N.; De Marco, M.F.; Madeo, D.; Martelli, M.; Nicoletti, I.; Rodeghiero, F. Aberrant cytoplasmic expression of C-terminal-truncated NPM leukaemic mutant is dictated by tryptophans loss and a new NES motif. Leukemia 2007, 21, 2052–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Den Besten, W.; Kuo, M.L.; Williams, R.T.; Sherr, C.J. Myeloid leukemia-associated nucleophosmin mutants perturb p53-dependent and independent activities of the Arf tumor suppressor protein. Cell Cycle 2005, 4, 1593–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brate, A.; Giannakakou, P. The importance of p53 location: Nuclear or cytoplasmic zip code? Drug Resist. Updat. 2003, 6, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Comel, A.; Sorrentino, G.; Capaci, V.; Del Sal, G. The cytoplasmic side of p53′s oncosuppressive activities. FEBS Lett. 2014, 588, 2600–2609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senapedis, W.T.; Baloglu, E.; Landesman, Y. Clinical translation of nuclear export inhibitors in cancer. Semin. Cancer Biol. 2014, 27, 74–86. [Google Scholar] [CrossRef]
- Lane, D.P.; Cheok, C.F.; Lain, S. P53-Based Cancer Therapy. Cold Spring Harb. Perspect. Biol. 2010, 2, a001222. [Google Scholar] [CrossRef] [Green Version]
- Marcus, J.M.; Burke, R.T.; Doak, A.E.; Park, S.; Orth, J.D. Loss of p53 expression in cancer cells alters cell cycle response after inhibition of exportin-1 but does not prevent cell death. Cell Cycle 2018, 17, 1329–1344. [Google Scholar] [CrossRef] [Green Version]
- Mao, L.; Yang, Y. Targeting the nuclear transport machinery by rational drug design. Curr. Pharm. Des. 2013, 19, 2318–2325. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.T.; Holloway, M.P.; Altura, R.A. The CRM1 nuclear export protein in normal development and disease. Int. J. Biochem. Mol. Biol. 2012, 3, 137–151. [Google Scholar] [PubMed]
- Das, A.; Wei, G.; Parikh, K.; Liu, D. Selective inhibitors of nuclear export (SINE) in hematological malignancies. Exp. Hematol. Oncol. 2015, 4. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xiong, Y. A p53 amino-terminal nuclear export signal inhibited by DNA damage-induced phosphorylation. Science 2001, 292, 1910–1915. [Google Scholar] [CrossRef] [Green Version]
- Stommel, J.M.; Marchenko, N.D.; Jimenez, G.S.; Moll, U.M.; Hope, T.J.; Wahl, G.M. A leucine-rich nuclear export signal in the p53 tetramerization domain: Regulation of subcellular localization and p53 activity by NES masking. EMBO J. 1999, 18, 1660–1672. [Google Scholar] [CrossRef] [Green Version]
- Turner, J.G.; Dawson, J.; Sullivan, D.M. Nuclear export of proteins and drug resistance in cancer. Biochem. Pharmacol. 2012, 83, 1021–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gravina, G.L.; Senapedis, W.; McCauley, D.; Baloglu, E.; Shacham, S.; Festuccia, C. Nucleo-cytoplasmic transport as a therapeutic target of cancer. J. Hematol. Oncol. 2014, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; Ebrahem, Q.; Mahfouz, R.Z.; Hasipek, M.; Enane, F.; Radivoyevitch, T.; Rapin, N.; Przychodzen, B.; Hu, Z.; Balusu, R.; et al. Leukemogenic nucleophosmin mutation disrupts the transcription factor hub that regulates granulomonocytic fates. J. Clin. Investig. 2018, 128, 4260–4279. [Google Scholar] [CrossRef] [Green Version]
- Kunchala, P.; Kuravi, S.; Jensen, R.; McGuirk, J.; Balusu, R. When the good go bad: Mutant NPM1 in acute myeloid leukemia. Blood Rev. 2018, 32, 167–183. [Google Scholar] [CrossRef]
- Brodska, B.; Holoubek, A.; Otevrelova, P.; Kuzelova, K. Low-Dose Actinomycin-D Induces Redistribution of Wild-Type and Mutated Nucleophosmin Followed by Cell Death in Leukemic Cells. J. Cell. Biochem. 2016, 117, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Grebenova, D.; Holoubek, A.; Roselova, P.; Obr, A.; Brodska, B.; Kuzelova, K. PAK1, PAK1 Delta 15, and PAK2: Similarities, differences and mutual interactions. Sci. Rep. 2019, 9, 17171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herman, P.; Holoubek, A.; Brodska, B. Lifetime-based photoconversion of EGFP as a tool for FLIM. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 266–277. [Google Scholar] [CrossRef]
- Patting, M. Evaluation of Time-Resolved Fluorescence Data: Typical Methods and Problems, Standardization and Quality Assurance in Fluorescence Measurements I. Springer Ser. Fluoresc. 2008, 5, 233–258. [Google Scholar]
- Strachotova, D.; Holoubek, A.; Kucerova, H.; Benda, A.; Humpolickova, J.; Vachova, L.; Palkova, Z. Ato protein interactions in yeast plasma membrane revealed by fluorescence lifetime imaging (FLIM). Biochim. Biophys. Acta 2012, 1818, 2126–2134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heikal, A.; Hess, S.; Webb, W. Multiphoton molecular spectroscopy and excited-state dynamics of enhanced green fluorescent proteins (EGFP): Acid-base specifity. Chem. Phys. 2001, 274, 37–55. [Google Scholar] [CrossRef]
- Hingorani, K.; Szebeni, A.; Olson, M.O. Mapping the functional domains of nucleolar protein B23. J. Biol. Chem. 2000, 275, 24451–24457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enomoto, T.; Lindstrom, M.S.; Jin, A.; Ke, H.; Zhang, Y. Essential role of the B23/NPM core domain in regulating ARF binding and B23 stability. J. Biol. Chem. 2006, 281, 18463–18472. [Google Scholar] [CrossRef] [Green Version]
- Wallrabe, H.; Periasamy, A. Imaging protein molecules using FRET and FLIM microscopy. Curr. Opin. Biotechnol. 2005, 16, 19–27. [Google Scholar] [CrossRef]
- Bastiaens, P.I.; Squire, A. Fluorescence lifetime imaging microscopy: Spatial resolution of biochemical processes in the cell. Trends Cell Biol. 1999, 9, 48–52. [Google Scholar] [CrossRef]
- Kenworthy, A.K. Molecular Imaging: FRET Microscopy and Spectroscopy. In Photobleaching FRET Microscopy; Periasamy, A., Day, R., Eds.; Oxford University Press: New York, NY, USA, 2005; p. 146. [Google Scholar]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Springer: New York, NY, USA, 2006. [Google Scholar]
- Suhling, K.; Siegel, J.; Phillips, D.; French, P.M.; Leveque-Fort, S.; Webb, S.E.; Davis, D.M. Imaging the environment of green fluorescent protein. Biophys. J. 2002, 83, 3589–3595. [Google Scholar] [CrossRef]
- Kojima, K.; Kornblau, S.M.; Ruvolo, V.; Dilip, A.; Duvvuri, S.; Davis, R.E.; Zhang, M.; Wang, Z.; Coombes, K.R.; Zhang, N.; et al. Prognostic impact and targeting of CRM1 in acute myeloid leukemia. Blood 2013, 121, 4166–4174. [Google Scholar] [CrossRef]
- Garzon, R.; Savona, M.; Baz, R.; Andreeff, M.; Gabrail, N.; Gutierrez, M.; Savoie, L.; Mau-Sorensen, P.M.; Wagner-Johnston, N.; Yee, K.; et al. A phase 1 clinical trial of single-agent selinexor in acute myeloid leukemia. Blood 2017, 129, 3165–3174. [Google Scholar] [CrossRef] [Green Version]
- Vousden, K.H.; Vande Woude, G.F. The ins and outs of p53. Nat. Cell Biol. 2000, 2, E178–E180. [Google Scholar] [CrossRef]
- Nakayama, R.; Zhang, Y.X.; Czaplinski, J.T.; Anatone, A.J.; Sicinska, E.T.; Fletcher, J.A.; Demetri, G.D.; Wagner, A.J. Preclinical activity of selinexor, an inhibitor of XPO1, in sarcoma. Oncotarget 2016, 7, 16581–16592. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, B.I.; Cautain, B.; Grenho, I.; Link, W. Small Molecule Inhibitors of CRM1. Front. Pharmacol. 2020, 11, 625. [Google Scholar] [CrossRef] [PubMed]
- Mahipal, A.; Malafa, M. Importins and exportins as therapeutic targets in cancer. Pharmacol. Ther. 2016, 164, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Otevrelova, P.; Brodska, B. Chemotherapy-induced survivin regulation in acute myeloid leukemia. Appl. Sci. 2021, 11, 460. [Google Scholar] [CrossRef]
- Russo, L.C.; Ferruzo, P.Y.M.; Forti, F.L. Nucleophosmin Protein Dephosphorylation by DUSP3 Is a Fine-Tuning Regulator of p53 Signaling to Maintain Genomic Stability. Front. Cell Dev. Biol. 2021, 9, 624933. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Construct | Abbreviation | Construct | Abbreviation | Construct | Abbreviation |
|---|---|---|---|---|---|
| eGFP_NPMwt | G_NPMwt | eGFP_Δ117NPMwt | G_Δ117wt | eGFP_p53wt | G_p53 |
| mRFP1_NPMwt | R_NPMwt | mRFP1_Δ117NPMwt | R_Δ117wt | mRFP1_p53wt | R_p53 |
| eGFP_NPMmutA 1 | G_NPMmut | eGFP_Δ117NPMmutA | G_Δ117mut | PAK2_eGFP 2 | G_PAK2 |
| mRFP1_NPMmutA | R_NPMmut | mRFP1_Δ117NPMmutA | R_Δ117mut |
| Interaction of p53 and NPM Variants | Localization of the Fusion Proteins | |||||
|---|---|---|---|---|---|---|
| Co-expressed fusion proteins | Immunoprecipitation results | FRET results | eGFP-tagged p53 | Cytoplasmic signal | mRFP1-tagged NPM variant | Cytoplasmic signal |
| G_p53 /R_NPMwt | 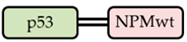 | ✓ |  | − |  | − |
| G_p53 /R_NPMmut | 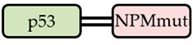 | ✓ |  | + |  | + + |
| G_p53 /R_ Δ 117wt |  | ✓ |  | − |  | − |
| G_p53 /R_ Δ 117mut | 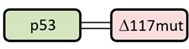 | X |  | + + | 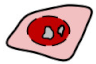 | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holoubek, A.; Strachotová, D.; Otevřelová, P.; Röselová, P.; Heřman, P.; Brodská, B. AML-Related NPM Mutations Drive p53 Delocalization into the Cytoplasm with Possible Impact on p53-Dependent Stress Response. Cancers 2021, 13, 3266. https://doi.org/10.3390/cancers13133266
Holoubek A, Strachotová D, Otevřelová P, Röselová P, Heřman P, Brodská B. AML-Related NPM Mutations Drive p53 Delocalization into the Cytoplasm with Possible Impact on p53-Dependent Stress Response. Cancers. 2021; 13(13):3266. https://doi.org/10.3390/cancers13133266
Chicago/Turabian StyleHoloubek, Aleš, Dita Strachotová, Petra Otevřelová, Pavla Röselová, Petr Heřman, and Barbora Brodská. 2021. "AML-Related NPM Mutations Drive p53 Delocalization into the Cytoplasm with Possible Impact on p53-Dependent Stress Response" Cancers 13, no. 13: 3266. https://doi.org/10.3390/cancers13133266
APA StyleHoloubek, A., Strachotová, D., Otevřelová, P., Röselová, P., Heřman, P., & Brodská, B. (2021). AML-Related NPM Mutations Drive p53 Delocalization into the Cytoplasm with Possible Impact on p53-Dependent Stress Response. Cancers, 13(13), 3266. https://doi.org/10.3390/cancers13133266