
Discovery of Spatial Peptide Signatures for Neuroblastoma Risk Assessment by MALDI Mass Spectrometry Imaging

 ,
,
Abstract
:Simple Summary
Abstract

1. Introduction
2. Results
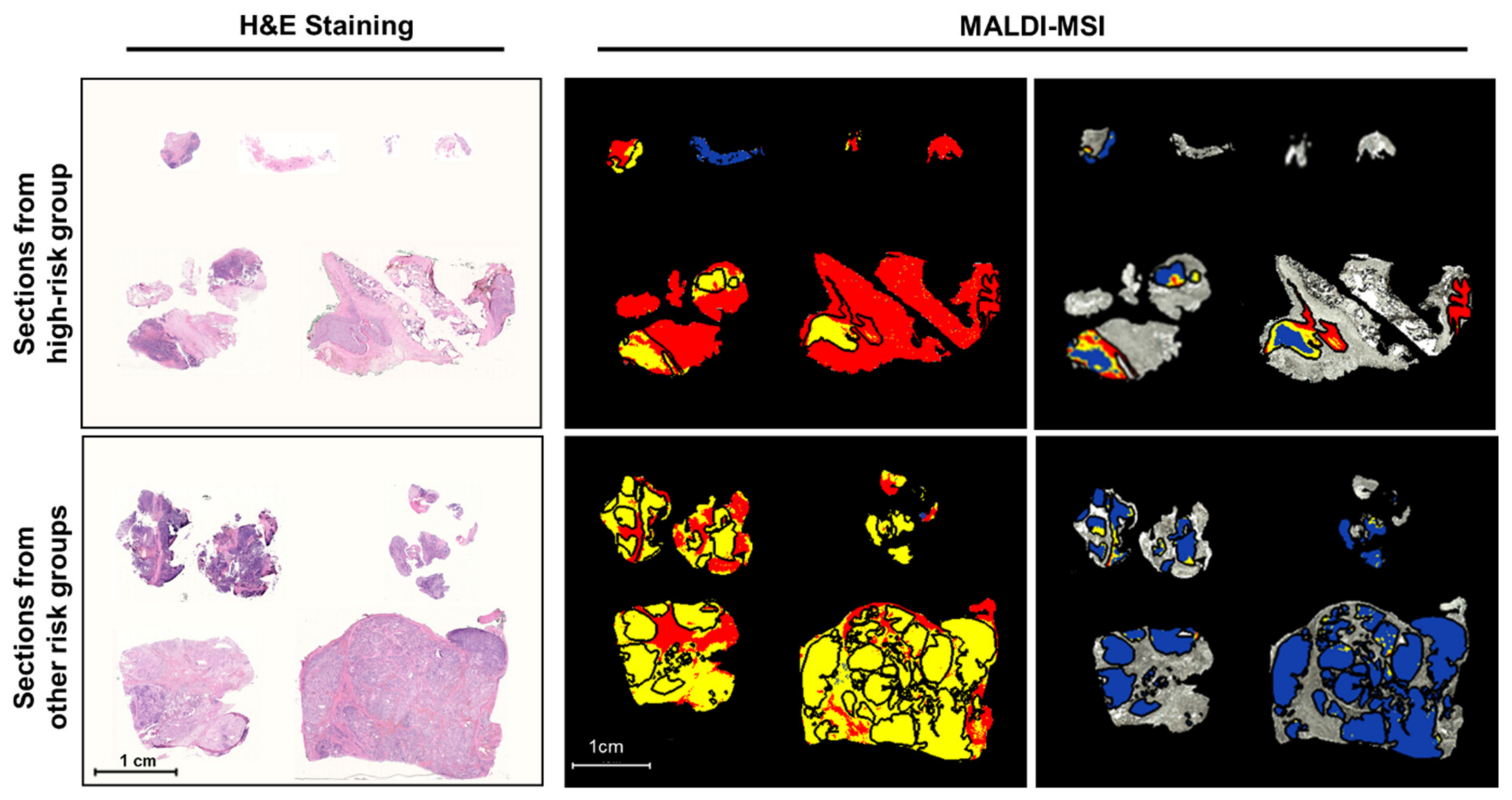
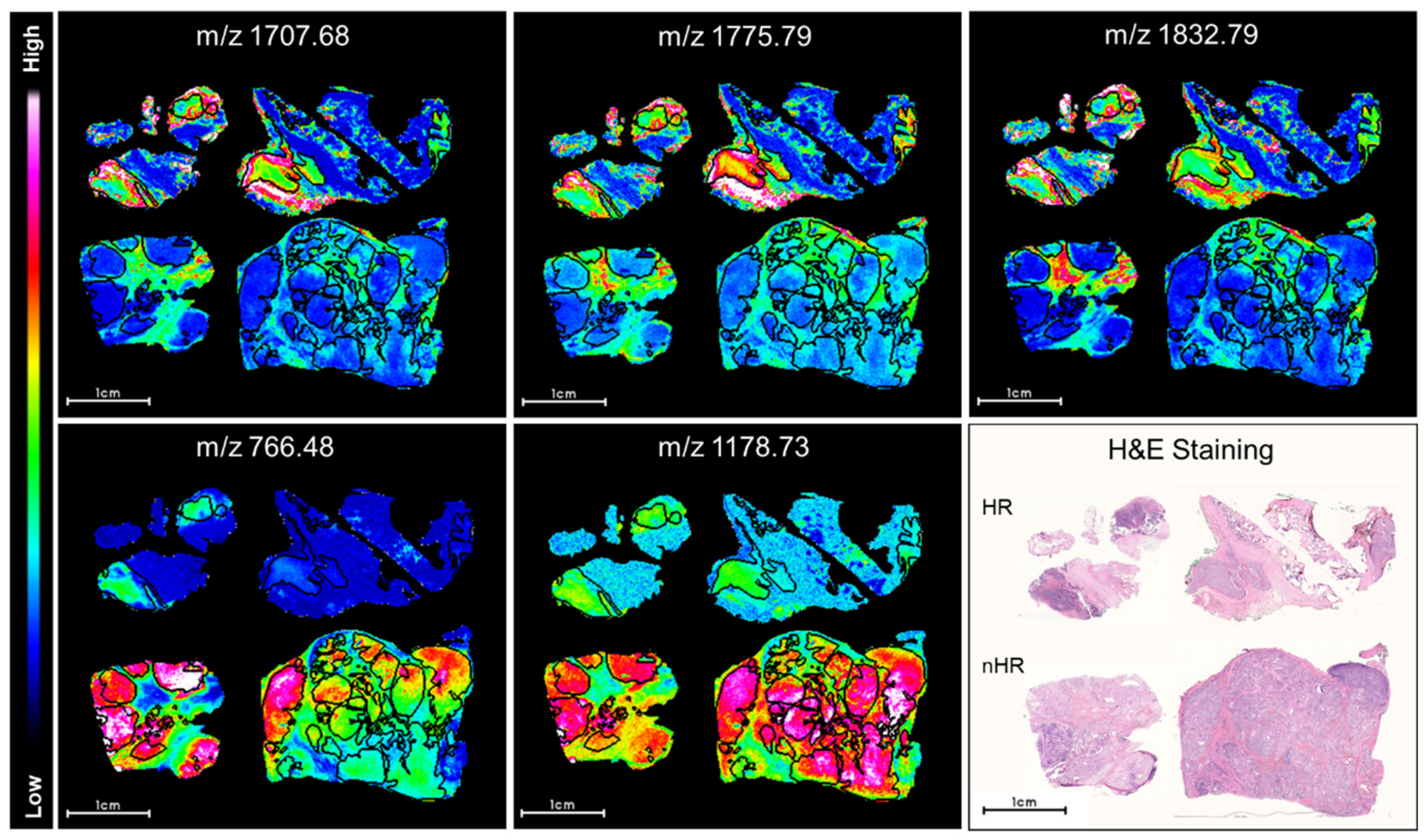
2.1. Discriminative Peptide Signatures Can Be Derived from MALDI-MSI Data to Identify Different Tumor Features
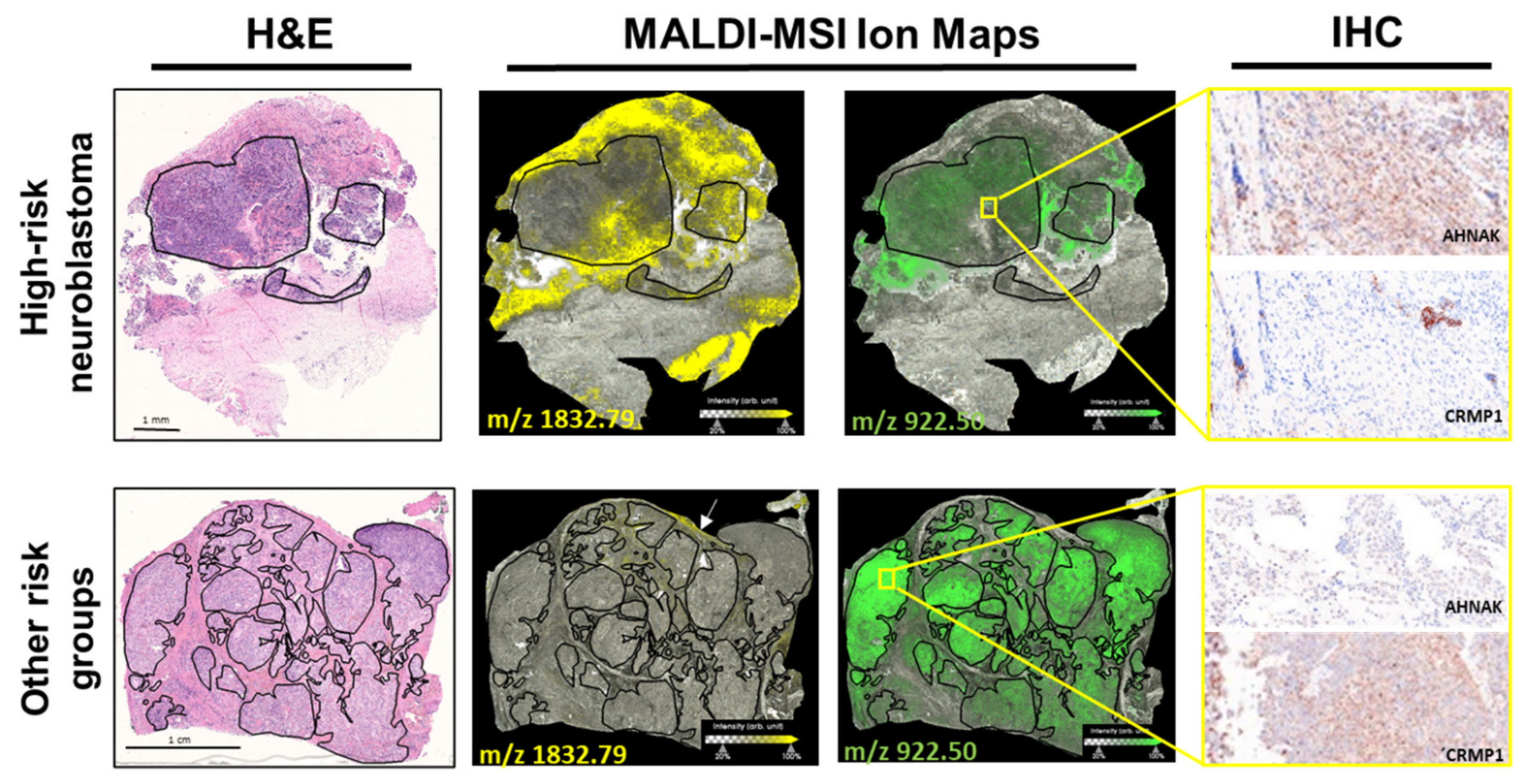
2.2. Discriminative Proteins Were Identified from Neuroblastoma Tissue Sections Based on MALDI-MSI Data
3. Discussion
4. Materials and Methods
4.1. Patient and Sample Cohort
4.2. Tissue Immunohistochemistry
4.3. MALDI-MSI
4.4. Protein Identification by Electrospray Ionization Tandem Mass Spectrometry
4.5. MALDI-MSI Data Processing for Statistical Analyses
4.6. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Husmann, G.; Kaatsch, P.; Katalinic, A.; Bertz, J.; Haberland, J.; Kraywinkel, K.; Wolf, U. Krebs in Deutschland 2005/2006. Häufigkeiten und Trends; Robert Koch. Institut: Berlin, Germany, 2010. [Google Scholar] [CrossRef]
- Maris, J.M. Recent Advances in Neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maris, J.M.; Hogarty, M.D.; Bagatell, R.; Cohn, S.L. Neuroblastoma. Lancet 2007, 369, 2106–2120. [Google Scholar] [CrossRef]
- Cheung, N.-K.V.; Dyer, M.A. Neuroblastoma: Developmental biology, cancer genomics and immunotherapy. Nat. Rev. Cancer 2013, 13, 397–411. [Google Scholar] [CrossRef] [Green Version]
- Simon, T.; Hero, B.; Schulte, J.H.; Deubzer, H.; Hundsdoerfer, P.; Von Schweinitz, D.; Fuchs, J.; Schmidt, M.; Prasad, V.; Krug, B.; et al. 2017 GPOH Guidelines for Diagnosis and Treatment of Patients with Neuroblastic Tumors. Klinische Pädiatrie 2017, 229, 147–167. [Google Scholar] [CrossRef] [PubMed]
- Cohn, S.L.; Pearson, A.D.J.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The international neuroblastoma risk group (INRG) classification system: An INRG task force report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef]
- Liang, W.H.; Federico, S.M.; London, W.B.; Naranjo, A.; Irwin, M.S.; Volchenboum, S.L.; Cohn, S.L. Tailoring Therapy for Children With Neuroblastoma on the Basis of Risk Group Classification: Past, Present, and Future. JCO Clin Cancer Inform 2020, 4, 895–905. [Google Scholar] [CrossRef]
- Øra, I.; Eggert, A. Progress in treatment and risk stratification of neuroblastoma: Impact on future clinical and basic research. Semin. Cancer Biol. 2011, 21, 217–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodeur, G.M.; Seeger, R.C.; Schwab, M.; E Varmus, H.; Bishop, J.M. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 1984, 224, 1121–1124. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.; Shyr, D.; Bagatell, R.; Fischer, M.; Nakagawara, A.; Nieto, A.C.; Brodeur, G.M.; Matthay, K.K.; London, W.B.; Dubois, S.G. Comprehensive evaluation of context dependence of the prognostic impact of MYCN amplification in neuroblastoma: A report from the International Neuroblastoma Risk Group (INRG) project. Pediatr. Blood Cancer 2019, 66, e27819. [Google Scholar] [CrossRef] [Green Version]
- Oberthuer, A.; Juraeva, D.; Hero, B.; Volland, R.; Sterz, C.; Schmidt, R.; Faldum, A.; Kahlert, Y.; Engesser, A.; Asgharzadeh, S.; et al. Revised Risk Estimation and Treatment Stratification of Low- and Intermediate-Risk Neuroblastoma Patients by Integrating Clinical and Molecular Prognostic Markers. Clin. Cancer Res. 2015, 21, 1904–1915. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, S.; Cartolano, M.; Hero, B.; Welte, A.; Kahlert, Y.; Roderwieser, A.; Bartenhagen, C.; Walter, E.; Gecht, J.; Kerschke, L.; et al. A mechanistic classification of clinical phenotypes in neuroblastoma. Science 2018, 362, 1165–1170. [Google Scholar] [CrossRef] [Green Version]
- Schramm, A.; Köster, J.; Assenov, Y.; Althoff, K.; Peifer, M.; Mahlow, E.; Odersky, A.; Beisser, D.; Ernst, C.; Henssen, A.; et al. Mutational dynamics between primary and relapse neuroblastomas. Nat. Genet. 2015, 47, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, J.; Valind, A.; Mengelbier, L.H.; Bredin, S.; Cornmark, L.; Jansson, C.; Wali, A.; Staaf, J.; Viklund, B.; Øra, I.; et al. Four evolutionary trajectories underlie genetic intratumoral variation in childhood cancer. Nat. Genet. 2018, 50, 944–950. [Google Scholar] [CrossRef]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013, 45, 279–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eleveld, T.F.; A Oldridge, D.; Bernard, V.; Koster, J.; Daâge, L.C.; Diskin, S.J.; Schild, L.; Bentahar, N.B.; Bellini, A.; Chicard, M.; et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat. Genet. 2015, 47, 864–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peifer, M.; Hertwig, F.; Roels, F.; Dreidax, D.; Gartlgruber, M.; Menon, R.; Krämer, A.; Roncaioli, J.L.; Sand, F.; Heuckmann, J.M.; et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nat. Cell Biol. 2015, 526, 700–704. [Google Scholar] [CrossRef]
- Molenaar, J.J.; Koster, J.; Zwijnenburg, D.A.; Van Sluis, P.; Valentijn, L.J.; Van Der Ploeg, I.; Hamdi, M.; Van Nes, J.; Westerman, B.A.; Van Arkel, J.; et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nat. Cell Biol. 2012, 483, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Gröbner, S.N.; Project, I.P.-S.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Brady, S.W.; Liu, Y.; Ma, X.; Gout, A.M.; Hagiwara, K.; Zhou, X.; Wang, J.; Macias, M.; Chen, X.; Easton, J.; et al. Pan-neuroblastoma analysis reveals age- and signature-associated driver alterations. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ohira, M.; Zhou, Y.; Xiong, T.; Luo, W.; Yang, C.; Li, X.; Gao, Z.; Zhou, R.; Nakamura, Y.; et al. Genomic analysis-integrated whole-exome sequencing of neuroblastomas identifies genetic mutations in axon guidance pathway. Oncotarget 2017, 8, 56684–56697. [Google Scholar] [CrossRef] [Green Version]
- de Sousa, V.M.L.; Carvalho, L. Heterogeneity in Lung Cancer. Pathobiology 2018, 85, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Heijs, B.; Holst-Bernal, S.; De Graaff, M.A.; Bruijn, I.H.B.-D.; Rodriguez-Girondo, M.; Van De Sande, M.A.J.; Wuhrer, M.; McDonnell, L.A.; Bovée, J.V.M.G. Molecular signatures of tumor progression in myxoid liposarcoma identified by N-glycan mass spectrometry imaging. Lab. Investig. 2020, 100, 1252–1261. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.; Roseborough, A.; Mor, R.; Yeung, K.K.-C.; Whitehead, S.N. Ganglioside Detection from Formalin-Fixed Human Brain Tissue Utilizing MALDI Imaging Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2020, 31, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Boyle, S.T.; Mittal, P.; Kaur, G.; Hoffmann, P.; Samuel, M.S.; Klingler-Hoffmann, M. Uncovering Tumor–Stroma Inter-relationships Using MALDI Mass Spectrometry Imaging. J. Proteome Res. 2020, 19, 4093–4103. [Google Scholar] [CrossRef]
- Aichler, M.; Walch, A. MALDI Imaging mass spectrometry: Current frontiers and perspectives in pathology research and practice. Lab. Investig. 2015, 95, 422–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermann, J.; Noels, H.; Theelen, W.; Lellig, M.; Orth-Alampour, S.; Boor, P.; Jankowski, V.; Jankowski, J. Sample preparation of formalin-fixed paraffin-embedded tissue sections for MALDI-mass spectrometry imaging. Anal. Bioanal. Chem. 2020, 412, 1263–1275. [Google Scholar] [CrossRef] [Green Version]
- Dilillo, M.; Ait-Belkacem, R.; Esteve, C.; Pellegrini, D.; Nicolardi, S.; Costa, M.; Vannini, E.; de Graaf, E.L.; Caleo, M.; McDonnell, L.A. Ultra-High Mass Resolution MALDI Imaging Mass Spectrometry of Proteins and Metabolites in a Mouse Model of Glioblastoma. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezger, S.T.P.; Mingels, A.M.A.; Bekers, O.; Heeren, R.M.A.; Cillero-Pastor, B. Mass Spectrometry Spatial-Omics on a Single Conductive Slide. Anal. Chem. 2021, 93, 2527–2533. [Google Scholar] [CrossRef]
- Spraggins, J.M.; Rizzo, D.G.; Moore, J.L.; Noto, M.J.; Skaar, E.P.; Caprioli, R.M. Next-generation technologies for spatial proteomics: Integrating ultra-high speed MALDI-TOF and high mass resolution MALDI FTICR imaging mass spectrometry for protein analysis. Proteomics 2016, 16, 1678–1689. [Google Scholar] [CrossRef] [Green Version]
- Kassuhn, W.; Klein, O.; Darb-Esfahani, S.; Lammert, H.; Handzik, S.; Taube, E.; Schmitt, W.; Keunecke, C.; Horst, D.; Dreher, F.; et al. Classification of Molecular Subtypes of High-Grade Serous Ovarian Cancer by MALDI-Imaging. Cancers 2021, 13, 1512. [Google Scholar] [CrossRef]
- Mascini, N.E.; Teunissen, J.; Noorlag, R.; Willems, S.M.; Heeren, R.M. Tumor classification with MALDI-MSI data of tissue microarrays: A case study. Methods 2018, 151, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Klein, O.; Fogt, F.; Hollerbach, S.; Nebrich, G.; Boskamp, T.; Wellmann, A. Classification of Inflammatory Bowel Disease from Formalin-Fixed, Paraffin-Embedded Tissue Biopsies via Imaging Mass Spectrometry. Proteom. Clin. Appl. 2020, 14, 1900131. [Google Scholar] [CrossRef]
- Neumann, J.M.; Niehaus, K.; Neumann, N.; Knobloch, H.C.; Bremmer, F.; Krafft, U.; Kellner, U.; Nyirády, P.; Szarvas, T.; Bednarz, H.; et al. A new technological approach in diagnostic pathology: Mass spectrometry imaging-based metabolomics for biomarker detection in urachal cancer. Lab. Investig. 2021, 1–8. [Google Scholar] [CrossRef]
- Meding, S.; Nitsche, U.; Balluff, B.; Elsner, M.; Rauser, S.; Schöne, C.; Nipp, M.; Maak, M.; Feith, M.; Ebert, M.P.; et al. Tumor Classification of Six Common Cancer Types Based on Proteomic Profiling by MALDI Imaging. J. Proteome Res. 2012, 11, 1996–2003. [Google Scholar] [CrossRef] [PubMed]
- Schwamborn, K.; Krieg, R.C.; Jirak, P.; Ott, G.; Knuchel, R.; Rosenwald, A.; Wellmann, A. Application of MALDI imaging for the diagnosis of classical Hodgkin lymphoma. J. Cancer Res. Clin. Oncol. 2010, 136, 1651–1655. [Google Scholar] [CrossRef]
- Rauser, S.; Marquardt, C.; Balluff, B.; Deininger, S.-O.; Albers, C.; Belau, E.; Hartmer, R.; Suckau, D.; Specht, K.; Ebert, M.P.; et al. Classification of HER2 Receptor Status in Breast Cancer Tissues by MALDI Imaging Mass Spectrometry. J. Proteome Res. 2010, 9, 1854–1863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balluff, B.; Rauser, S.; Meding, S.; Elsner, M.; Schöne, C.; Feuchtinger, A.; Schuhmacher, C.; Novotny, A.; Jütting, U.; Maccarrone, G.; et al. MALDI Imaging Identifies Prognostic Seven-Protein Signature of Novel Tissue Markers in Intestinal-Type Gastric Cancer. Am. J. Pathol. 2011, 179, 2720–2729. [Google Scholar] [CrossRef]
- Bauer, J.A.; Chakravarthy, A.B.; Rosenbluth, J.M.; Mi, D.; Seeley, E.H.; Granja-Ingram, N.D.M.; Olivares, M.G.; Kelley, M.C.; Mayer, I.A.; Meszoely, I.M.; et al. Identification of Markers of Taxane Sensitivity Using Proteomic and Genomic Analyses of Breast Tumors from Patients Receiving Neoadjuvant Paclitaxel and Radiation. Clin. Cancer Res. 2010, 16, 681–690. [Google Scholar] [CrossRef] [Green Version]
- Cillero-Pastor, B.; Heeren, R.M.A. Matrix-Assisted Laser Desorption Ionization Mass Spectrometry Imaging for Peptide and Protein Analyses: A Critical Review of On-Tissue Digestion. J. Proteome Res. 2013, 13, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.; Loos, B.; Engelbrecht, A.-M. AHNAK: The giant jack of all trades. Cell. Signal. 2014, 26, 2683–2693. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, X.; Huang, R.; Liu, X.; Liang, Z.; Liu, T. Upregulation of nucleoprotein AHNAK is associated with poor outcome of pancreatic ductal adenocarcinoma prognosis via mediating epithelial-mesenchymal transition. J. Cancer 2019, 10, 3860–3870. [Google Scholar] [CrossRef] [Green Version]
- Hartomo, T.B.; Kozaki, A.; Hasegawa, D.; Pham, T.V.H.; Yamamoto, N.; Saitoh, A.; Ishida, T.; Kawasaki, K.; Kosaka, Y.; Ohashi, H.; et al. Minimal residual disease monitoring in neuroblastoma patients based on the expression of a set of real-time RT-PCR markers in tumor-initiating cells. Oncol. Rep. 2013, 29, 1629–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirase, S.; Saitoh, A.; Hartomo, T.B.; Kozaki, A.; Yanai, T.; Hasegawa, D.; Kawasaki, K.; Kosaka, Y.; Matsuo, M.; Yamamoto, N.; et al. Early detection of tumor relapse/regrowth by consecutive minimal residual disease monitoring in high-risk neuroblastoma patients. Oncol. Lett. 2016, 12, 1119–1123. [Google Scholar] [CrossRef]
- Yamamoto, N.; Kozaki, A.; Hartomo, T.B.; Yanai, T.; Hasegawa, D.; Kawasaki, K.; Kosaka, Y.; Matsuo, M.; Hirase, S.; Mori, T.; et al. Differential expression of minimal residual disease markers in peripheral blood and bone marrow samples from high-risk neuroblastoma patients. Oncol. Lett. 2015, 10, 3228–3232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thwin, K.K.; Ishida, T.; Uemura, S.; Yamamoto, N.; Lin, K.S.; Tamura, A.; Kozaki, A.; Saito, A.; Kishimoto, K.; Mori, T.; et al. Level of Seven Neuroblastoma-Associated mRNAs Detected by Droplet Digital PCR Is Associated with Tumor Relapse/Regrowth of High-Risk Neuroblastoma Patients. J. Mol. Diagn. 2020, 22, 236–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pappin, D.; Højrup, P.; Bleasby, A. Rapid identification of proteins by peptide-mass fingerprinting. Curr. Biol. 1993, 3, 327–332. [Google Scholar] [CrossRef]
- Klein, O.; Kanter, F.; Kulbe, H.; Jank, P.; Denkert, C.; Nebrich, G.; Schmitt, W.D.; Wu, Z.; Kunze, C.A.; Sehouli, J.; et al. MALDI-Imaging for Classification of Epithelial Ovarian Cancer Histotypes from a Tissue Microarray Using Machine Learning Methods. Proteom. Clin. Appl. 2019, 13, e1700181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulbe, H.; Klein, O.; Wu, Z.; Taube, E.; Kassuhn, W.; Horst, D.; Darb-Esfahani, S.; Jank, P.; Abobaker, S.; Ringel, F.; et al. Discovery of Prognostic Markers for Early-Stage High-Grade Serous Ovarian Cancer by Maldi-Imaging. Cancers 2020, 12, 2000. [Google Scholar] [CrossRef] [PubMed]
- Casadonte, R.; Longuespee, R.; Kriegsmann, J.; Kriegsmann, M. MALDI IMS and Cancer Tissue Microarrays. Adv. Cancer Res. 2017, 134, 173–200. [Google Scholar] [CrossRef] [PubMed]
- Kriegsmann, J.; Kriegsmann, M.; Casadonte, R. MALDI TOF imaging mass spectrometry in clinical pathology: A valuable tool for cancer diagnostics (Review). Int. J. Oncol. 2014, 46, 893–906. [Google Scholar] [CrossRef] [Green Version]
- Longuespee, R.; Baiwir, D.; Mazzucchelli, G.; Smargiasso, N.; De Pauw, E. Laser Microdissection-Based Microproteomics of Formalin-Fixed and Paraffin-Embedded (FFPE) Tissues. Methods Mol. Biol. 2018, 1723, 19–31. [Google Scholar] [CrossRef]
- Giordano, S.; Zucchetti, M.; Decio, A.; Cesca, M.; Nerini, I.F.; Maiezza, M.; Ferrari, M.; Licandro, S.A.; Frapolli, R.; Giavazzi, R.; et al. Heterogeneity of paclitaxel distribution in different tumor models assessed by MALDI mass spectrometry imaging. Sci. Rep. 2016, 6, 39284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schöne, C.; Höfler, H.; Walch, A. MALDI imaging mass spectrometry in cancer research: Combining proteomic profiling and histological evaluation. Clin. Biochem. 2013, 46, 539–545. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.R.; Song, Y.K.; Yu, L.R.; Wei, J.S.; Chung, J.Y.; Hewitt, S.M.; Veenstra, T.D.; Khan, J. Global genomic and proteomic analysis identifies biological pathways related to high-risk neuroblastoma. J. Proteome Res. 2010, 9, 373–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.; Zhu, X.; Feng, C.; Wang, T.; Hong, Q.; Liu, Z.; Tang, S. Proteomics-based identification of spontaneous regression-associated proteins in neuroblastoma. J. Pediatr. Surg. 2011, 46, 1948–1955. [Google Scholar] [CrossRef] [PubMed]
- Tan, F.; Thiele, C.J.; Li, Z. Collapsin response mediator proteins: Potential diagnostic and prognostic biomarkers in cancers (Review). Oncol. Lett. 2014, 7, 1333–1340. [Google Scholar] [CrossRef]
- Cheng, F.; Liu, C.; Lin, C.-C.; Zhao, J.; Jia, P.; Li, W.-H.; Zhao, Z. A Gene Gravity Model for the Evolution of Cancer Genomes: A Study of 3,000 Cancer Genomes across 9 Cancer Types. PLoS Comput. Biol. 2015, 11, e1004497. [Google Scholar] [CrossRef]
- Xiang, X.; Langlois, S.; St-Pierre, M.-E.; Blinder, A.; Charron, P.; Graber, T.E.; Fowler, S.L.; Baird, S.D.; Bennett, S.A.L.; Alain, T.; et al. Identification of pannexin 1-regulated genes, interactome, and pathways in rhabdomyosarcoma and its tumor inhibitory interaction with AHNAK. Oncogene 2021, 40, 1868–1883. [Google Scholar] [CrossRef]
- Soini, T.; Eloranta, K.; Pihlajoki, M.; Kyronlahti, A.; Akinrinade, O.; Andersson, N.; Lohi, J.; Pakarinen, M.P.; Wilson, D.B.; Heikinheimo, M. Transcription factor GATA4 associates with mesenchymal-like gene expression in human hepatoblastoma cells. Tumor Biol. 2018, 40, 1010428318785498. [Google Scholar] [CrossRef] [Green Version]
- Jaskiewicz, N.M.; Townson, D.H. Hyper-O-GlcNAcylation promotes epithelial-mesenchymal transition in endometrial cancer cells. Oncotarget 2019, 10, 2899–2910. [Google Scholar] [CrossRef] [Green Version]
- Sohn, M.; Shin, S.; Yoo, J.Y.; Goh, Y.; Lee, I.H.; Bae, Y.S. Ahnak promotes tumor metastasis through transforming growth factor-beta-mediated epithelial-mesenchymal transition. Sci. Rep. 2018, 8, 14379. [Google Scholar] [CrossRef] [Green Version]
- Shen, E.; Wang, X.; Liu, X.; Lv, M.; Zhang, L.; Zhu, G.; Sun, Z. MicroRNA-93-5p promotes epithelial-mesenchymal transition in gastric cancer by repressing tumor suppressor AHNAK expression. Cancer Cell Int. 2020, 20, 76. [Google Scholar] [CrossRef]
- Zhao, Z.; Xiao, S.; Yuan, X.; Yuan, J.; Zhang, C.; Li, H.; Su, J.; Wang, X.; Liu, Q. AHNAK as a Prognosis Factor Suppresses the Tumor Progression in Glioma. J. Cancer 2017, 8, 2924–2932. [Google Scholar] [CrossRef] [Green Version]
- Cimas, F.J.; Manzano, A.; Baliu-Piqué, M.; García-Gil, E.; Pérez-Segura, P.; Nagy, Ádám; Pandiella, A.; Győrffy, B.; Ocana, A. Genomic Mapping Identifies Mutations in RYR2 and AHNAK as Associated with Favorable Outcome in Basal-Like Breast Tumors Expressing PD1/PD-L1. Cancers 2020, 12, 2243. [Google Scholar] [CrossRef] [PubMed]
- Klein, O.; Strohschein, K.; Nebrich, G.; Oetjen, J.; Trede, D.; Thiele, H.; Alexandrov, T.; Giavalisco, P.; Duda, G.N.; von Roth, P.; et al. MALDI imaging mass spectrometry: Discrimination of pathophysiological regions in traumatized skeletal muscle by characteristic peptide signatures. Proteomics 2014, 14, 2249–2260. [Google Scholar] [CrossRef]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, T.; Becker, M.; Deininger, S.-O.; Ernst, G.; Wehder, L.; Grasmair, M.; Von Eggeling, F.; Thiele, H.; Maass, P. Spatial Segmentation of Imaging Mass Spectrometry Data with Edge-Preserving Image Denoising and Clustering. J. Proteome Res. 2010, 9, 6535–6546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, T.; Becker, M.; Guntinas-Lichius, O.; Ernst, G.; von Eggeling, F. MALDI-imaging segmentation is a powerful tool for spatial functional proteomic analysis of human larynx carcinoma. J. Cancer Res. Clin. Oncol. 2013, 139, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Trede, D.; Schiffler, S.; Becker, M.; Wirtz, S.; Steinhorst, K.; Strehlow, J.; Aichler, M.; Kobarg, J.H.; Oetjen, J.; Dyatlov, A.; et al. Exploring Three-Dimensional Matrix-Assisted Laser Desorption/Ionization Imaging Mass Spectrometry Data: Three-Dimensional Spatial Segmentation of Mouse Kidney. Anal. Chem. 2012, 84, 6079–6087. [Google Scholar] [CrossRef]
- McDonnell, L.A.; van Remoortere, A.; van Zeijl, R.J.M.; Deelder, A.M. Mass spectrometry image correlation: Quantifying colocalization. J. Proteome Res. 2008, 7, 3619–3627. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| INSS/INRG Staging | Age at Diagnosis (Months) | MYCN Status | Chromosome 1p Status | Treatment Risk Group |
|---|---|---|---|---|
| 1 | not amplified | normal | Low | |
| amplified | High | |||
| 2 | not amplified | normal | Low | |
| deletion/imbalance | intermediate | |||
| amplified | High | |||
| 3 | <24 | not amplified | normal | Low |
| ≥24 | not amplified | normal | intermediate | |
| not amplified | deletion/imbalance | |||
| amplified | High | |||
| 4s/MS | <18 | not amplified | normal | Low |
| amplified | High | |||
| 4/M | <18 | not amplified | intermediate | |
| ≥18 | amplified | High |
| MALDI IMS m/z Value | ROC [AUC] for High Versus other Risk * | ROC [AUC] HR/nHR TMA † | Significance Rating-WRS | LC-MS/MS [Mr + H+ cal.] | Scores § | Deviation [Da] | Correlation Coefficient | Protein Symbol | Protein |
|---|---|---|---|---|---|---|---|---|---|
| 868.4930 | 0.85 | 0.73 | <0.001 | 868.46 | 48.1 | 0.03 | 0.38 | COL1A2 | Collagen type I alpha 2 chain |
| 1562.7700 | 0.91 | 0.74 | <0.001 | 1562.79 | 127. | 0.02 | 0.64 | ||
| 2026.9100 | 0.86 | 0.73 | <0.001 | 2027.02 | 65.8 | 0.11 | 0.36 | ||
| 1459.8500 | 0.72 | 0.66 | <0.001 | 1459.86 | 40.5 | 0.01 | 0.38 | COL6A3 | Collagen type VI alpha 3 chain |
| 2056.9200 | 0.88 | 0.63 | <0.001 | 2057.04 | 59.4 | 0.12 | 0.32 | ||
| 766.4820 | 0.08 | 0.28 | <0.001 | 766.46 | 21.7 | 0.03 | 0.44 | CRMP1 | Collapsin response mediator protein 1 |
| 922.4990 | 0.14 | 0.34 | <0.001 | 922.51 | 22.3 | 0.02 | 0.40 | ||
| 1833.9900 | 0.87 | 0.67 | <0.001 | 1833.91 | 65.1 | 0.08 | 0.40 | HSPA5 | Heat shock protein family A (Hsp70) member 5 |
| 2042.2200 | 0.85 | 0.73 | <0.001 | 2042.05 | 25.6 | 0.17 | 0.32 | ||
| 1477.8600 | 0.90 | 0.75 | <0.001 | 1477.79 | 28.1 | 0.07 | 0.41 | HIST1H2BC | H2B clustered histone 4 |
| 1743.6800 | 0.82 | 0.58 | <0.001 | 1743.82 | 96.2 | 0.14 | 0.58 | ||
| 1775.7900 | 0.90 | 0.70 | <0.001 | 1775.81 | 123. | 0.02 | 0.55 | ||
| 1586.7700 | 0.90 | 0.74 | <0.001 | 1586.77 | 89.4 | 0.00 | 0.47 | KRT9 | Keratin 9 |
| 2705.2800 | 0.86 | 0.78 | <0.001 | 2705.16 | 67.9 | 0.12 | 0.44 | ||
| 1267.5000 | 0.87 | 0.74 | <0.001 | 1267.65 | 63.9 | 0.12 | 0.38 | AHNAK | AHNAK nucleoprotein |
| 1832.7900 | 0.92 | 0.70 | <0.001 | 1832.88 | 44.7 | 0.09 | 0.39 | ||
| 1706.7800 | 0.87 | 0.74 | <0.001 | 1706.78 | 31.2 | 0.00 | 0.31 | NID2 | Nidogen 2 |
| 2455.3600 | 0.79 | 0.72 | <0.001 | 2455.17 | 34.9 | 0.19 | 0.33 |
| ID | Sex | Age (Years) | INSS Stage | MYCN Amplification | Risk Classification (at Diagnosis) | Disease Recurrence | Death | Metastasis |
|---|---|---|---|---|---|---|---|---|
| 1 | F | 0.3 | 3 | + | high | - | - | No |
| 2 | M | 0.6 | 2 | + | high | - | - | No |
| 3 | M | 1 | 3 | + | high | - | + | No † |
| 4 | M | 1.4 | 4 | + | high | - | - | Yes |
| 5 | F | 1.2 | 4 | + | high | + | + | Yes |
| 6 | M | 2.8 | 4 | + | high | + | + | Yes |
| 7 | M | 7.8 | 4 | - | high | - | - | Yes |
| 8 | F | 8 | 4 | + | high | - | - | Yes |
| 9 ‡ | M | 1.2 | 3 | - | high | + | - | No ‡ |
| 10 | F | 2.4 | 1 | - | low | - | - | No |
| 11 | F | 0.8 | 4 | - | intermediate | - | - | Yes |
| 12 | M | 0.1 | 4s | - | low | - | - | Yes |
| 13 | F | 0.1 | 3 | mosaic | low | - | - | No |
| 14 | F | 5.9 | 3 | - | intermediate | - | - | No |
| 15 | M | 1.9 | 2 | - | low | - | - | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Z.; Hundsdoerfer, P.; Schulte, J.H.; Astrahantseff, K.; Boral, S.; Schmelz, K.; Eggert, A.; Klein, O. Discovery of Spatial Peptide Signatures for Neuroblastoma Risk Assessment by MALDI Mass Spectrometry Imaging. Cancers 2021, 13, 3184. https://doi.org/10.3390/cancers13133184
Wu Z, Hundsdoerfer P, Schulte JH, Astrahantseff K, Boral S, Schmelz K, Eggert A, Klein O. Discovery of Spatial Peptide Signatures for Neuroblastoma Risk Assessment by MALDI Mass Spectrometry Imaging. Cancers. 2021; 13(13):3184. https://doi.org/10.3390/cancers13133184
Chicago/Turabian StyleWu, Zhiyang, Patrick Hundsdoerfer, Johannes H. Schulte, Kathy Astrahantseff, Senguel Boral, Karin Schmelz, Angelika Eggert, and Oliver Klein. 2021. "Discovery of Spatial Peptide Signatures for Neuroblastoma Risk Assessment by MALDI Mass Spectrometry Imaging" Cancers 13, no. 13: 3184. https://doi.org/10.3390/cancers13133184