
Distinct Characteristics and Clinical Outcomes to Predict the Emergence of MET Amplification in Patients with Non-Small Cell Lung Cancer Who Developed Resistance after Treatment with Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Patients
2.2. Assessments
2.3. EGFR Mutation and MET ISH Analyses
2.4. Statistical Analysis
3. Results
3.1. Clinicopathological Characteristics of the Study Population
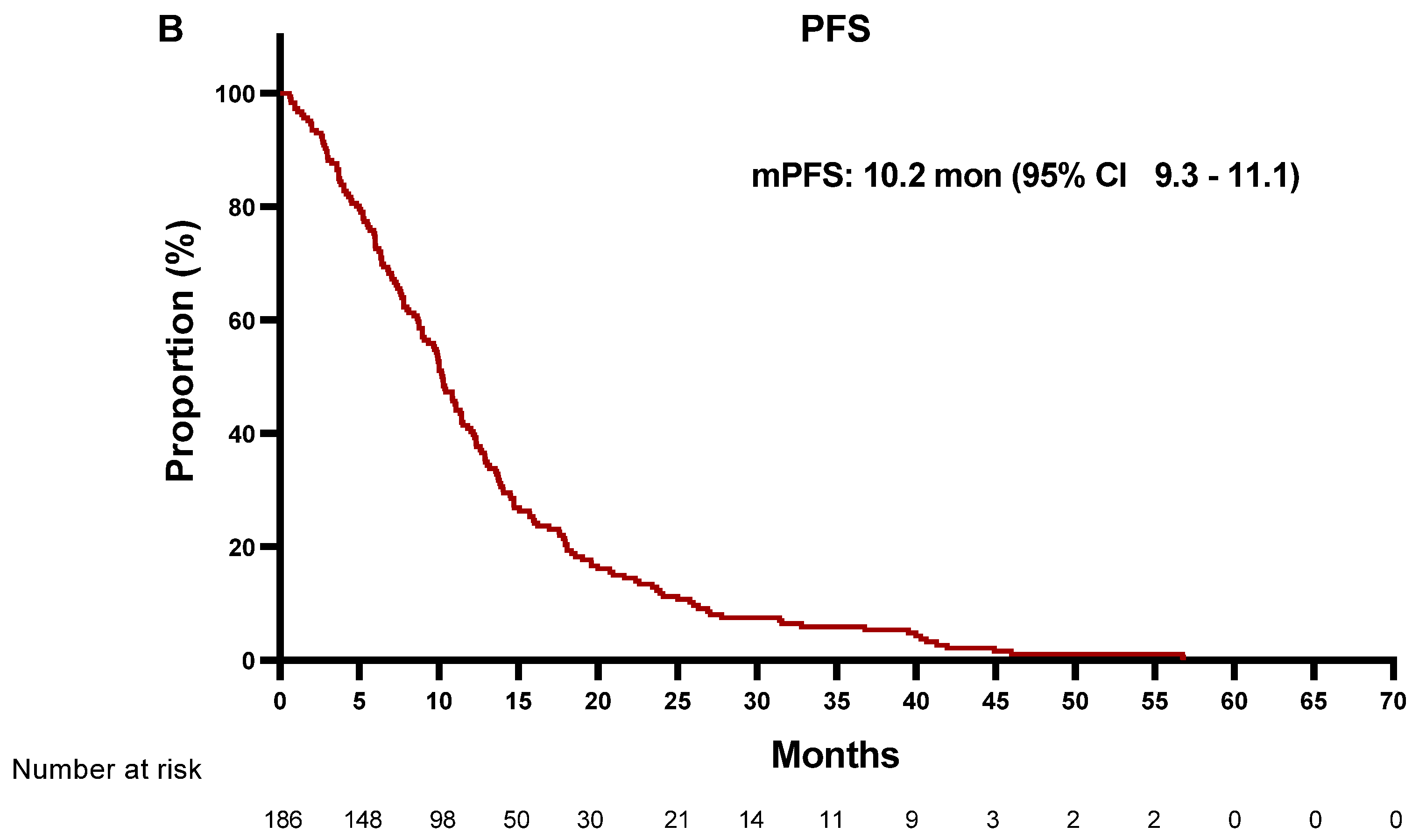
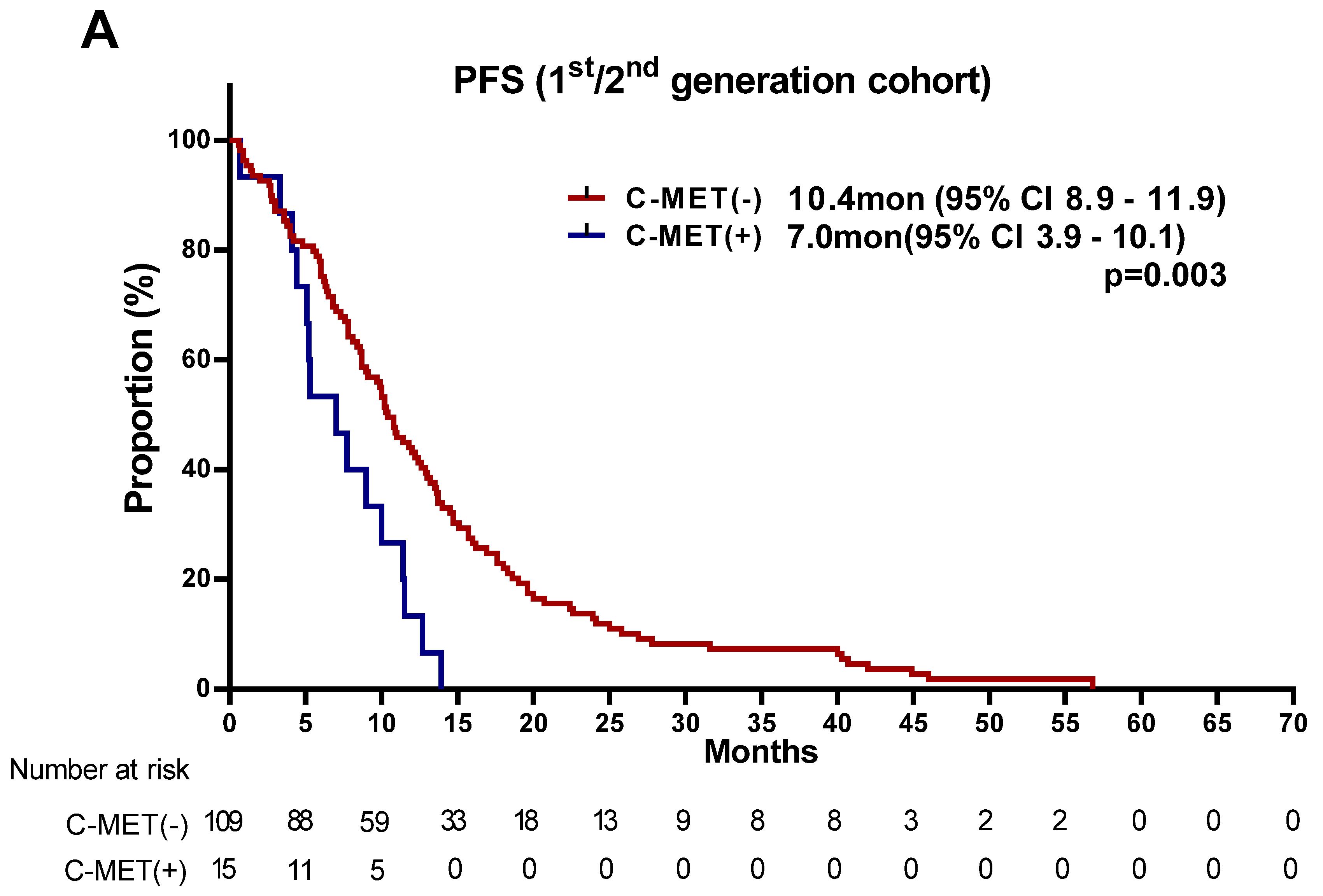
3.2. Treatment Outcomes of EGFR-TKIs According to MET Status
3.3. Potential Predictors for MET Amplification
3.4. Characteristics of Patients with MET Amplification
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rosell, R.; Bivona, T.G.; Karachaliou, N. Genetics and biomarkers in personalisation of lung cancer treatment. Lancet 2013, 382, 720–731. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.; Thongprasert, S.; Yang, C.; Chu, D.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Nishiwaki, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Park, K.; Tan, E.H.; O’Byrne, K.; Zhang, L.; Boyer, M.; Mok, T.; Hirsh, V.; Yang, J.h.; Lee, K.H.; Lu, S.; et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): A phase 2B, open-label, randomised controlled trial. Lancet Oncol. 2016, 17, 577–589. [Google Scholar] [CrossRef]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oxnard, G.R.; Hu, Y.; Mileham, K.F.; Husain, H.; Costa, D.B.; Tracy, P.; Feeney, N.; Sholl, L.M.; Dahlberg, S.E.; Redig, A.J.; et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients With EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 2018, 4, 1527–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.M.; Syn, N.L.; Cho, B.C.; Soo, R.A. Acquired resistance to EGFR targeted therapy in non-small cell lung cancer: Mechanisms and therapeutic strategies. Cancer Treat. Rev. 2018, 65, 1–10. [Google Scholar] [CrossRef]
- Gristina, V.; La Mantia, M.; Galvano, A.; Cutaia, S.; Barraco, N.; Castiglia, M.; Perez, A.; Bono, M.; Iacono, F.; Greco, M. Non-Small Cell Lung Cancer Harboring Concurrent EGFR Genomic Alterations: A Systematic Review and Critical Appraisal of the Double Dilemma. J. Mol. Pathol. 2021, 2, 173–196. [Google Scholar] [CrossRef]
- Gristina, V.; Malapelle, U.; Galvano, A.; Pisapia, P.; Pepe, F.; Rolfo, C.; Tortorici, S.; Bazan, V.; Troncone, G.; Russo, A. The significance of epidermal growth factor receptor uncommon mutations in non-small cell lung cancer: A systematic review and critical appraisal. Cancer Treat. Rev. 2020, 85, 101994. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; Vande Woude, G. Targeting MET in cancer: Rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef] [Green Version]
- Ramalingam, S.S.; Cheng, Y.; Zhou, C.; Ohe, Y.; Imamura, F.; Cho, B.C.; Lin, M.; Majem, M.; Shah, R.; Rukazenkov, Y.; et al. Mechanisms of acquired resistance to first-line osimertinib: Preliminary data from the phase III FLAURA study. Ann. Oncol. 2018, 29 (Suppl. 9), 173–178. [Google Scholar] [CrossRef]
- Gan, H.K.; Millward, M.J.; Hua, Y.; Qi, C.; Sai, Y.; Su, W.; Wang, J.; Zhang, L.; Frigault, M.M.; Morgan, S.; et al. First-in-Human Phase I Study of the Selective MET Inhibitor, Savolitinib, in Patients with Advanced Solid Tumors: Safety, Pharmacokinetics and Anti-Tumor Activity. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Ahn, M.-J.; Kim, S.-W.; Cho, B.C.; Sequist, L.; Orlov, S.; Ottesen, L.H.; Verheijen, R.B.; Mellemgaard, A.; Wessen, J.; et al. Abstract CT032: TATTON Phase Ib expansion cohort: Osimertinib plus savolitinib for patients (pts) with EGFR-mutant, MET-amplified NSCLC after progression on prior first/second-generation epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI). In Proceedings of the AACR Annual Meeting, Atlanta, GA, USA, 29 March–3 April 2019. [Google Scholar]
- Lee, Y.J.; Kim, J.H.; Kim, S.K.; Ha, S.J.; Mok, T.S.; Mitsudomi, T.; Cho, B.C. Lung cancer in never smokers: Change of a mindset in the molecular era. Lung Cancer 2011, 72, 9–15. [Google Scholar] [CrossRef]
- Shaw, A.T.; Yeap, B.Y.; Mino-Kenudson, M.; Digumarthy, S.R.; Costa, D.B.; Heist, R.S.; Solomon, B.; Stubbs, H.; Admane, S.; McDermott, U.; et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J. Clin. Oncol. 2009, 27, 4247–4253. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Yan, B.; Zhang, Y.; Xu, J.; Qiao, R.; Dong, Y.; Zhang, B.; Zhao, Y.; Zhang, L.; Qian, J.; et al. Different characteristics and survival in non-small cell lung cancer patients with primary and acquired EGFR T790M mutation. Int. J. Cancer 2019, 144, 2880–2886. [Google Scholar] [CrossRef] [PubMed]
- Oxnard, G.R.; Yang, J.C.H.; Yu, H.; Kim, S.W.; Saka, H.; Horn, L.; Goto, K.; Ohe, Y.; Mann, H.; Thress, K.S.; et al. TATTON: A multi-arm, phase Ib trial of osimertinib combined with selumetinib, savolitinib, or durvalumab in EGFR-mutant lung cancer. Ann. Oncol. 2020, 31, 507–516. [Google Scholar] [CrossRef] [Green Version]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Arcila, M.E.; Oxnard, G.R.; Nafa, K.; Riely, G.J.; Solomon, S.B.; Zakowski, M.F.; Kris, M.G.; Pao, W.; Miller, V.A.; Ladanyi, M. Rebiopsy of Lung Cancer Patients with Acquired Resistance to EGFR Inhibitors and Enhanced Detection of the T790M Mutation Using a Locked Nucleic Acid-Based Assay. Clin. Cancer Res. 2011, 17, 1169–1180. [Google Scholar] [CrossRef] [Green Version]
- Cross, D.A.E.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.V.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an Irreversible EGFR TKI, Overcomes T790M-Mediated Resistance to EGFR Inhibitors in Lung Cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR Mutation and Resistance of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Nagano, T.; Tachihara, M.; Nishimura, Y. Mechanism of Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors and a Potential Treatment Strategy. Cells 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Cuaran, S.; Scheffler, M.; Plenker, D.; Dahmen, l.; Scheel, A.H.; Fernandez-Cuesta, L.; Meder, L.; Lovly, C.M.; Persigehl, T.; Merkelbach-Bruse, S.; et al. Heterogeneous Mechanisms of Primary and Acquired Resistance to Third-Generation EGFR Inhibitors. Clin. Cancer Res. 2016, 22, 4837–4847. [Google Scholar] [CrossRef] [Green Version]
- Loriot, Y.; Planchard, D.; André, F.; Gobert, A.; Soria, J.C.; Auger, N.; Lacroix, L. EGFR-independent mechanisms of acquired resistance to AZD9291 in EGFR T790M-positive NSCLC patients. Ann. Oncol. 2015, 26, 2073–2078. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L.; et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef] [Green Version]
- Bergethon, K.; Shaw, A.T.; Ou, S.H.; Katayama, R.; Lovly, C.M.; McDonald, N.T.; Massion, P.P.; Siwak-Tapp, C.; Gonzalez, A.; Fang, R.; et al. ROS1 rearrangements define a unique molecular class of lung cancers. J. Clin. Oncol. 2012, 30, 863–870. [Google Scholar] [CrossRef] [Green Version]
- Schrock, A.B.; Frampton, G.M.; Suh, J.; Chalmers, Z.R.; Rosenzweig, M.; Erlich, R.L.; Halmos, B.; Goldman, J.; Forde, P.; Leuenberger, K.; et al. Characterization of 298 Patients with Lung Cancer Harboring MET Exon 14 Skipping Alterations. J. Thorac. Oncol. 2016, 11, 1493–1502. [Google Scholar] [CrossRef] [Green Version]
- Awad, M.M.; Oxnard, G.R.; Jackman, D.M.; Savukoski, D.O.; Hall, D.; Shivdasani, P.; Heng, J.C.; Dahlberg, S.E.; Janne, P.A.; Verma, S.; et al. MET Exon 14 Mutations in Non-Small-Cell Lung Cancer Are Associated With Advanced Age and Stage-Dependent MET Genomic Amplification and c-Met Overexpression. J. Clin. Oncol. 2016, 34, 721–730. [Google Scholar] [CrossRef] [Green Version]
- Bubendorf, L.; Dafni, U.; Schöbel, M.; Finn, S.P.; Tischler, V.; Sejda, A.; Marchetti, A.; Thunnissen, E.; Verbeken, E.K.; Warth, A.; et al. Prevalence and clinical association of MET gene overexpression and amplification in patients with NSCLC: Results from the European Thoracic Oncology Platform (ETOP) Lungscape project. Lung Cancer 2017, 111, 143–149. [Google Scholar] [CrossRef]
- Sørensen, J.B.; Hansen, H.H.; Hansen, M.; Dombernowsky, P. Brain metastases in adenocarcinoma of the lung: Frequency, risk groups, and prognosis. J. Clin. Oncol. 1988, 6, 1474–1480. [Google Scholar] [CrossRef]
- Villano, J.L.; Durbin, E.B.; Normandeau, C.; Thakkar, J.P.; Moirangthem, V.; Davis, F.G. Incidence of brain metastasis at initial presentation of lung cancer. Neuro Oncol. 2014, 17, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Soria, J.C.; Tan, D.S.W.; Chiari, R.; Wu, Y.L.; Paz-Ares, L.; Wolf, J.; Geater, S.L.; Orlov, S.; Cortinovis, D.; Yu, C.J.; et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): A randomised, open-label, phase 3 study. Lancet 2017, 389, 917–929. [Google Scholar] [CrossRef]
- Gainor, J.F.; Tseng, D.; Yoda, S.; Dagogo-Jack, I.; Friboulet, L.; Lin, J.J.; Hubbeling, H.G.; Dardaei, L.; Farago, A.F.; Schultz, K.R.; et al. Patterns of Metastatic Spread and Mechanisms of Resistance to Crizotinib in ROS1-Positive Non-Small-Cell Lung Cancer. JCO Precis. Oncol. 2017, 2017. [Google Scholar] [CrossRef]
- Park, S.; Ahn, B.C.; Lim, S.W.; Sun, J.M.; Kim, H.R.; Hong, M.H.; Lee, S.H.; Ahn, J.S.; Park, K.; Choi, Y.; et al. Characteristics and Outcome of ROS1-Positive Non-Small Cell Lung Cancer Patients in Routine Clinical Practice. J. Thorac. Oncol. 2018, 13, 1373–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, G.G.Y.; Lim, T.H.; Lim, J.; Liew, P.J.R.; Kwang, X.L.; Nahar, R.; Aung, Z.W.; Takano, A.; Lee, Y.Y.; Lau, D.P.X.; et al. Clonal MET Amplification as a Determinant of Tyrosine Kinase Inhibitor Resistance in Epidermal Growth Factor Receptor-Mutant Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2019, 37, 876–884. [Google Scholar] [CrossRef]
- Catenacci, D.V.; Ang, A.; Liao, W.L.; Shen, J.; O’Day, E.; Loberg, R.D.; Cecchi, F.; Hembrough, T.; Ruzzo, A.; Graziano, F. MET tyrosine kinase receptor expression and amplification as prognostic biomarkers of survival in gastroesophageal adenocarcinoma. Cancer 2017, 123, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Go, H.; Jeon, Y.K.; Park, H.J.; Sung, S.W.; Seo, J.W.; Chung, D.H. High MET gene copy number leads to shorter survival in patients with non-small cell lung cancer. J. Thorac. Oncol. 2010, 5, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.H.; Yeung, S.F.; Chan, A.W.H.; Chung, L.Y.; Chau, S.L.; Lung, R.W.M.; Tong, C.Y.; Chow, C.; Tin, E.K.Y.; Yu, Y.H.; et al. MET Amplification and Exon 14 Splice Site Mutation Define Unique Molecular Subgroups of Non–Small Cell Lung Carcinoma with Poor Prognosis. Clin. Cancer Res. 2016, 22, 3048–3056. [Google Scholar] [CrossRef] [Green Version]
- Spigel, D.R.; Edelman, M.J.; O’Byrne, K.; Paz-Ares, L.; Mocci, S.; Phan, S.; Shames, D.S.; Smith, D.; Yu, W.; Paton, V.E.; et al. Results From the Phase III Randomized Trial of Onartuzumab Plus Erlotinib Versus Erlotinib in Previously Treated Stage IIIB or IV Non-Small-Cell Lung Cancer: METLung. J. Clin. Oncol. 2017, 35, 412–420. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.L.; Zhang, L.; Kim, D.W.; Liu, X.; Lee, D.H.; Yang, J.C.; Ahn, M.J.; Vansteenkiste, J.F.; Su, W.C.; Felip, E.; et al. Phase Ib/II Study of Capmatinib (INC280) Plus Gefitinib After Failure of Epidermal Growth Factor Receptor (EGFR) Inhibitor Therapy in Patients With EGFR-Mutated, MET Factor-Dysregulated Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 3101–3109. [Google Scholar] [CrossRef]
- Onozato, R.; Kosaka, T.; Kuwano, H.; Sekido, Y.; Yatabe, Y.; Mitsudomi, T. Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers. J. Thorac. Oncol. 2009, 4, 5–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuda, K.; Sasaki, H.; Yukiue, H.; Yano, M.; Fujii, Y. Met gene copy number predicts the prognosis for completely resected non-small cell lung cancer. Cancer Sci. 2008, 99, 2280–2285. [Google Scholar] [CrossRef] [PubMed]
- Cappuzzo, F.; Marchetti, A.; Skokan, M.; Rossi, E.; Gajapathy, S.; Felicioni, L.; Grammastro, M.d.; Sciarrotta, M.G.; Buttitta, F.; Incarbone, M.; et al. Increased MET Gene Copy Number Negatively Affects Survival of Surgically Resected Non–Small-Cell Lung Cancer Patients. J. Clin. Oncol. 2009, 27, 1667–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noonan, S.A.; Berry, L.; Lu, X.; Gao, D.; Barón, A.E.; Chesnut, P.; Sheren, J.; Aisner, D.L.; Merrick, D.; Doebele, R.C.; et al. Identifying the Appropriate FISH Criteria for Defining MET Copy Number–Driven Lung Adenocarcinoma through Oncogene Overlap Analysis. J. Thorac. Oncol. 2016, 11, 1293–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MET − (N = 156) | MET + (N = 30) | ||
|---|---|---|---|
| N (%) | N (%) | p Value | |
| Median age, years (range) | 61 (28–84) | 59 (28–76) | 0.362 |
| Sex | |||
| Male | 57 (36.5%) | 13 (43.3%) | |
| Female | 99 (63.5%) | 17 (56.7%) | 0.482 |
| Smoking status | |||
| Current smoker | 8 (5.1%) | 1 (3.3%) | |
| Ex-smoker | 37 (23.7%) | 15 (50.0%) | |
| Never smoker | 111 (71.2%) | 14 (46.7%) | 0.013 |
| Median pack-year of smoking (interquartile range) | 0.0 (0.0–9.5) | 2.5 (0–21.0) | 0.785 |
| Previous TKI generation | |||
| First/second generation | 109 (69.9%) | 15 (50%) | |
| Third generation | 47 (30.1%) | 15 (50%) | 0.034 |
| Previous TKI line | |||
| First line | 93 (59.6%) | 11 (36.7%) | |
| Second line | 58 (37.2%) | 14 (46.7%) | |
| Third line | 5 (3.2%) | 5 (16.7%) | 0.003 |
| Founder EGFR mutation | |||
| Exon 19 deletion | 88 (56.4%) | 19 (63.3%) | |
| L858R | 59 (37.8%) | 10 (33.3%) | |
| Other mutations ** | 9 (5.8%) | 1 (3.3%) | 0.732 |
| Liver metastases | |||
| No metastases | 125 (80.1%) | 18 (60.0%) | |
| Baseline metastases without progression | 1 (0.6%) | 3 (10.0%) | |
| Baseline metastases with progression | 4 (2.6%) | 7 (23.3%) | |
| Progression with new lesion | 26 (16.7%) | 2 (6.7%) | 0.185 * |
| Brain metastases | |||
| No metastases | 44 (28.2%) | 16 (53.3%) | |
| Baseline metastases without progression | 7 (4.5%) | 7 (23.3%) | |
| Baseline metastases with progression | 59 (37.8%) | 5 (16.7%) | |
| Progression with new lesion | 37 (23.7%) | 2 (6.7%) | <0.001 * |
| Not evaluated | 9 (5.8%) | 0 (0.0%) |
| Category | Univariate | Multivariate | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | p Value | HR | 95% CI | p Value | |
| Age | 0.983 | 0.948–1.020 | 0.360 | - | - | - |
| Sex (male versus female) | 0.753 | 0.341–1.663 | 0.483 | - | - | - |
| Smoking status (never versus ex- or current smoker) | 2.819 | 1.271–6.252 | 0.011 | 3.346 | 1.326–8.442 | 0.011 |
| TKI generation (first/second versus third) | 2.319 | 1.049–5.126 | 0.038 | 2.618 | 1.044–6.565 | 0.040 |
| Baseline EGFR mutation site | ||||||
| Exon 19 deletion | 1 | |||||
| L858R | 0.785 | 0.341–1.807 | 0.569 | - | - | - |
| Other mutations * | 0.515 | 0.061–4.307 | 0.540 | - | - | - |
| PFS of most recent TKI | 0.930 | 0.875–0.988 | 0.019 | 0.898 | 0.835–0.965 | 0.004 |
| Liver metastases (no PD versus PD) | 1.800 | 0.749–4.325 | 0.189 | - | - | - |
| Brain metastases (no PD versus PD) | 0.162 | 0.065–0.402 | <0.001 | 0.139 | 0.052–0.373 | <0.001 |
| Treatment Response | PD Site | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Tyrosine Kinase Inhibitor (TKI) | n | Treatment Line | Best Response ORR DCR | Median PFS(95% CI) | Median OS (95% CI) | Primary Lung | Intrathoracic | Liver | Bone | Extrathoracic | Brain |
| First-generation TKI | 8 | 1L 6 Pts (75.0%) 2L 1 Pts (12.5%) 3L 1 Pts (12.5%) | PR 6 Pts (75.0%) SD 1 Pts (12.5%) PD 1 Pts (12.5%) ORR 75.0%/DCR 87.5% | 5.1 months (4.0–6.1) | 81.7 months (NR) | 7 (87.5%) | 7 (87.5%) | 4 (50.0%) | 1 (12.5%) | 1 (12.5%) | 0 |
| Gefitinib | 5 | ORR 60.0%/DCR 80.0% | 5.1 months (3.7–6.4) | NR | 4 (80.0%) | 5 (100%) | 3 (60.0%) | 0 | 1 (20%) | 1 (20%) | |
| Erlotinib | 3 | ORR 100%/DCR 100% | 5.2 months (3.5–6.9) | 18.7 months (NR) | 3 (100%) | 2 (66.7%) | 1 (33.3%) | 1 (33.3%) | 0 | 0 | |
| Second-generation TKI Afatinib | 7 | 1L 5 Pts (71.4%) 2L 2 Pts (28.6%) | PR 5 Pts (71.4%) SD 2 Pts (28.6%) ORR 71.4%/DCR 100% | 7.7 months (6.0–9.4) | 26.5 months (0–71.3) | 4 (57.1%) | 3 (42.9%) | 2 (28.6%) | 0 | 1 (14.3%) | 2 (28.6%) |
| Third-generation TKI | 15 | 2L 12 Pts (80.0%) 3L 3 Pts (20.0%) | PR 8 Pts (53.3%) SD 4 Pts (26.7%) PD 3 Pts (20.0%) ORR 53.3%/DCR 80.0% | 7.2 months (3.5–10.8) | 38.8 months (31.0–46.6) | 12 (80%) | 7 (46.7%) | 3 (20%) | 3 (20%) | 6 (40%) | 4 (26.7%) |
| Osimertinib | 12 | ORR 58.3%/DCR 83.3% | 7.2 months (4.8–9.6) | 34.7 months (27.6–42.0) | 9 (13.3%) | 6 (50%) | 2 (16.7%) | 3 (25%) | 5 (41.7%) | 3 (25%) | |
| Olmutinib | 1 | ORR 100%/DCR 100% | 18.0 months | 74.0 months | 1 (100%) | 1 (100%) | 1 (100%) | 0 | 0 | 1 (100%) | |
| Lazertinib | 2 | ORR 0%/DCR 50.0% | 1.7 months (NR) | 17.0 months (NR) | 2 (100%) | 0 | 0 | 0 | 1 (50%) | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahn, B.-C.; Lee, J.H.; Kim, M.H.; Pyo, K.-H.; Lee, C.-k.; Lim, S.M.; Kim, H.R.; Cho, B.C.; Hong, M.H. Distinct Characteristics and Clinical Outcomes to Predict the Emergence of MET Amplification in Patients with Non-Small Cell Lung Cancer Who Developed Resistance after Treatment with Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors. Cancers 2021, 13, 3096. https://doi.org/10.3390/cancers13123096
Ahn B-C, Lee JH, Kim MH, Pyo K-H, Lee C-k, Lim SM, Kim HR, Cho BC, Hong MH. Distinct Characteristics and Clinical Outcomes to Predict the Emergence of MET Amplification in Patients with Non-Small Cell Lung Cancer Who Developed Resistance after Treatment with Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors. Cancers. 2021; 13(12):3096. https://doi.org/10.3390/cancers13123096
Chicago/Turabian StyleAhn, Beung-Chul, Ji Hyun Lee, Min Hwan Kim, Kyoung-Ho Pyo, Choong-kun Lee, Sun Min Lim, Hye Ryun Kim, Byoung Chul Cho, and Min Hee Hong. 2021. "Distinct Characteristics and Clinical Outcomes to Predict the Emergence of MET Amplification in Patients with Non-Small Cell Lung Cancer Who Developed Resistance after Treatment with Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors" Cancers 13, no. 12: 3096. https://doi.org/10.3390/cancers13123096
APA StyleAhn, B.-C., Lee, J. H., Kim, M. H., Pyo, K.-H., Lee, C.-k., Lim, S. M., Kim, H. R., Cho, B. C., & Hong, M. H. (2021). Distinct Characteristics and Clinical Outcomes to Predict the Emergence of MET Amplification in Patients with Non-Small Cell Lung Cancer Who Developed Resistance after Treatment with Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors. Cancers, 13(12), 3096. https://doi.org/10.3390/cancers13123096