Immunoglobulin M Paraproteinaemias

 , , and
, , and
Abstract
1. Introduction
2. Summary of WHO and IMWG (International Myeloma Working Group) Defined Disease Categories Associated with IgM Paraproteins
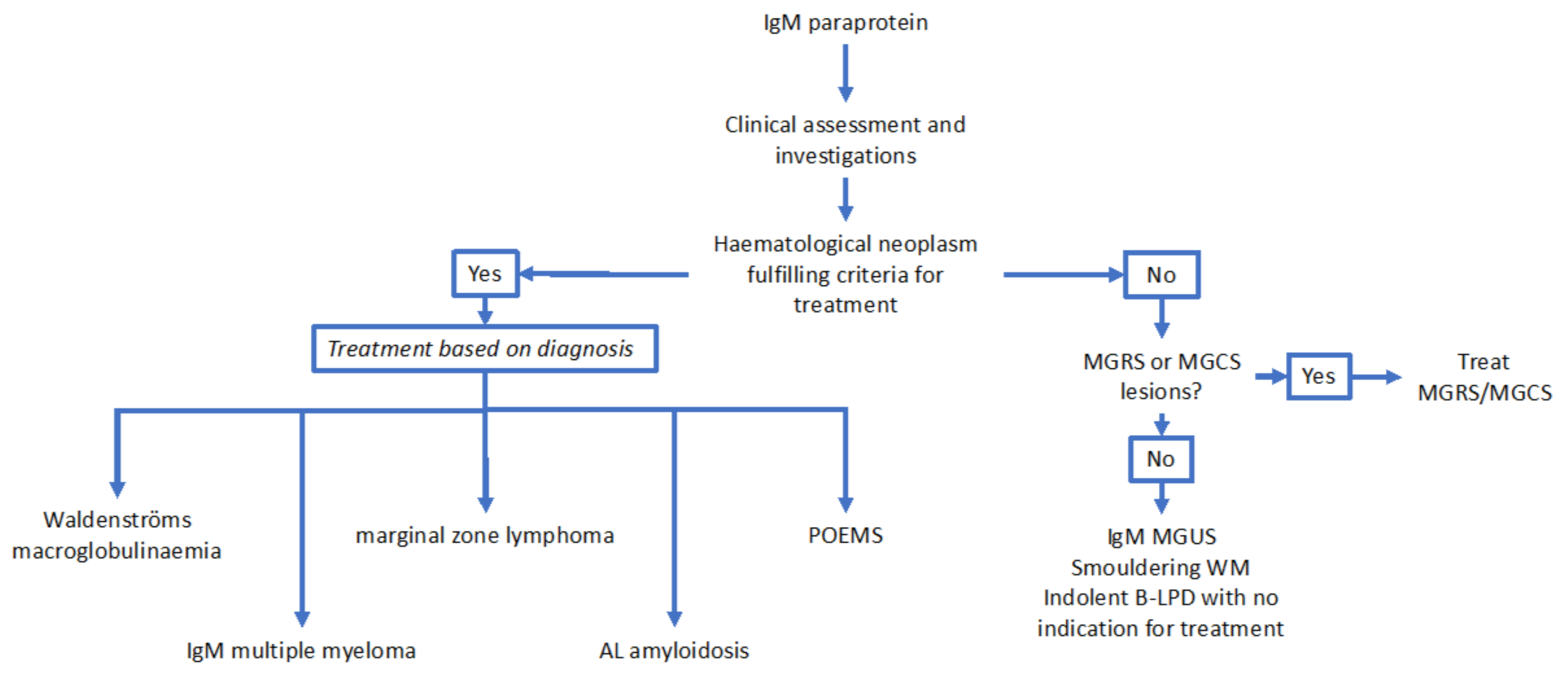
2.1. Immunoglobulin M Monoclonal Gammopathy of Uncertain Significance
2.2. Waldenströms Macroglobulinaemia
2.3. Immunoglobulin M Multiple Myeloma
2.4. Marginal Zone Lymphoma
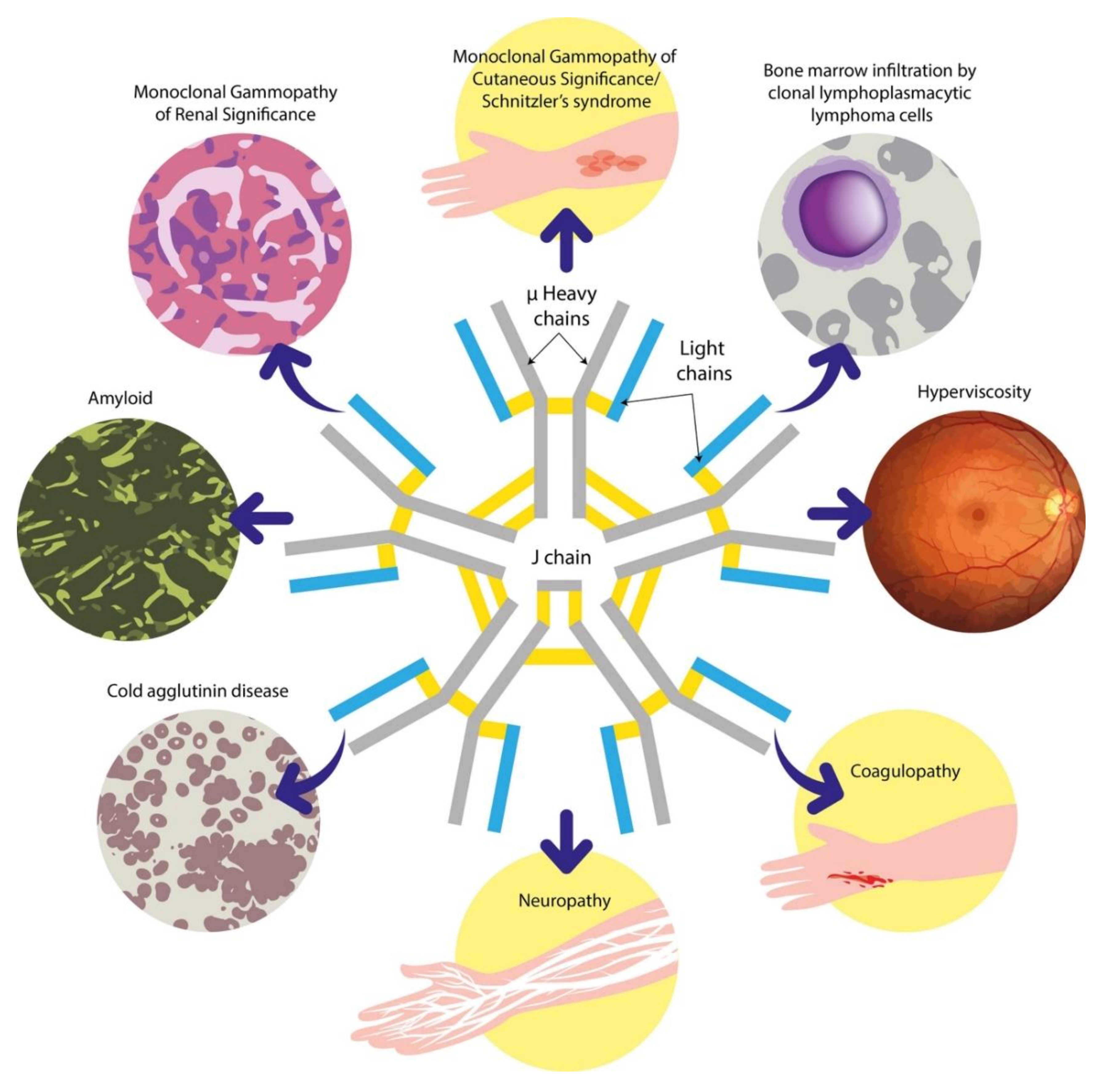
2.5. Other B-cell and Plasma Cell Dyscrasias Associated with IgM Paraproteins
3. Approach to the Clinical Evaluation and Initial Investigation of Patients Presenting with IgM Paraproteins
4. Overview of Laboratory Techniques Used to Diagnose IgM Paraproteinaemias
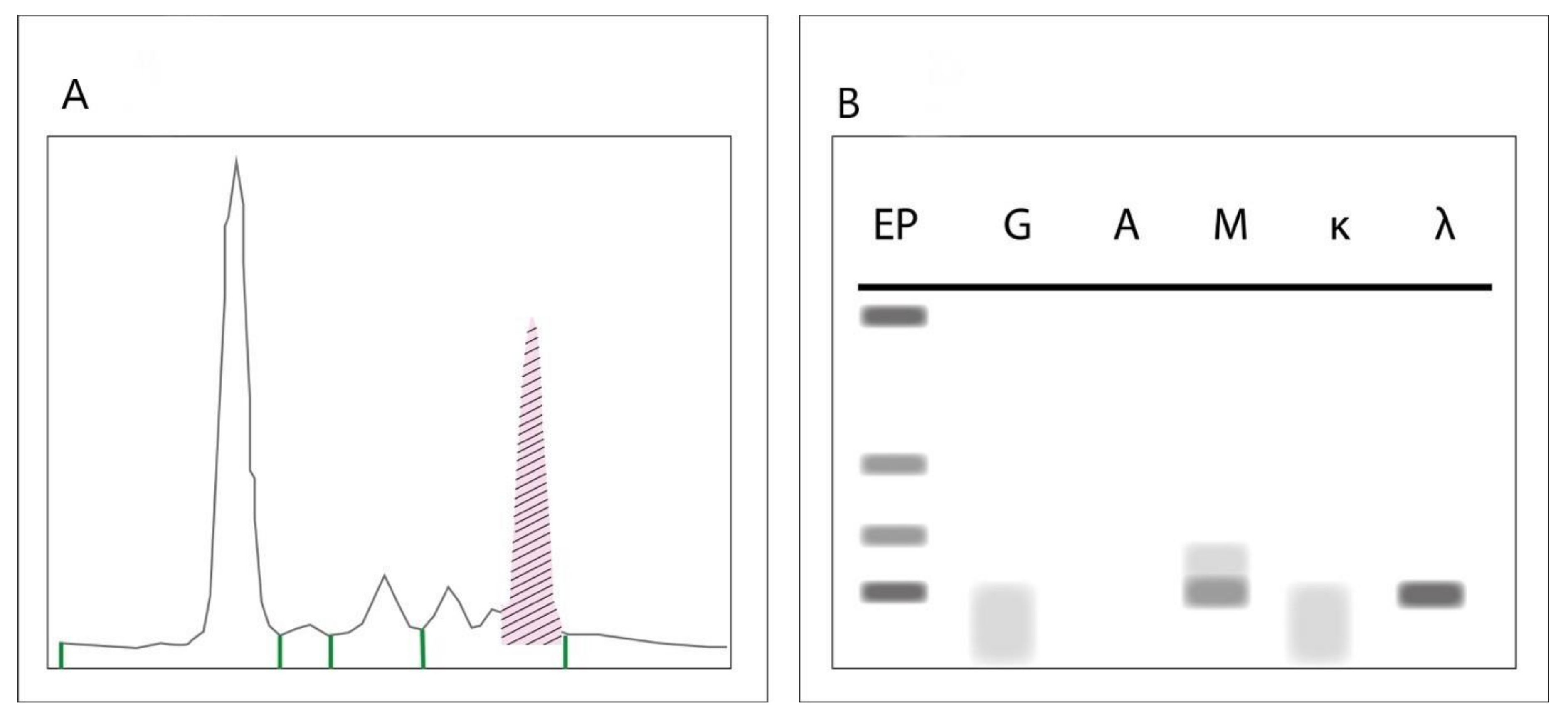
4.1. Serum Protein Electrophoresis, Immunofixation Electrophoresis and Serum Free Light Chain Assays
4.2. “M Protein” Quantification
4.3. Bone Marrow Studies
4.4. Flow Cytometry
4.5. Karyotyping and Fluorescent In Situ Hybridisation
4.6. AL Amyloidosis-Specific Investigations
5. Utility of Imaging in the Evaluation of Patients with IgM Paraproteins
6. Challenges Associated with IgM Paraproteinaemias
6.1. The Distinction Between LPL/WM and MZL
6.2. Clinical Utility of MYD88 L265P Mutation Screening
6.3. Neuropathies Associated with IgM Paraproteins
6.4. Evaluation of the Patient with IgM Paraproteinaemia and Renal Impairment
6.5. Autoimmune Manifestations of IgM Paraproteinaemias
6.6. Evaluation of the Patient with IgM Paraproteinaemia and Cutaneous Lesions
7. Future Directions
7.1. Genomic Categorisation of WM
7.2. Novel Techniques to Identify and Quantify Paraproteins
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tate, J.R. The Paraprotein—an Enduring Biomarker. Clin. Biochem. Rev. 2019, 40, 5–22. [Google Scholar] [PubMed]
- Kyle, R.A.; Durie, B.G.M.; Rajkumar, S.V.; Landgren, O.; Blade, J.; Merlini, G.; Kröger, N.; Einsele, H.; Vesole, D.H.; On Behalf of the International Myeloma Working Group; et al. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia 2010, 24, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.; Fonseca, R.; Rajkumar, S.V. Waldenström’s Macroglobulinemia. Oncology 2000, 5, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Owen, R.G.; Parapia, L.A.; Higginson, J.; Misbah, S.A.; Child, J.A.; Morgan, G.; Jack, A.S. Clinicopathological Correlates of IgM Paraproteinemias. Clin. Lymphoma 2000, 1, 39–43. [Google Scholar] [CrossRef]
- Katzmann, J.A.; Kyle, R.A.; Benson, J.; Larson, D.R.; Snyder, M.R.; Lust, J.A.; Rajkumar, S.V.; Dispenzieri, A. Screening Panels for Detection of Monoclonal Gammopathies. Clin. Chem. 2009, 55, 1517–1522. [Google Scholar] [CrossRef]
- Dispenzieri, A.; Kyle, R.; Merlini, G.; San-Miguel, J.F.; Ludwig, H.; Hájek, R.; Palumbo, A.; Jagannath, S.; Blade, J.; on Behalf of the International Myeloma Working Group; et al. International Myeloma Working Group guidelines for serum-free light chain analysis in multiple myeloma and related disorders. Leukemia 2008, 23, 215–224. [Google Scholar] [CrossRef]
- Bird, J.; Behrens, J.; Westin, J.; Turesson, I.; Drayson, M.; Beetham, R.; D’Sa, S.; Soutar, R.; Waage, A.; Gulbrandsen, N.; et al. UK Myeloma Forum (UKMF) and Nordic Myeloma Study Group (NMSG): Guidelines for the investigation of newly detected M-proteins and the management of monoclonal gammopathy of undetermined significance (MGUS). Br. J. Haematol. 2009, 147, 22–42. [Google Scholar] [CrossRef]
- Kyle, R.A.; Anderson, K.C. A Tribute to Jan Gosta Waldenström. Blood 1997, 89, 4245–4247. [Google Scholar] [CrossRef]
- Go, R.S.; Rajkumar, S.V. How I manage monoclonal gammopathy of undetermined significance. Blood 2018, 131, 163–173. [Google Scholar] [CrossRef]
- Kyle, R.A.; Therneau, T.M.; Rajkumar, S.V.; Larson, D.R.; Plevak, M.F.; Offord, J.R.; Dispenzieri, A.; A Katzmann, J.; Melton, L.J. Prevalence of Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 2006, 354, 1362–1369. [Google Scholar] [CrossRef]
- Roberts-Thomson, P.; Nikoloutsopoulos, T.; Smith, A.J. IgM paraproteinaemia: Disease associations and laboratory features. Pathology 2002, 34, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M. Acute hyperviscosity: Syndromes and management. Blood 2018, 132, 1379–1385. [Google Scholar] [CrossRef] [PubMed]
- Rison, R.; Beydoun, S.R. Paraproteinemic neuropathy: A practical review. BMC Neurol. 2016, 16, 13. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; A Dimopoulos, M.; Palumbo, A.; Bladé, J.; Merlini, G.; Mateos, M.-V.; Rajkumar, S.V.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Landgren, O.; Weiss, B.M. Patterns of monoclonal gammopathy of undetermined significance and multiple myeloma in various ethnic/racial groups: Support for genetic factors in pathogenesis. Leukemia 2009, 23, 1691–1697. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Therneau, T.M.; Rajkumar, S.V.; Remstein, E.D.; Offord, J.R.; Larson, D.R.; Plevak, M.F.; Melton, L.J. Long-term follow-up of IgM monoclonal gammopathy of undetermined significance. Blood 2003, 102, 3759–3764. [Google Scholar] [CrossRef] [PubMed]
- Varettoni, M.; Zibellini, S.; Arcaini, L.; Boveri, E.; Rattotti, S.; Pascutto, C.; Mangiacavalli, S.; Gotti, M.; Pochintesta, L.; Paulli, M.; et al. MYD88 (L265P) mutation is an independent risk factor for progression in patients with IgM monoclonal gammopathy of undetermined significance. Blood 2013, 122, 2284–2285. [Google Scholar] [CrossRef]
- Sabattini, E.; Bacci, F.; Sagramoso, C.; Pileri, S.A. WHO classification of tumours of haematopoietic and lymphoid tissues in 2008: An overview. Pathology 2010, 102, 83–87. [Google Scholar]
- Gertz, M. Waldenström macroglobulinemia treatment algorithm 2018. Blood Cancer J. 2018, 8, 40. [Google Scholar] [CrossRef]
- Schop, R.F.J.; Kuehl, W.; Van Wier, S.A.; Ahmann, G.J.; Price-Troska, T.; Bailey, R.J.; Jalal, S.M.; Qi, Y.; Kyle, R.A.; Greipp, P.R.; et al. Waldenström macroglobulinemia neoplastic cells lack immunoglobulin heavy chain locus translocations but have frequent 6q deletions. Blood 2002, 100, 2996–3001. [Google Scholar] [CrossRef]
- Treon, S.P.; Xu, L.; Yang, G.; Zhou, Y.; Liu, X.; Cao, Y.; Sheehy, P.; Manning, R.J.; Patterson, C.J.; Tripsas, C.; et al. MYD88 L265P Somatic Mutation in Waldenström’s Macroglobulinemia. N. Engl. J. Med. 2012, 367, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Abeykoon, J.P.; Zanwar, S.; Ansell, S.M.; Winters, J.; Gertz, M.; King, R.L.; Murray, D.; Habermann, T.; Dingli, D.; Muchtar, E.; et al. Predictors of symptomatic hyperviscosity in Waldenström macroglobulinemia. Am. J. Hematol. 2018, 93, 1384–1393. [Google Scholar] [CrossRef] [PubMed]
- Atrash, S.; Zhang, Q.; Papanikolaou, X.; Stein, C.; Abdallah, A.-O.; Barlogie, B. Clinical Presentation and Gene Expression Profiling of Immunoglobulin M Multiple Myeloma Compared with Other Myeloma Subtypes and Waldenström Macroglobulinemia. J. Glob. Oncol. 2018, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hunter, Z.R.; Manning, R.J.; Hanzis, C.; Ciccarelli, B.T.; Ioakimidis, L.; Patterson, C.J.; Lewicki, M.C.; Tseng, H.; Gong, P.; Liu, X.; et al. IgA and IgG hypogammaglobulinemia in Waldenström’s macroglobulinemia. Haematology 2009, 95, 470–475. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Anagnostopoulos, A.; Kyrtsonis, M.-C.; Zervas, K.; Tsatalas, C.; Kokkinis, G.; Repoussis, P.; Symeonidis, A.; Delimpasi, S.; Katodritou, E.; et al. Primary Treatment of Waldenström Macroglobulinemia With Dexamethasone, Rituximab, and Cyclophosphamide. J. Clin. Oncol. 2007, 25, 3344–3349. [Google Scholar] [CrossRef] [PubMed]
- Rummel, M.; Niederle, N.; Maschmeyer, G.; Banat, G.A.; Von Grünhagen, U.; Losem, C.; Kofahl-Krause, D.; Heil, G.; Welslau, M.; Balser, C.; et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: An open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 2013, 381, 1203–1210. [Google Scholar] [CrossRef]
- Treon, S.P.; Ioakimidis, L.; Soumerai, J.D.; Patterson, C.J.; Sheehy, P.; Nelson, M.; Willen, M.; Matous, J.; Mattern, J.; Diener, J.G.; et al. Primary Therapy of Waldenström Macroglobulinemia with Bortezomib, Dexamethasone, and Rituximab: WMCTG Clinical Trial 05-180. J. Clin. Oncol. 2009, 27, 3830–3835. [Google Scholar] [CrossRef]
- Leblond, V.; Kastritis, E.; Advani, R.; Ansell, S.M.; Buske, C.; Castillo, J.J.; García-Sanz, R.; Gertz, M.; Kimby, E.; Kyriakou, C.; et al. Treatment recommendations from the Eighth International Workshop on Waldenström’s Macroglobulinemia. Blood 2016, 128, 1321–1328. [Google Scholar] [CrossRef]
- Treon, S.P.; Tripsas, C.K.; Meid, K.; Warren, D.; Varma, G.; Green, R.; Argyropoulos, K.V.; Yang, G.; Cao, Y.; Xu, L.; et al. Ibrutinib in Previously Treated Waldenström’s Macroglobulinemia. N. Engl. J. Med. 2015, 372, 1430–1440. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Tedeschi, A.; Trotman, J.; García-Sanz, R.; Macdonald, D.; Leblond, V.; Mahé, B.; Herbaux, C.; Tam, C.; Orsucci, L.; et al. Phase 3 Trial of Ibrutinib plus Rituximab in Waldenström’s Macroglobulinemia. N. Engl. J. Med. 2018, 378, 2399–2410. [Google Scholar] [CrossRef]
- Kyle, R.A.; A Gertz, M.; Witzig, T.E.; Lust, J.A.; Lacy, M.Q.; Dispenzieri, A.; Fonseca, R.; Rajkumar, S.V.; Offord, J.R.; Larson, D.R.; et al. Review of 1027 Patients With Newly Diagnosed Multiple Myeloma. Mayo Clin. Proc. 2003, 78, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.R.; Rajkumar, S.V.; Dispenzieri, A.; Morice, W.; Aspitia, A.M.; Ansell, S.; Kyle, R.; Mikhael, J. IgM multiple myeloma: Disease definition, prognosis, and differentiation from Waldenstrom’s macroglobulinemia. Am. J. Hematol. 2010, 85, 853–855. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kaufman, J.L.; Gasparetto, C.; Mikhael, J.; Vij, R.; Pegourie, B.; Benboubker, L.; Facon, T.; Amiot, M.; Moreau, P.; et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood 2017, 130, 2401–2409. [Google Scholar] [CrossRef] [PubMed]
- Zucca, E.; Bertoni, F. The spectrum of MALT lymphoma at different sites: Biological and therapeutic relevance. Blood 2016, 127, 2082–2092. [Google Scholar] [CrossRef]
- Cerutti, A.; Cols, M.; Puga, I. Marginal zone B cells: Virtues of innate-like antibody-producing lymphocytes. Nat. Rev. Immunol. 2013, 13, 118–132. [Google Scholar] [CrossRef]
- Lin, P.; Hao, S.; Handy, B.C.; Bueso-Ramos, C.E.; Medeiros, L.J. Lymphoid Neoplasms Associated With IgM Paraprotein A Study of 382 Patients. Am. J. Clin. Pathol. 2005, 123, 200–205. [Google Scholar] [CrossRef]
- Bassarova, A.; Trøen, G.; Spetalen, S.; Micci, F.; Tierens, A.; Delabie, J. Lymphoplasmacytic Lymphoma and Marginal Zone Lymphoma in the Bone Marrow. Am. J. Clin. Pathol. 2015, 143, 797–806. [Google Scholar] [CrossRef]
- Pangalis, G.A.; Kyrtsonis, M.-C.; Kontopidou, F.N.; Siakantaris, M.P.; Dimopoulou, M.N.; Vassilakopoulos, T.P.; Tzenou, T.; Kokoris, S.; Dimitriadou, E.; Kalpadakis, C.; et al. Differential Diagnosis of Waldenström’s Macroglobulinemia and Other B-Cell Disorders. Clin. Lymphoma 2005, 5, 235–240. [Google Scholar] [CrossRef]
- Sinclair, D.; Dagg, J.H.; Mowat, A.M.; Parrott, D.M.; I Stott, D. Serum paraproteins in chronic lymphocytic leukaemia. J. Clin. Pathol. 1984, 37, 463–466. [Google Scholar] [CrossRef]
- Yin, C.C.; Lin, P.; Carney, D.A.; Handy, B.C.; Rassidakis, G.Z.; Admirand, J.H.; Keating, M.J.; Medeiros, L.J. Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma Associated with IgM Paraprotein: A Clinicopathologic Study of 26 Cases. Am. J. Clin. Pathol. 2005, 123, 594–602. [Google Scholar] [CrossRef]
- Laurenti, L.; Corbingi, A.; Innocenti, I.; Tomasso, A.; Pasquale, R.; Visentin, A.; Varettoni, M.; Autore, F.; Morelli, F.; Trentin, L.; et al. Impact of Serum Immunoglobulin Subsets and Levels on Chronic Lymphocytic Leukemia Natural History: A Retrospective Multicentric Italian Experience. Blood 2019, 134, 3026. [Google Scholar] [CrossRef]
- Cox, M.C.; Di Napoli, A.; Scarpino, S.; Salerno, G.; Tatarelli, C.; Talerico, C.; Lombardi, M.; Monarca, B.; Amadori, S.; Ruco, L. Clinicopathologic Characterization of Diffuse-Large-B-Cell Lymphoma with an Associated Serum Monoclonal IgM Component. PLoS ONE 2014, 9, e93903. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, L.; Zhu, H.-Y.; Liang, J.-H.; Wu, W.; Wu, J.-Z.; Xia, Y.; Fan, L.; Li, J.-Y.; Xu, W. Prognostic significance of serum immunoglobulin paraprotein in patients with diffuse large B cell lymphoma. Br. J. Haematol. 2017, 182, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Sidana, S.; Larson, D.P.; Greipp, P.T.; He, R.; McPhail, E.D.; Dispenzieri, A.; Murray, D.L.; Dasari, S.; Ansell, S.M.; Muchtar, E.; et al. IgM AL amyloidosis: Delineating disease biology and outcomes with clinical, genomic and bone marrow morphological features. Leukemia 2019, 34, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Wechalekar, A.D.; Lachmann, H.J.; Goodman, H.J.B.; Bradwell, A.R.; Hawkins, P.N.; Gillmore, J.D. AL amyloidosis associated with IgM paraproteinemia: Clinical profile and treatment outcome. Blood 2008, 112, 4009–4016. [Google Scholar] [CrossRef]
- Cao, X.-X.; Meng, Q.; Mao, Y.-Y.; Su, W.; Zhen, J.-F.; Shen, K.-N.; Zhang, C.-L.; Huang, X.-F.; Duan, M.-H.; Zhang, W.; et al. The clinical spectrum of IgM monoclonal gammopathy: A single center retrospective study of 377 patients. Leuk. Res. 2016, 46, 85–88. [Google Scholar] [CrossRef]
- Tate, J.; Caldwell, G.; Daly, J.; Gillis, D.; Jenkins, M.; Jovanovich, S.; Martin, H.; Steele, R.; Wienholt, L.; Mollee, P.; et al. Recommendations for standardized reporting of protein electrophoresis in Australia and New Zealand. Ann. Clin. Biochem. Int. J. Lab. Med. 2012, 49, 242–256. [Google Scholar] [CrossRef]
- Guinan, J.E.C.; Kenny, D.F.; Gatenby, P.A. Detection and typing of paraproteins: Comparison of different methods in a routine diagnostic laboratory. Pathology 1989, 21, 35–41. [Google Scholar] [CrossRef]
- González-Calle, V.; Mateos, M.-V. Monoclonal gammopathies of unknown significance and smoldering myeloma: Assessment and management of the elderly patients. Eur. J. Intern. Med. 2018, 58, 57–63. [Google Scholar] [CrossRef]
- Durot, E.; Tomowiak, C.; Michallet, A.-S.; Dupuis, J.; Hivert, B.; Lepretre, S.; Toussaint, E.; Godet, S.; Merabet, F.; Neste, E.V.D.; et al. Transformed Waldenström macroglobulinaemia: Clinical presentation and outcome. A multi-institutional retrospective study of 77 cases from the French Innovative Leukemia Organization (FILO). Br. J. Haematol. 2017, 179, 439–448. [Google Scholar] [CrossRef]
- Merlini, G. AL amyloidosis: From molecular mechanisms to targeted therapies. Haematology 2017, 2017, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Treon, S.P. How I treat Waldenström macroglobulinemia. Blood 2015, 126, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Sokumbi, O.; Drage, L.A.; Peters, M.S. Clinical and histopathologic review of Schnitzler syndrome: The Mayo Clinic experience (1972–2011). J. Am. Acad. Dermatol. 2012, 67, 1289–1295. [Google Scholar] [CrossRef]
- Castillo, J.J.; Treon, S.P. How we manage Bing–Neel syndrome. Br. J. Haematol. 2019, 187, 277–285. [Google Scholar] [CrossRef]
- Jenkins, M.A. Serum and Urine Electrophoresis for Detection and Identification of Monoclonal Proteins. Clin. Biochem. Rev. 2009, 30, 119–122. [Google Scholar] [PubMed]
- Dasgupta, A. Chapter 22—Protein Electrophoresis and Immunofixation. In Clinical Chemistry, Immunology and Laboratory Quality Control; Dasgupta, A., Wahed, A., Eds.; Elsevier: San Diego, CA, USA, 2014; pp. 391–406. [Google Scholar]
- Van Dongen, J.J.; Lhermitte, L.; Böttcher, S.; Almeida, J.; Van Der Velden, V.H.J.; Flores-Montero, J.; Rawstron, A.C.; Asnafi, V.; Lécrevisse, Q.; Lucio, P.; et al. EuroFlow antibody panels for standardized n-dimensional flow cytometric immunophenotyping of normal, reactive and malignant leukocytes. Leukemia 2012, 26, 1908–1975. [Google Scholar] [CrossRef]
- Paiva, B.; Montes, M.C.; García-Sanz, R.; Ocio, E.M.; Heras, N.D.L.; Escalante, F.; Cuello, R.; De Coca, A.G.; Galende, J.; Hernández, J.; et al. Multiparameter flow cytometry for the identification of the Waldenström’s clone in IgM-MGUS and Waldenström’s Macroglobulinemia: New criteria for differential diagnosis and risk stratification. Leukemia 2013, 28, 166–173. [Google Scholar] [CrossRef]
- Seegmiller, A.C.; Hsi, E.D.; Craig, F.E. The current role of clinical flow cytometry in the evaluation of mature B-cell neoplasms. Cytom. Part. B Clin. Cytom. 2018, 96, 20–29. [Google Scholar] [CrossRef]
- Gertz, M.; Dispenzieri, A.; Muchtar, E. Importance of FISH genetics in light chain amyloidosis. Oncotarget 2017, 8, 81735–81736. [Google Scholar] [CrossRef]
- Schop, R.F.; Van Wier, S.A.; Xu, R.; Ghobrial, I.; Ahmann, G.J.; Greipp, P.R.; Kyle, R.A.; Dispenzieri, A.; Lacy, M.Q.; Rajkumar, S.V.; et al. 6q deletion discriminates Waldenström macroglobulinemia from IgM monoclonal gammopathy of undetermined significance. Cancer Genet. Cytogenet. 2006, 169, 150–153. [Google Scholar] [CrossRef] [PubMed]
- Grogan, M.; Dispenzieri, A.; A Gertz, M. Light-chain cardiac amyloidosis: Strategies to promote early diagnosis and cardiac response. Heart 2017, 103, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Sucker, C.; Hetzel, G.; Grabensee, B.; Stockschlaeder, M.; Scharf, R. Amyloidosis and Bleeding: Pathophysiology, Diagnosis, and Therapy. Am. J. Kidney Dis. 2006, 47, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Hillengass, J.; Usmani, S.; Rajkumar, S.V.; Durie, B.G.M.; Mateos, M.-V.; Lonial, S.; Joao, C.; Anderson, K.C.; García-Sanz, R.; Riva, E.; et al. International myeloma working group consensus recommendations on imaging in monoclonal plasma cell disorders. Lancet Oncol. 2019, 20, e302–e312. [Google Scholar] [CrossRef]
- Cheson, B.D.; Fisher, R.I.; Barrington, S.F.; Cavalli, F.; Schwartz, L.H.; Zucca, E.; Lister, T.A.; Alliance, Australasian Leukaemia and Lymphoma Group; Eastern Cooperative Oncology Group. Recommendations for Initial Evaluation, Staging, and Response Assessment of Hodgkin and Non-Hodgkin Lymphoma: The Lugano Classification. J. Clin. Oncol. 2014, 32, 3059–3067. [Google Scholar] [CrossRef]
- Sachchithanantham, S.; Wechalekar, A.D. Imaging in systemic amyloidosis. Br. Med. Bull. 2013, 107, 41–56. [Google Scholar] [CrossRef]
- Lin, P.; Molina, T.J.; Cook, J.R.; Swerdlow, S.H. Lymphoplasmacytic Lymphoma and Other Non–Marginal Zone Lymphomas with Plasmacytic Differentiation. Am. J. Clin. Pathol. 2011, 136, 195–210. [Google Scholar] [CrossRef]
- Thieblemont, C.; Cascione, L.; Conconi, A.; Kiesewetter, B.; Raderer, M.; Gaidano, G.; Martelli, M.; Laszlo, D.; Coiffier, B.; Guillermo, A.L.; et al. A MALT lymphoma prognostic index. Blood 2017, 130, 1409–1417. [Google Scholar] [CrossRef]
- Morel, P.; Duhamel, A.; Gobbi, P.; Dimopoulos, M.A.; Dhodapkar, M.V.; McCoy, J.; Crowley, J.; Ocio, E.M.; García-Sanz, R.; Treon, S.P.; et al. International prognostic scoring system for Waldenström macroglobulinemia. Blood 2009, 113, 4163–4170. [Google Scholar] [CrossRef]
- Noy, A.; De Vos, S.; Thieblemont, C.; Martin, P.; Flowers, C.R.; Morschhauser, F.; Collins, G.; Ma, S.; Coleman, M.; Peles, S.; et al. Targeting Bruton tyrosine kinase with ibrutinib in relapsed/refractory marginal zone lymphoma. Blood 2017, 129, 2224–2232. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Kuzu, I.; Dogan, A.; Dirnhofer, S.; Chan, J.K.C.; Sander, B.; Ott, G.; Xerri, L.; Quintanilla-Martinez, L.; Campo, E. The many faces of small B cell lymphomas with plasmacytic differentiation and the contribution of MYD88 testing. Virchows Arch. 2015, 468, 259–275. [Google Scholar] [CrossRef] [PubMed]
- Deguine, J.; Barton, G.M. MyD88: A central player in innate immune signaling. F1000Prime Rep. 2014, 6, 97. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, W.; Deng, Q.; Li, L.; Hsi, E.D.; Young, K.H.; Zhang, M.; Li, Y. MYD88L265P Mutation in Lymphoid Malignancies. Cancer Res. 2018, 78, 2457–2462. [Google Scholar] [CrossRef] [PubMed]
- Ondrejka, S.L.; Lin, J.J.; Warden, D.W.; Durkin, L.; Cook, J.R.; Hsi, E.D. MYD88 L265P Somatic Mutation. Am. J. Clin. Pathol. 2013, 140, 387–394. [Google Scholar] [CrossRef]
- Landgren, O.; Staudt, L. MYD88 L265P somatic mutation in IgM MGUS. N. Engl. J. Med. 2012, 367, 2255–2257. [Google Scholar] [CrossRef]
- Fermand, J.-P.; Bridoux, F.; Dispenzieri, A.; Jaccard, A.; Kyle, R.A.; Leung, N.; Merlini, G. Monoclonal gammopathy of clinical significance: A novel concept with therapeutic implications. Blood 2018, 132, 1478–1485. [Google Scholar] [CrossRef]
- Nobile-Orazio, E.; Marmiroli, P.; Baldini, L.; Spagnol, G.; Barbieri, S.; Moggio, M.; Polli, N.; Polli, E.; Scarlato, G. Peripheral neuropathy in macroglobulinemia: Incidence and antigen-specificity of M proteins. Neurology 1987, 37, 1506. [Google Scholar] [CrossRef]
- Ramchandren, S.; Lewis, R.A. An Update on Monoclonal Gammopathy and Neuropathy. Curr. Neurol. Neurosci. Rep. 2011, 12, 102–110. [Google Scholar] [CrossRef]
- Stork, A.C.; Jacobs, B.C.; Tio-Gillen, A.P.; Eurelings, M.; Jansen, M.; Berg, L.H.V.D.; Notermans, N.C.; Van Der Pol, W.-L. Prevalence, specificity and functionality of anti-ganglioside antibodies in neuropathy associated with IgM monoclonal gammopathy. J. Neuroimmunol. 2014, 268, 89–94. [Google Scholar] [CrossRef]
- Vital, C.; Vital, A.; Deminiere, C.; Julien, J.; Lagueny, A.; Steck, A.J. Myelin modifications in 8 cases of peripheral neuropathy with Waldenström’s macroglobulinemia and anti-MAG activity. Ultrastruct. Pathol. 1997, 21, 509–516. [Google Scholar] [CrossRef]
- Dellagi, K.; Dupouey, P.; Brouet, J.C.; Billecocq, A.; Gomez, D.; Clauvel, J.P.; Seligmann, M. Waldenström’s macroglobulinemia and peripheral neuropathy: A clinical and immunologic study of 25 patients. Blood 1983, 62, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, H.M.; Mauermann, M.L.; Rajkumar, S.V. Monoclonal Gammopathy-Associated Peripheral Neuropathy: Diagnosis and Management. Mayo Clin. Proc. 2017, 92, 838–850. [Google Scholar] [CrossRef] [PubMed]
- Léger, J.M.; Younes-Chennoufi, A.B.; Chassande, B.; Davila, G.; Bouche, P.; Baumann, N.; Brunet, P. Human immunoglobulin treatment of multifocal motor neuropathy and polyneuropathy associated with monoclonal gammopathy. J. Neurol. Neurosurg. Psychiatry 1994, 57, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Živković, S.A. Rituximab in the treatment of peripheral neuropathy associated with monoclonal gammopathy. Expert Rev. Neurother. 2006, 6, 1267–1274. [Google Scholar] [CrossRef] [PubMed]
- Goldfarb, A.R.; Weimer, L.H.; Brannagan, T.H. Rituximab treatment of an IgM monoclonal autonomic and sensory neuropathy. Muscle Nerve 2005, 31, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Dyck, P.J.; Low, P.A.; Windebank, A.; Jaradeh, S.S.; Gosselin, S.; Bourque, P.; Smith, B.E.; Kratz, K.M.; Karnes, J.L.; Evans, B.A.; et al. Plasma Exchange in Polyneuropathy Associated with Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 1991, 325, 1482–1486. [Google Scholar] [CrossRef]
- Wilson, H.C.; Lunn, M.P.T.; Schey, S.; Hughes, R.A.C. Successful treatment of IgM paraproteinaemic neuropathy with fludarabine. J. Neurol. Neurosurg. Psychiatry 1999, 66, 575–580. [Google Scholar] [CrossRef]
- Leung, N.; Bridoux, F.; Batuman, V.; Chaidos, A.; Cockwell, P.; D’Agati, V.D.; Dispenzieri, A.; Fervenza, F.C.; Fermand, J.-P.; Gibbs, S.; et al. Publisher Correction: The evaluation of monoclonal gammopathy of renal significance: A consensus report of the International Kidney and Monoclonal Gammopathy Research Group. Nat. Rev. Nephrol. 2019, 15, 121. [Google Scholar] [CrossRef]
- Nasr, S.; Satoskar, A.; Markowitz, G.S.; Valeri, A.M.; Appel, G.B.; Stokes, M.B.; Nadasdy, T.; D’Agati, V.D. Proliferative glomerulonephritis with monoclonal IgG deposits. J. Am. Soc. Nephrol. 2009, 20, 2055–2064. [Google Scholar] [CrossRef]
- Jain, A.; Haynes, R.; Kothari, J.; Khera, A.; Soares, M.; Ramasamy, K. Pathophysiology and management of monoclonal gammopathy of renal significance. Blood Adv. 2019, 3, 2409–2423. [Google Scholar] [CrossRef]
- Swiecicki, P.L.; Hegerova, L.T.; Gertz, M.A. Cold agglutinin disease. Blood 2013, 122, 1114–1121. [Google Scholar] [CrossRef] [PubMed]
- Karunaratne, S.; Weerasinghe, S.; Govindapala, D.; Fernando, H.; Jayaratne, B. Cold autoimmune haemolytic anaemia secondary to Epstein Barr virus infection presenting with peripheral gangrene; case report. Thromb. J. 2012, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.Y.; A Yassin, M. Mycoplasma pneumoniae associated with severe autoimmune hemolytic anemia: Case report and literature review. Braz. J. Infect. Dis. 2009, 13, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Berentsen, S.; Tjonnfjord, G.E.; Brudevold, R.; Gjertsen, B.T.; Langholm, R.; Lokkevik, E.; Sorbo, J.H.; Ulvestad, E. Favourable response to therapy with the anti-CD20 monoclonal antibody rituximab in primary chronic cold agglutinin disease. Br. J. Haematol. 2001, 115, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Casals, M.; Stone, J.H.; Cid, M.C.; Bosch, X. The cryoglobulinaemias. Lancet 2012, 379, 348–360. [Google Scholar] [CrossRef]
- Muchtar, E.; Magen, H.; Gertz, M. How I treat cryoglobulinemia. Blood 2017, 129, 289–298. [Google Scholar] [CrossRef]
- Rossi, D.; De Paoli, L.; Franceschetti, S.; Capello, D.; Vendramin, C.; Lunghi, M.; Conconi, A.; Magnani, C.; Gaïdano, G. Prevalence and clinical characteristics of immune thrombocytopenic purpura in a cohort of monoclonal gammopathy of uncertain significance. Br. J. Haematol. 2007, 138, 249–252. [Google Scholar] [CrossRef]
- Mayerhofer, M.; Haushofer, A.; Kyrle, P.A.; Chott, A.; Mullner, C.; Quehenberger, P.; Worel, N.; Traby, L.; Eichinger, S. Mechanisms underlying acquired von Willebrand syndrome associated with an IgM paraprotein. Eur. J. Clin. Investig. 2009, 39, 833–836. [Google Scholar] [CrossRef]
- Lipsker, D. Monoclonal gammopathy of cutaneous significance: Review of a relevant concept. J. Eur. Acad. Dermatol. Venereol. 2016, 31, 45–52. [Google Scholar] [CrossRef]
- George, C.; Deroide, F.; Rustin, M. Pyoderma gangrenosum—A guide to diagnosis and management. Clin. Med. 2019, 19, 224–228. [Google Scholar] [CrossRef]
- Alegría-Landa, V.; Cerroni, L.; Kutzner, H.; Requena, L. Paraprotein deposits in the skin. J. Am. Acad. Dermatol. 2017, 77, 1145–1158. [Google Scholar] [CrossRef] [PubMed]
- Harel, S.; Mohr, M.; Jahn, I.; Aucouturier, F.; Galicier, L.; Asli, B.; Malphettes, M.; Szalat, R.; Brouet, J.-C.; Lipsker, D.; et al. Clinico-biological characteristics and treatment of type I monoclonal cryoglobulinaemia: A study of 64 cases. Br. J. Haematol. 2014, 168, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Camp, B.J.; Magro, C.M. Cutaneous macroglobulinosis: A case series. J. Cutan. Pathol. 2012, 39, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Lipsker, D. The Schnitzler syndrome. Orphanet J. Rare Dis. 2010, 5, 38. [Google Scholar] [CrossRef] [PubMed]
- Rowczenio, D.M.; Pathak, S.; Arostegui, J.I.; Mensa-Vilaro, A.; Omoyinmi, E.; Brogan, P.; Lipsker, D.; Scambler, T.; Owen, R.; Trojer, H.; et al. Molecular genetic investigation, clinical features, and response to treatment in 21 patients with Schnitzler syndrome. Blood 2018, 131, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Treon, S.P.; Cao, Y.; Xu, L.; Yang, G.; Liu, X.; Hunter, Z.R. Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenström macroglobulinemia. Blood 2014, 123, 2791–2796. [Google Scholar] [CrossRef] [PubMed]
- Zanwar, S.; Abeykoon, J.P.; Durot, E.; King, R.; M.S., G.E.P.B.; Kumar, S.; Gertz, M.; Quinquenel, A.; Delmer, A.; Gonsalves, W.I.; et al. Impact of MYD88 L265P mutation status on histological transformation of Waldenström Macroglobulinemia. Am. J. Hematol. 2019, 95, 274–281. [Google Scholar] [CrossRef]
- Abeykoon, J.P.; Paludo, J.; King, R.L.; Ansell, S.M.; Gertz, M.A.; LaPlant, B.R.; Halvorson, A.E.; Gonsalves, W.I.; Dingli, D.; Fang, H.; et al. MYD88 mutation status does not impact overall survival in Waldenström macroglobulinemia. Am. J. Hematol. 2017, 93, 187–194. [Google Scholar] [CrossRef]
- Treon, S.P.; Xu, L.; Hunter, Z. MYD88 mutations and response to ibrutinib in Waldenström’s macroglobulinemia. N. Engl. J. Med. 2015, 373, 584–586. [Google Scholar] [CrossRef]
- Abeykoon, J.P.; Zanwar, S.; Ansell, S.M.; Gertz, M.A.; Kumar, S.; Manske, M.; Novak, A.J.; King, R.; Greipp, P.; Go, R.; et al. Ibrutinib monotherapy outside of clinical trial setting in Waldenström macroglobulinaemia: Practice patterns, toxicities and outcomes. Br. J. Haematol. 2019, 188, 394–403. [Google Scholar] [CrossRef]
- Sklavenitis-Pistofidis, R.; Capelletti, M.; Liu, C.-J.; Reidy, M.; Zavidij, O.; Huynh, D.; Henrick, P.; Savell, A.; Reyes, K.; Rivotto, B.; et al. Bortezomib overcomes the negative impact of CXCR4 mutations on survival of Waldenstrom macroglobulinemia patients. Blood 2018, 132, 2608–2612. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.J.; Hunter, Z.R.; Yang, G.; Treon, S.P. Novel approaches to targeting MYD88 in Waldenström macroglobulinemia. Expert Rev. Hematol. 2017, 10, 739–744. [Google Scholar] [CrossRef]
- Xu, L.; Hunter, Z.R.; Yang, G.; Cao, Y.; Liu, X.; Manning, R.; Tripsas, C.; Chen, J.; Patterson, C.J.; Kluk, M.; et al. Detection of MYD88 L265P in peripheral blood of patients with Waldenström’s Macroglobulinemia and IgM monoclonal gammopathy of undetermined significance. Leukemia 2014, 28, 1698–1704. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Paiva, B.; Anderson, K.C.; Durie, B.; Landgren, O.; Moreau, P.; Munshi, N.; Lonial, S.; Bladé, J.; Mateos, M.-V.; et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016, 17, e328–e346. [Google Scholar] [CrossRef]
- Boyle, E.; Manier, S.; Hennache, B. IgMκ and IgMλ measurements for the assessment of patients with Waldenström’s macroglobulinaemia. Clin. Cancer Res. 2016, 22, 5152–5158. [Google Scholar] [CrossRef]
- Keren, D.F. Heavy/Light-Chain Analysis of Monoclonal Gammopathies. Clin. Chem. 2009, 55, 1606–1608. [Google Scholar] [CrossRef]
- Zajec, M.; Langerhorst, P.; VanDuijn, M.M.; Gloerich, J.; Russcher, H.; Van Gool, A.J.; Luider, T.M.; Joosten, I.; De Rijke, Y.B.; Jacobs, J.F.M. Mass Spectrometry for Identification, Monitoring, and Minimal Residual Disease Detection of M-Proteins. Clin. Chem. 2020, 66, 421–433. [Google Scholar] [CrossRef]
- Barnidge, D.R.; Dasari, S.; Botz, C.M.; Murray, D.H.; Snyder, M.R.; Katzmann, J.A.; Dispenzieri, A.; Murray, D.L. Using Mass Spectrometry to Monitor Monoclonal Immunoglobulins in Patients with a Monoclonal Gammopathy. J. Proteome Res. 2014, 13, 1419–1427. [Google Scholar] [CrossRef]
- Bondarenko, P.V.; Second, T.P.; Zabrouskov, V.; Makarov, A.A.; Zhang, Z. Mass measurement and top-down HPLC/MS analysis of intact monoclonal antibodies on a hybrid linear quadrupole ion trap-orbitrap mass spectrometer. J. Am. Soc. Mass Spectrom. 2009, 20, 1415–1424. [Google Scholar] [CrossRef]
- Falick, A.M.; Shackleton, C.H.L.; Green, B.N.; Witkowska, H.E. Tandem mass spectrometry in the clinical analysis of variant hemoglobins. Rapid Commun. Mass Spectrom. 1990, 4, 396–400. [Google Scholar] [CrossRef]
- Kishikawa, M.; Nakanishi, T.; Miyazaki, A.; Shimizu, A.; Nakazato, M.; Kangawa, K.; Matsuo, H. Simple detection of abnormal serum transthyretin from patients with familial amyloidotic polyneuropathy by high-performance liquid chromatography/electrospray ionization mass spectrometry using material precipitated with specific antiserum. J. Mass Spectrom. 1996, 31, 112–114. [Google Scholar] [CrossRef]
- Abeykoon, J.P.; Murray, D.L.; Murray, I.; Jevremovic, A.; Dispenzieri, A.; Arendt, B.K.; Dasari, S.; A Gertz, M.; I Gonsalves, W.; Kourelis, T.; et al. Prognostic Implications of Serum Monoclonal Protein Positivity By Mass-Fix in Bone Marrow Minimal Residual Disease Negative (MRD-) Patients with Multiple Myeloma. Blood 2019, 134, 4386. [Google Scholar] [CrossRef]
- Bradwell, A.R.; Harding, S.J.; Fourrier, N.J.; Wallis, G.L.; Drayson, M.T.; Carr-Smith, H.D.; Mead, G.P. Assessment of Monoclonal Gammopathies by Nephelometric Measurement of Individual Immunoglobulin κ/λ Ratios. Clin. Chem. 2009, 55, 1646–1655. [Google Scholar] [CrossRef] [PubMed]
- A Katzmann, J.; Willrich, M.A.V.; Kohlhagen, M.C.; A Kyle, R.; Murray, D.L.; Snyder, M.R.; Rajkumar, S.V.; Dispenzieri, A. Monitoring IgA Multiple Myeloma: Immunoglobulin Heavy/Light Chain Assays. Clin. Chem. 2015, 61, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, H.; Milosavljevic, D.; Zojer, N.; Faint, J.M.; Bradwell, A.R.; Hübl, W.; Harding, S.J. Immunoglobulin heavy/light chain ratios improve paraprotein detection and monitoring, identify residual disease and correlate with survival in multiple myeloma patients. Leukemia 2013, 27, 996. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Diagnosis | Clinical Presentation | Median Serum IgM Level (mg/dL) | Morphology | Immunophenotype | Cytogenetics | MYD88 L265P Mutation |
|---|---|---|---|---|---|---|
| Waldenströms macroglobulinaemia | Hyperviscosity syndrome Symptomatic anaemia | 3215 | Lymphoplasmacytic lymphoma/plasma cell | LPL: CD5−/CD10−/CD20+ PC: CD56− | 6q deletion in 40% | 80–100% |
| IgM MGUS | Asymptomatic | 840 | Lymphoplasmacytic lymphoma/plasma cell | LPL phenotype as in WM | Unknown | 10–40% |
| IgM myeloma | CRAB symptoms | 4660 | Plasma cell | PC: CD38++/CD138+/CD56+ | t(11;14) in 40% | 0% |
| Marginal zone lymphoma | Varies by subtype | 285 | Marginal zone B-cell | CD5−/CD10−/CD20+ | t(11;18) in MALT | 6–13% |
| Chronic lymphocytic leukaemia | Often asymptomatic Lymphadenopathy Splenomegaly Anaemia Thrombocytopenia Autoimmunity | 400 | Small mature lymphocytes | CD5+/CD10−/CD23+ | 11q, 13q abnormalities, 17p deletion | 0% |
| Light Chain Amyloidosis | Nephrotic syndrome, cardiac failure | 800 | Plasma cells or mature B-cells | Clonal PC may have a phenotype similar to MM | t(11;14) in 50% | 0% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Girard, L.-P.; Soekojo, C.Y.; Ooi, M.; Poon, L.M.; Chng, W.-J.; de Mel, S. Immunoglobulin M Paraproteinaemias. Cancers 2020, 12, 1688. https://doi.org/10.3390/cancers12061688
Girard L-P, Soekojo CY, Ooi M, Poon LM, Chng W-J, de Mel S. Immunoglobulin M Paraproteinaemias. Cancers. 2020; 12(6):1688. https://doi.org/10.3390/cancers12061688
Chicago/Turabian StyleGirard, Louis-Pierre, Cinnie Yentia Soekojo, Melissa Ooi, Li Mei Poon, Wee-Joo Chng, and Sanjay de Mel. 2020. "Immunoglobulin M Paraproteinaemias" Cancers 12, no. 6: 1688. https://doi.org/10.3390/cancers12061688
APA StyleGirard, L.-P., Soekojo, C. Y., Ooi, M., Poon, L. M., Chng, W.-J., & de Mel, S. (2020). Immunoglobulin M Paraproteinaemias. Cancers, 12(6), 1688. https://doi.org/10.3390/cancers12061688