Telomerase and CD4 T Cell Immunity in Cancer


Abstract
1. Introduction
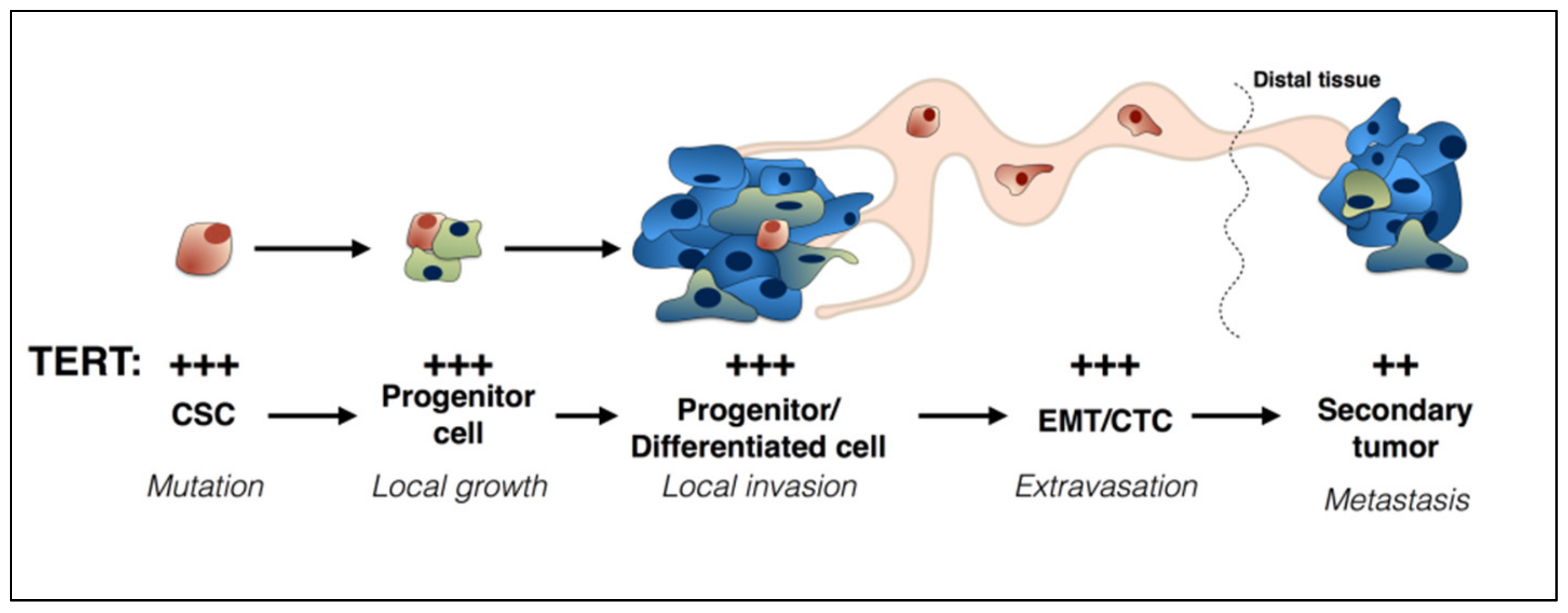
2. TERT and Cancer
3. TERT-Specific CD4 Th1 Cells as Pivotal Modulator of the Anti-Tumor Immune Response
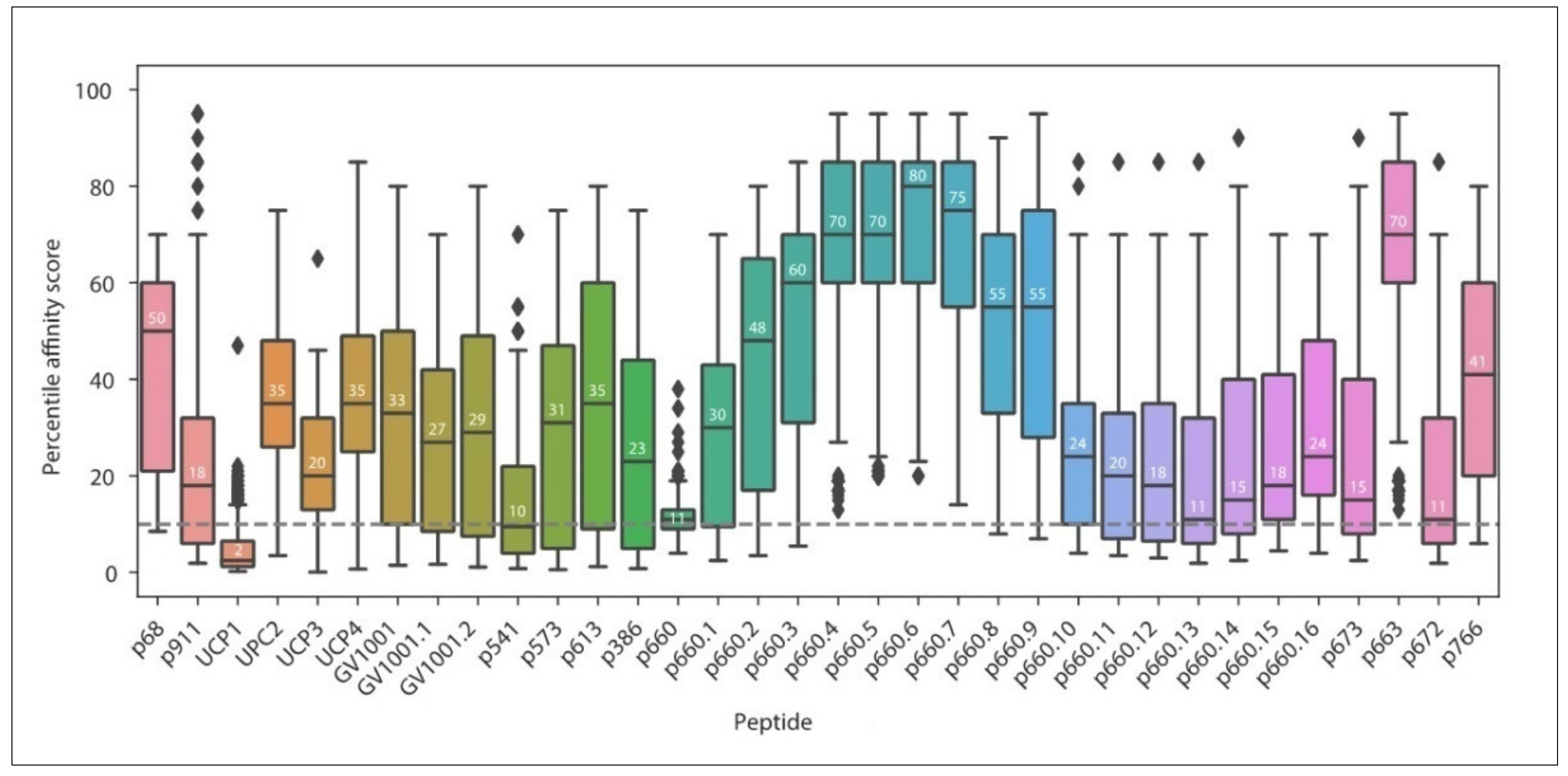
3.1. Current MHC-II Restricted TERT Peptides
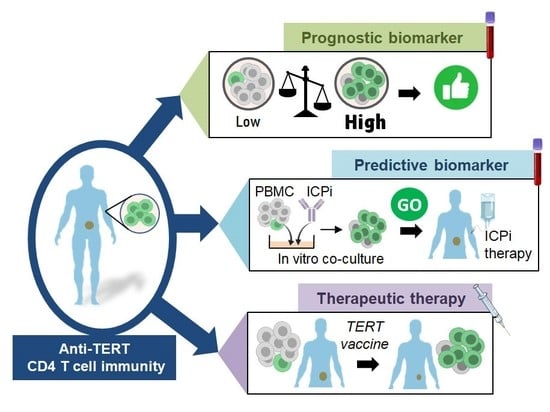
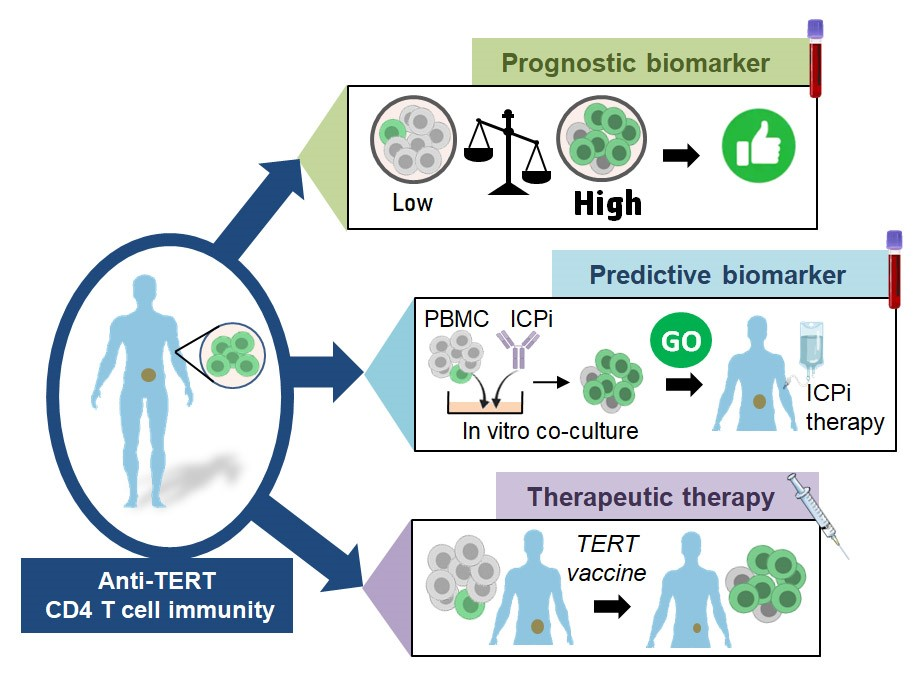
3.2. Prognostic Value of Systemic Anti-TERT CD4 T Cell Immunity in Cancer
3.3. Past and Current Therapeutic Approaches Targeting Anti-TERT CD4 Th1 Cell Immunity
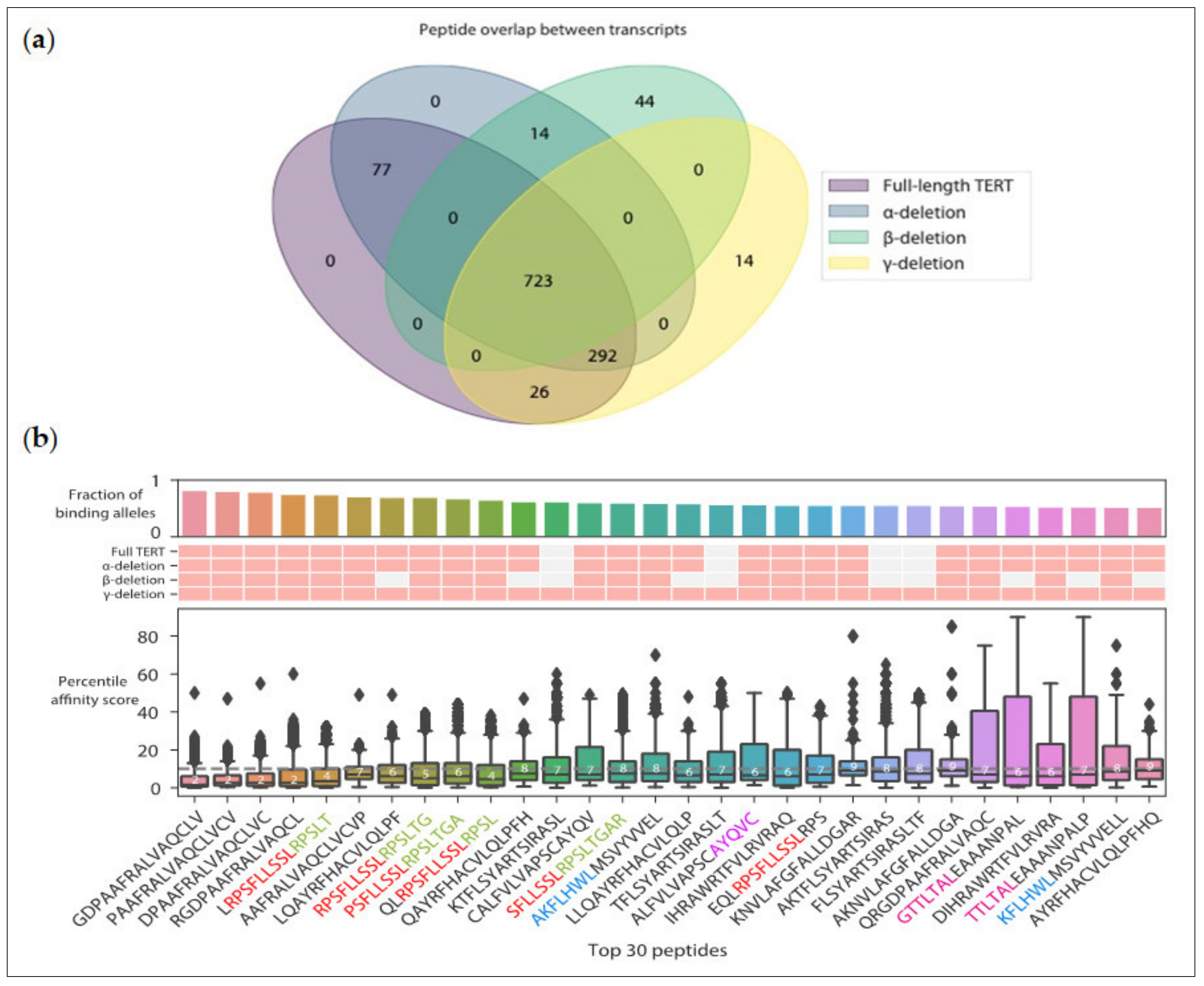
4. Prospects for the Identification of Novel Immunogenic TERT CD4 Epitopes
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Burnet, F.M. Immunological surveillance in neoplasia. Transplant. Rev. 1971, 7, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Brossart, P.; Stuhler, G.; Flad, T.; Stevanovic, S.; Rammensee, H.G.; Kanz, L.; Brugger, W. Her-2/neu-derived peptides are tumor-associated antigens expressed by human renal cell and colon carcinoma lines and are recognized by in vitro induced specific cytotoxic T lymphocytes. Cancer Res. 1998, 58, 732–736. [Google Scholar]
- Molldrem, J.J.; Lee, P.P.; Wang, C.; Felio, K.; Kantarjian, H.M.; Champlin, R.E.; Davis, M.M. Evidence that specific T lymphocytes may participate in the elimination of chronic myelogenous leukemia. Nat. Med. 2000, 6, 1018–1023. [Google Scholar] [CrossRef]
- Filaci, G.; Fravega, M.; Setti, M.; Traverso, P.; Millo, E.; Fenoglio, D.; Negrini, S.; Ferrera, F.; Romagnoli, A.; Basso, M.; et al. Frequency of telomerase-specific CD8+ T lymphocytes in patients with cancer. Blood 2006, 107, 1505–1512. [Google Scholar] [CrossRef]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Restifo, N.P. Cancer immunotherapy: Moving beyond current vaccines. Nat. Med. 2004, 10, 909–915. [Google Scholar] [CrossRef]
- Klebanoff, C.A.; Acquavella, N.; Yu, Z.; Restifo, N.P. Therapeutic cancer vaccines: Are we there yet? Immunol. Rev. 2011, 239, 27–44. [Google Scholar] [CrossRef] [PubMed]
- Koretz, K.; Moldenhauer, G.; Majdic, O.; Möller, P. Correlation of HLA-D/Ii antigen expression in breast carcinoma with local lymphohistiocytic infiltration reveals considerable dysregulation in a subset of tumors. Int. J. Cancer 1989, 44, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Marincola, F.M.; Jaffee, E.M.; Hicklin, D.J.; Ferrone, S. Escape of human solid tumors from T-cell recognition: Molecular mechanisms and functional significance. Adv. Immunol. 2000, 74, 181–273. [Google Scholar] [CrossRef] [PubMed]
- Möller, P.; Mattfeldt, T.; Gross, C.; Schlosshauer, P.; Koch, A.; Koretz, K.; Moldenhauer, G.; Kaufmann, M.; Otto, H.F. Expression of HLA-A, -B, -C, -DR, -DP, -DQ, and of HLA-D-associated invariant chain (Ii) in non-neoplastic mammary epithelium, fibroadenoma, adenoma, and carcinoma of the breast. Am. J. Pathol. 1989, 135, 73–83. [Google Scholar] [PubMed]
- Mitchison, N.A. The carrier effect in the secondary response to hapten-protein conjugates. II. Cellular cooperation. Eur. J. Immunol. 1971, 1, 18–27. [Google Scholar] [CrossRef]
- Cassell, D.; Forman, J. Linked recognition of helper and cytotoxic antigenic determinants for the generation of cytotoxic T lymphocytes. Ann. N. Y. Acad. Sci. 1988, 532, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Gerloni, M.; Xiong, S.; Mukerjee, S.; Schoenberger, S.P.; Croft, M.; Zanetti, M. Functional cooperation between T helper cell determinants. Proc. Natl. Acad. Sci. USA 2000, 97, 13269–13274. [Google Scholar] [CrossRef] [PubMed]
- Janssen, E.M.; Lemmens, E.E.; Wolfe, T.; Christen, U.; von Herrath, M.G.; Schoenberger, S.P. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 2003, 421, 852–856. [Google Scholar] [CrossRef] [PubMed]
- Shedlock, D.J.; Shen, H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science 2003, 300, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.C.; Bevan, M.J. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science 2003, 300, 339–342. [Google Scholar] [CrossRef]
- Langlade-Demoyen, P.; Garcia-Pons, F.; Castiglioni, P.; Garcia, Z.; Cardinaud, S.; Xiong, S.; Gerloni, M.; Zanetti, M. Role of T cell help and endoplasmic reticulum targeting in protective CTL response against influenza virus. Eur. J. Immunol. 2003, 33, 720–728. [Google Scholar] [CrossRef]
- Sakaguchi, S. Regulatory T cells: Key controllers of immunologic self-tolerance. Cell 2000, 101, 455–458. [Google Scholar] [CrossRef]
- Korn, T.; Oukka, M.; Kuchroo, V.; Bettelli, E. Th17 cells: Effector T cells with inflammatory properties. Semin. Immunol. 2007, 19, 362–371. [Google Scholar] [CrossRef]
- Kim, H.-J.; Cantor, H. CD4 T-cell subsets and tumor immunity: The helpful and the not-so-helpful. Cancer Immunol Res. 2014, 2, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, M. Tapping CD4 T cells for cancer immunotherapy: The choice of personalized genomics. J. Immunol. 2015, 194, 2049–2056. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, P.D.; Cheever, M.A.; Fefer, A. Eradication of disseminated murine leukemia by chemoimmunotherapy with cyclophosphamide and adoptively transferred immune syngeneic Lyt-1+2- lymphocytes. J. Exp. Med. 1981, 154, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, H.; Fukuzawa, M.; Yoshioka, T.; Nakajima, H.; Hamaoka, T. The role of tumor-specific Lyt-1+2- T cells in eradicating tumor cells in vivo. I. Lyt-1+2- T cells do not necessarily require recruitment of host’s cytotoxic T cell precursors for implementation of in vivo immunity. J. Immunol. 1984, 133, 1671–1676. [Google Scholar] [PubMed]
- Hock, H.; Dorsch, M.; Diamantstein, T.; Blankenstein, T. Interleukin 7 induces CD4+ T cell-dependent tumor rejection. J. Exp. Med. 1991, 174, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- Lauritzsen, G.F.; Weiss, S.; Dembic, Z.; Bogen, B. Naive idiotype-specific CD4+ T cells and immunosurveillance of B-cell tumors. Proc. Natl. Acad. Sci. USA 1994, 91, 5700–5704. [Google Scholar] [CrossRef]
- Hung, K.; Hayashi, R.; Lafond-Walker, A.; Lowenstein, C.; Pardoll, D.; Levitsky, H. The central role of CD4(+) T cells in the antitumor immune response. J. Exp. Med. 1998, 188, 2357–2368. [Google Scholar] [CrossRef]
- Mumberg, D.; Monach, P.A.; Wanderling, S.; Philip, M.; Toledano, A.Y.; Schreiber, R.D.; Schreiber, H. CD4(+) T cells eliminate MHC class II-negative cancer cells in vivo by indirect effects of IFN-gamma. Proc. Natl. Acad. Sci. USA 1999, 96, 8633–8638. [Google Scholar] [CrossRef]
- Tempero, R.M.; VanLith, M.L.; Morikane, K.; Rowse, G.J.; Gendler, S.J.; Hollingsworth, M.A. CD4+ lymphocytes provide MUC1-specific tumor immunity in vivo that is undetectable in vitro and is absent in MUC1 transgenic mice. J. Immunol. 1998, 161, 5500–5506. [Google Scholar]
- Greenberg, P.D. Adoptive T cell therapy of tumors: Mechanisms operative in the recognition and elimination of tumor cells. Adv. Immunol. 1991, 49, 281–355. [Google Scholar] [CrossRef]
- Dighe, A.S.; Richards, E.; Old, L.J.; Schreiber, R.D. Enhanced in vivo growth and resistance to rejection of tumor cells expressing dominant negative IFN gamma receptors. Immunity 1994, 1, 447–456. [Google Scholar] [CrossRef]
- Williamson, B.D.; Carswell, E.A.; Rubin, B.Y.; Prendergast, J.S.; Old, L.J. Human tumor necrosis factor produced by human B-cell lines: Synergistic cytotoxic interaction with human interferon. Proc. Natl. Acad. Sci. USA 1983, 80, 5397–5401. [Google Scholar] [CrossRef] [PubMed]
- Fransen, L.; Van der Heyden, J.; Ruysschaert, R.; Fiers, W. Recombinant tumor necrosis factor: Its effect and its synergism with interferon-gamma on a variety of normal and transformed human cell lines. Eur. J. Cancer Clin. Oncol. 1986, 22, 419–426. [Google Scholar] [CrossRef]
- Coughlin, C.M.; Salhany, K.E.; Gee, M.S.; LaTemple, D.C.; Kotenko, S.; Ma, X.; Gri, G.; Wysocka, M.; Kim, J.E.; Liu, L.; et al. Tumor cell responses to IFNgamma affect tumorigenicity and response to IL-12 therapy and antiangiogenesis. Immunity 1998, 9, 25–34. [Google Scholar] [CrossRef]
- Qin, Z.; Blankenstein, T. CD4+ T cell--mediated tumor rejection involves inhibition of angiogenesis that is dependent on IFN gamma receptor expression by nonhematopoietic cells. Immunity 2000, 12, 677–686. [Google Scholar] [CrossRef]
- Corthay, A.; Skovseth, D.K.; Lundin, K.U.; Røsjø, E.; Omholt, H.; Hofgaard, P.O.; Haraldsen, G.; Bogen, B. Primary antitumor immune response mediated by CD4+ T cells. Immunity 2005, 22, 371–383. [Google Scholar] [CrossRef]
- Haabeth, O.A.W.; Lorvik, K.B.; Hammarström, C.; Donaldson, I.M.; Haraldsen, G.; Bogen, B.; Corthay, A. Inflammation driven by tumour-specific Th1 cells protects against B-cell cancer. Nat. Commun. 2011, 2, 240. [Google Scholar] [CrossRef]
- Xie, Y.; Akpinarli, A.; Maris, C.; Hipkiss, E.L.; Lane, M.; Kwon, E.-K.M.; Muranski, P.; Restifo, N.P.; Antony, P.A. Naive tumor-specific CD4(+) T cells differentiated in vivo eradicate established melanoma. J. Exp. Med. 2010, 207, 651–667. [Google Scholar] [CrossRef]
- Quezada, S.A.; Simpson, T.R.; Peggs, K.S.; Merghoub, T.; Vider, J.; Fan, X.; Blasberg, R.; Yagita, H.; Muranski, P.; Antony, P.A.; et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J. Exp. Med. 2010, 207, 637–650. [Google Scholar] [CrossRef]
- Braumüller, H.; Wieder, T.; Brenner, E.; Aßmann, S.; Hahn, M.; Alkhaled, M.; Schilbach, K.; Essmann, F.; Kneilling, M.; Griessinger, C.; et al. T-helper-1-cell cytokines drive cancer into senescence. Nature 2013, 494, 361–365. [Google Scholar] [CrossRef]
- Horna, P.; Cuenca, A.; Cheng, F.; Brayer, J.; Wang, H.-W.; Borrello, I.; Levitsky, H.; Sotomayor, E.M. In vivo disruption of tolerogenic cross-presentation mechanisms uncovers an effective T-cell activation by B-cell lymphomas leading to antitumor immunity. Blood 2006, 107, 2871–2878. [Google Scholar] [CrossRef] [PubMed]
- Perez-Diez, A.; Joncker, N.T.; Choi, K.; Chan, W.F.N.; Anderson, C.C.; Lantz, O.; Matzinger, P. CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood 2007, 109, 5346–5354. [Google Scholar] [CrossRef] [PubMed]
- Thomas, W.D.; Hersey, P. TNF-related apoptosis-inducing ligand (TRAIL) induces apoptosis in Fas ligand-resistant melanoma cells and mediates CD4 T cell killing of target cells. J. Immunol. 1998, 161, 2195–2200. [Google Scholar] [PubMed]
- Lundin, K.U.; Screpanti, V.; Omholt, H.; Hofgaard, P.O.; Yagita, H.; Grandien, A.; Bogen, B. CD4+ T cells kill Id+ B-lymphoma cells: FasLigand-Fas interaction is dominant in vitro but is redundant in vivo. Cancer Immunol. Immunother. 2004, 53, 1135–1145. [Google Scholar] [CrossRef]
- Southwood, S.; Sidney, J.; Kondo, A.; del Guercio, M.F.; Appella, E.; Hoffman, S.; Kubo, R.T.; Chesnut, R.W.; Grey, H.M.; Sette, A. Several common HLA-DR types share largely overlapping peptide binding repertoires. J. Immunol. 1998, 160, 3363–3373. [Google Scholar] [PubMed]
- Consogno, G.; Manici, S.; Facchinetti, V.; Bachi, A.; Hammer, J.; Conti-Fine, B.M.; Rugarli, C.; Traversari, C.; Protti, M.P. Identification of immunodominant regions among promiscuous HLA-DR-restricted CD4+ T-cell epitopes on the tumor antigen MAGE-3. Blood 2003, 101, 1038–1044. [Google Scholar] [CrossRef]
- Neumann, F.; Wagner, C.; Stevanovic, S.; Kubuschok, B.; Schormann, C.; Mischo, A.; Ertan, K.; Schmidt, W.; Pfreundschuh, M. Identification of an HLA-DR-restricted peptide epitope with a promiscuous binding pattern derived from the cancer testis antigen HOM-MEL-40/SSX2. Int. J. Cancer 2004, 112, 661–668. [Google Scholar] [CrossRef]
- Wang, X.-F.; Kerzerho, J.; Adotevi, O.; Nuyttens, H.; Badoual, C.; Munier, G.; Oudard, S.; Tu, S.; Tartour, E.; Maillère, B. Comprehensive analysis of HLA-DR- and HLA-DP4-restricted CD4+ T cell response specific for the tumor-shared antigen survivin in healthy donors and cancer patients. J. Immunol. 2008, 181, 431–439. [Google Scholar] [CrossRef]
- Zeng, G.; Touloukian, C.E.; Wang, X.; Restifo, N.P.; Rosenberg, S.A.; Wang, R.F. Identification of CD4+ T cell epitopes from NY-ESO-1 presented by HLA-DR molecules. J. Immunol. 2000, 165, 1153–1159. [Google Scholar] [CrossRef]
- Zeng, G.; Wang, X.; Robbins, P.F.; Rosenberg, S.A.; Wang, R.F. CD4(+) T cell recognition of MHC class II-restricted epitopes from NY-ESO-1 presented by a prevalent HLA DP4 allele: Association with NY-ESO-1 antibody production. Proc. Natl. Acad. Sci. USA 2001, 98, 3964–3969. [Google Scholar] [CrossRef]
- Campi, G.; Crosti, M.; Consogno, G.; Facchinetti, V.; Conti-Fine, B.M.; Longhi, R.; Casorati, G.; Dellabona, P.; Protti, M.P. CD4(+) T cells from healthy subjects and colon cancer patients recognize a carcinoembryonic antigen-specific immunodominant epitope. Cancer Res. 2003, 63, 8481–8486. [Google Scholar] [PubMed]
- Kudela, P.; Sun, Z.; Fourcade, J.; Janjic, B.; Kirkwood, J.M.; Maillere, B.; Zarour, H.M. Epitope hierarchy of spontaneous CD4+ T cell responses to LAGE-1. J. Immunol. 2011, 186, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Ohue, Y.; Eikawa, S.; Okazaki, N.; Mizote, Y.; Isobe, M.; Uenaka, A.; Fukuda, M.; Old, L.J.; Oka, M.; Nakayama, E. Spontaneous antibody, and CD4 and CD8 T-cell responses against XAGE-1b (GAGED2a) in non-small cell lung cancer patients. Int. J. Cancer 2012, 131, E649–E658. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, T.; Matsuzaki, J.; Ritter, E.; Miliotto, A.; Ritter, G.; Odunsi, K.; Old, L.J.; Gnjatic, S. Split T cell tolerance against a self/tumor antigen: Spontaneous CD4+ but not CD8+ T cell responses against p53 in cancer patients and healthy donors. PLoS ONE 2011, 6, e23651. [Google Scholar] [CrossRef]
- Munir, S.; Larsen, S.K.; Iversen, T.Z.; Donia, M.; Klausen, T.W.; Svane, I.M.; Straten, P.T.; Andersen, M.H. Natural CD4+ T-cell responses against indoleamine 2,3-dioxygenase. PLoS ONE 2012, 7, e34568. [Google Scholar] [CrossRef]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef]
- Yoshida, N.; Abe, H.; Ohkuri, T.; Wakita, D.; Sato, M.; Noguchi, D.; Miyamoto, M.; Morikawa, T.; Kondo, S.; Ikeda, H.; et al. Expression of the MAGE-A4 and NY-ESO-1 cancer-testis antigens and T cell infiltration in non-small cell lung carcinoma and their prognostic significance. Int. J. Oncol. 2006, 28, 1089–1098. [Google Scholar] [CrossRef]
- Ayyoub, M.; Pignon, P.; Classe, J.-M.; Odunsi, K.; Valmori, D. CD4+ T effectors specific for the tumor antigen NY-ESO-1 are highly enriched at ovarian cancer sites and coexist with, but are distinct from, tumor-associated Treg. Cancer Immunol Res. 2013, 1, 303–308. [Google Scholar] [CrossRef]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Fridman, W.H.; Zitvogel, L.; Sautès-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef]
- Zanetti, M.; Hernandez, X.; Langlade-Demoyen, P. Telomerase reverse transcriptase as target for anti-tumor T cell responses in humans. Springer Semin. Immunopathol. 2005, 27, 87–104. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, M. A second chance for telomerase reverse transcriptase in anticancer immunotherapy. Nat. Rev. Clin. Oncol. 2017, 14, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Hayflick, L. The Limited In Vitro Lifetime of Human Diploid Cell Strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Hahn, W.C.; Counter, C.M.; Lundberg, A.S.; Beijersbergen, R.L.; Brooks, M.W.; Weinberg, R.A. Creation of human tumour cells with defined genetic elements. Nature 1999, 400, 464–468. [Google Scholar] [CrossRef]
- Blackburn, E.H. Telomerases. Annu. Rev. Biochem. 1992, 61, 113–129. [Google Scholar] [CrossRef]
- Shay, J.W.; Wright, W.E. Telomeres and telomerase: Three decades of progress. Nat. Rev. Genet. 2019, 20, 299–309. [Google Scholar] [CrossRef]
- Blackburn, E.H.; Epel, E.S.; Lin, J. Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science 2015, 350, 1193–1198. [Google Scholar] [CrossRef]
- Kim, N.W. Clinical implications of telomerase in cancer. Eur. J. Cancer 1997, 33, 781–786. [Google Scholar] [CrossRef]
- Nakamura, T.M.; Morin, G.B.; Chapman, K.B.; Weinrich, S.L.; Andrews, W.H.; Lingner, J.; Harley, C.B.; Cech, T.R. Telomerase catalytic subunit homologs from fission yeast and human. Science 1997, 277, 955–959. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Reddel, R.R.; Wright, W.E. Cancer. Cancer and telomeres—An ALTernative to telomerase. Science 2012, 336, 1388–1390. [Google Scholar] [CrossRef] [PubMed]
- Low, K.C.; Tergaonkar, V. Telomerase: Central regulator of all of the hallmarks of cancer. Trends Biochem. Sci. 2013, 38, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Hannen, R.; Bartsch, J.W. Essential roles of telomerase reverse transcriptase hTERT in cancer stemness and metastasis. FEBS Lett. 2018, 592, 2023–2031. [Google Scholar] [CrossRef] [PubMed]
- Flores, I.; Blasco, M.A. A p53-dependent response limits epidermal stem cell functionality and organismal size in mice with short telomeres. PLoS ONE 2009, 4, e4934. [Google Scholar] [CrossRef]
- Goldman, F.D.; Aubert, G.; Klingelhutz, A.J.; Hills, M.; Cooper, S.R.; Hamilton, W.S.; Schlueter, A.J.; Lambie, K.; Eaves, C.J.; Lansdorp, P.M. Characterization of primitive hematopoietic cells from patients with dyskeratosis congenita. Blood 2008, 111, 4523–4531. [Google Scholar] [CrossRef]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.M.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells—Perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef]
- Beier, F.; Beier, C.P.; Aschenbrenner, I.; Hildebrandt, G.C.; Brümmendorf, T.H.; Beier, D. Identification of CD133(-)/telomerase(low) progenitor cells in glioblastoma-derived cancer stem cell lines. Cell Mol. Neurobiol. 2011, 31, 337–343. [Google Scholar] [CrossRef]
- Xu, T.; He, K.; Wang, L.; Goldkorn, A. Prostate tumor cells with cancer progenitor properties have high telomerase activity and are rapidly killed by telomerase interference. Prostate 2011, 71, 1390–1400. [Google Scholar] [CrossRef]
- Fiñones, R.R.; Yeargin, J.; Lee, M.; Kaur, A.P.; Cheng, C.; Sun, P.; Wu, C.; Nguyen, C.; Wang-Rodriguez, J.; Meyer, A.N.; et al. Early human prostate adenocarcinomas harbor androgen-independent cancer cells. PLoS ONE 2013, 8, e74438. [Google Scholar] [CrossRef]
- Goldkorn, A.; Ely, B.; Tangen, C.M.; Tai, Y.-C.; Xu, T.; Li, H.; Twardowski, P.; Veldhuizen, P.J.V.; Agarwal, N.; Carducci, M.A.; et al. Circulating tumor cell telomerase activity as a prognostic marker for overall survival in SWOG 0421: A phase III metastatic castration resistant prostate cancer trial. Int. J. Cancer 2015, 136, 1856–1862. [Google Scholar] [CrossRef]
- Ito, H.; Inoue, H.; Kimura, S.; Ohmori, T.; Ishikawa, F.; Gohda, K.; Sato, J. Prognostic impact of the number of viable circulating cells with high telomerase activity in gastric cancer patients: A prospective study. Int. J. Oncol. 2014, 45, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, Q.; Li, K.; Chen, L.; Li, W.; Hou, M.; Liu, T.; Yang, J.; Lindvall, C.; Björkholm, M.; et al. Telomerase reverse transcriptase promotes epithelial-mesenchymal transition and stem cell-like traits in cancer cells. Oncogene 2013, 32, 4203–4213. [Google Scholar] [CrossRef] [PubMed]
- Galaine, J.; Turco, C.; Vauchy, C.; Royer, B.; Mercier-Letondal, P.; Queiroz, L.; Loyon, R.; Mouget, V.; Boidot, R.; Laheurte, C.; et al. CD4 T cells target colorectal cancer antigens upregulated by oxaliplatin. Int. J. Cancer 2019, 145, 3112–3125. [Google Scholar] [CrossRef] [PubMed]
- Tatsumoto, N.; Hiyama, E.; Murakami, Y.; Imamura, Y.; Shay, J.W.; Matsuura, Y.; Yokoyama, T. High telomerase activity is an independent prognostic indicator of poor outcome in colorectal cancer. Clin. Cancer Res. 2000, 6, 2696–2701. [Google Scholar]
- Bertorelle, R.; Briarava, M.; Rampazzo, E.; Biasini, L.; Agostini, M.; Maretto, I.; Lonardi, S.; Friso, M.L.; Mescoli, C.; Zagonel, V.; et al. Telomerase is an independent prognostic marker of overall survival in patients with colorectal cancer. Br. J. Cancer 2013, 108, 278–284. [Google Scholar] [CrossRef]
- Marchetti, A.; Bertacca, G.; Buttitta, F.; Chella, A.; Quattrocolo, G.; Angeletti, C.A.; Bevilacqua, G. Telomerase activity as a prognostic indicator in stage I non-small cell lung cancer. Clin. Cancer Res. 1999, 5, 2077–2081. [Google Scholar]
- Poremba, C.; Heine, B.; Diallo, R.; Heinecke, A.; Wai, D.; Schaefer, K.-L.; Braun, Y.; Schuck, A.; Lanvers, C.; Bànkfalvi, A.; et al. Telomerase as a prognostic marker in breast cancer: High-throughput tissue microarray analysis of hTERT and hTR. J. Pathol. 2002, 198, 181–189. [Google Scholar] [CrossRef]
- Elkak, A.; Mokbel, R.; Wilson, C.; Jiang, W.G.; Newbold, R.F.; Mokbel, K. hTERT mRNA expression is associated with a poor clinical outcome in human breast cancer. Anticancer Res. 2006, 26, 4901–4904. [Google Scholar]
- Vinagre, J.; Almeida, A.; Pópulo, H.; Batista, R.; Lyra, J.; Pinto, V.; Coelho, R.; Celestino, R.; Prazeres, H.; Lima, L.; et al. Frequency of TERT promoter mutations in human cancers. Nat. Commun. 2013, 4, 2185. [Google Scholar] [CrossRef]
- Weinhold, N.; Jacobsen, A.; Schultz, N.; Sander, C.; Lee, W. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat. Genet. 2014, 46, 1160–1165. [Google Scholar] [CrossRef] [PubMed]
- Borah, S.; Xi, L.; Zaug, A.J.; Powell, N.M.; Dancik, G.M.; Cohen, S.B.; Costello, J.C.; Theodorescu, D.; Cech, T.R. Cancer. TERT promoter mutations and telomerase reactivation in urothelial cancer. Science 2015, 347, 1006–1010. [Google Scholar] [CrossRef] [PubMed]
- Gourd, E. TERT mutations in urine could predict bladder cancer recurrence. Lancet Oncol. 2017, 18, e443. [Google Scholar] [CrossRef]
- Myung, J.K.; Kwak, B.K.; Lim, J.A.; Lee, M.-C.; Kim, M.J. TERT Promoter Mutations and Tumor Persistence/Recurrence in Papillary Thyroid Cancer. Cancer Res. Treat. 2016, 48, 942–947. [Google Scholar] [CrossRef]
- Descotes, F.; Kara, N.; Decaussin-Petrucci, M.; Piaton, E.; Geiguer, F.; Rodriguez-Lafrasse, C.; Terrier, J.E.; Lopez, J.; Ruffion, A. Non-invasive prediction of recurrence in bladder cancer by detecting somatic TERT promoter mutations in urine. Br. J. Cancer 2017, 117, 583–587. [Google Scholar] [CrossRef]
- Heidenreich, B.; Kumar, R. Altered TERT promoter and other genomic regulatory elements: Occurrence and impact. Int. J. Cancer 2017, 141, 867–876. [Google Scholar] [CrossRef]
- Minev, B.; Hipp, J.; Firat, H.; Schmidt, J.D.; Langlade-Demoyen, P.; Zanetti, M. Cytotoxic T cell immunity against telomerase reverse transcriptase in humans. Proc. Natl. Acad. Sci. USA 2000, 97, 4796–4801. [Google Scholar] [CrossRef]
- Vonderheide, R.H. Universal tumor antigens for cancer vaccination: Targeting telomerase for immunoprevention. Discov. Med. 2007, 7, 103–108. [Google Scholar]
- Schroers, R.; Huang, X.F.; Hammer, J.; Zhang, J.; Chen, S.-Y. Identification of HLA DR7-restricted epitopes from human telomerase reverse transcriptase recognized by CD4+ T-helper cells. Cancer Res. 2002, 62, 2600–2605. [Google Scholar]
- Schroers, R.; Shen, L.; Rollins, L.; Rooney, C.M.; Slawin, K.; Sonderstrup, G.; Huang, X.F.; Chen, S.-Y. Human telomerase reverse transcriptase-specific T-helper responses induced by promiscuous major histocompatibility complex class II-restricted epitopes. Clin. Cancer Res. 2003, 9, 4743–4755. [Google Scholar]
- Brunsvig, P.F.; Aamdal, S.; Gjertsen, M.K.; Kvalheim, G.; Markowski-Grimsrud, C.J.; Sve, I.; Dyrhaug, M.; Trachsel, S.; Møller, M.; Eriksen, J.A.; et al. Telomerase peptide vaccination: A phase I/II study in patients with non-small cell lung cancer. Cancer Immunol. Immunother. 2006, 55, 1553–1564. [Google Scholar] [CrossRef] [PubMed]
- Bernardeau, K.; Kerzhero, J.; Fortun, A.; Moreau-Aubry, A.; Favry, E.; Echasserieau, K.; Tartour, E.; Maillère, B.; Lang, F. A simple competitive assay to determine peptide affinity for HLA class II molecules: A useful tool for epitope prediction. J. Immunol. Methods 2011, 371, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Kyte, J.A.; Gaudernack, G.; Dueland, S.; Trachsel, S.; Julsrud, L.; Aamdal, S. Telomerase peptide vaccination combined with temozolomide: A clinical trial in stage IV melanoma patients. Clin. Cancer Res. 2011, 17, 4568–4580. [Google Scholar] [CrossRef] [PubMed]
- Suso, E.M.I.; Dueland, S.; Rasmussen, A.-M.; Vetrhus, T.; Aamdal, S.; Kvalheim, G.; Gaudernack, G. hTERT mRNA dendritic cell vaccination: Complete response in a pancreatic cancer patient associated with response against several hTERT epitopes. Cancer Immunol. Immunother. 2011, 60, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Godet, Y.; Fabre, E.; Dosset, M.; Lamuraglia, M.; Levionnois, E.; Ravel, P.; Benhamouda, N.; Cazes, A.; Le Pimpec-Barthes, F.; Gaugler, B.; et al. Analysis of spontaneous tumor-specific CD4 T-cell immunity in lung cancer using promiscuous HLA-DR telomerase-derived epitopes: Potential synergistic effect with chemotherapy response. Clin. Cancer Res. 2012, 18, 2943–2953. [Google Scholar] [CrossRef] [PubMed]
- Dosset, M.; Vauchy, C.; Beziaud, L.; Adotevi, O.; Godet, Y. Universal tumor-reactive helper peptides from telomerase as new tools for anticancer vaccination. Oncoimmunology 2013, 2, e23430. [Google Scholar] [CrossRef] [PubMed]
- Laheurte, C.; Galaine, J.; Beziaud, L.; Dosset, M.; Kerzerho, J.; Jacquemard, C.; Gaugler, B.; Ferrand, C.; Dormoy, A.; Aubin, F.; et al. Immunoprevalence and magnitude of HLA-DP4 versus HLA-DR-restricted spontaneous CD4(+) Th1 responses against telomerase in cancer patients. Oncoimmunology 2016, 5, e1137416. [Google Scholar] [CrossRef]
- Kumagai, M.; Mizukoshi, E.; Tamai, T.; Kitahara, M.; Yamashita, T.; Arai, K.; Terashima, T.; Iida, N.; Fushimi, K.; Kaneko, S. Immune response to human telomerase reverse transcriptase-derived helper T cell epitopes in hepatocellular carcinoma patients. Liver Int. 2018, 38, 1635–1645. [Google Scholar] [CrossRef]
- Melief, C.J.M.; van der Burg, S.H. Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nat. Rev. Cancer 2008, 8, 351–360. [Google Scholar] [CrossRef]
- Laheurte, C.; Dosset, M.; Vernerey, D.; Boullerot, L.; Gaugler, B.; Gravelin, E.; Kaulek, V.; Jacquin, M.; Cuche, L.; Eberst, G.; et al. Distinct prognostic value of circulating anti-telomerase CD4+ Th1 immunity and exhausted PD-1+/TIM-3+ T cells in lung cancer. Br. J. Cancer 2019, 121, 405–416. [Google Scholar] [CrossRef]
- Kim, S.; François, E.; André, T.; Samalin, E.; Jary, M.; El Hajbi, F.; Baba-Hamed, N.; Pernot, S.; Kaminsky, M.-C.; Bouché, O.; et al. Docetaxel, cisplatin, and fluorouracil chemotherapy for metastatic or unresectable locally recurrent anal squamous cell carcinoma (Epitopes-HPV02): A multicentre, single-arm, phase 2 study. Lancet Oncol. 2018, 19, 1094–1106. [Google Scholar] [CrossRef]
- Beziaud, L.; Mansi, L.; Ravel, P.; Marie-Joseph, E.L.; Laheurte, C.; Rangan, L.; Bonnefoy, F.; Pallandre, J.-R.; Boullerot, L.; Gamonet, C.; et al. Rapalogs Efficacy Relies on the Modulation of Antitumor T-cell Immunity. Cancer Res. 2016, 76, 4100–4112. [Google Scholar] [CrossRef] [PubMed]
- Dosset, M.; Godet, Y.; Vauchy, C.; Beziaud, L.; Lone, Y.C.; Sedlik, C.; Liard, C.; Levionnois, E.; Clerc, B.; Sandoval, F.; et al. Universal cancer peptide-based therapeutic vaccine breaks tolerance against telomerase and eradicates established tumor. Clin. Cancer Res. 2012, 18, 6284–6295. [Google Scholar] [CrossRef] [PubMed]
- Zarour, H.M.; Maillere, B.; Brusic, V.; Coval, K.; Williams, E.; Pouvelle-Moratille, S.; Castelli, F.; Land, S.; Bennouna, J.; Logan, T.; et al. NY-ESO-1 119-143 is a promiscuous major histocompatibility complex class II T-helper epitope recognized by Th1- and Th2-type tumor-reactive CD4+ T cells. Cancer Res. 2002, 62, 213–218. [Google Scholar]
- Mandic, M.; Castelli, F.; Janjic, B.; Almunia, C.; Andrade, P.; Gillet, D.; Brusic, V.; Kirkwood, J.M.; Maillere, B.; Zarour, H.M. One NY-ESO-1-derived epitope that promiscuously binds to multiple HLA-DR and HLA-DP4 molecules and stimulates autologous CD4+ T cells from patients with NY-ESO-1-expressing melanoma. J. Immunol. 2005, 174, 1751–1759. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Huang, A.C.; Postow, M.A.; Orlowski, R.J.; Mick, R.; Bengsch, B.; Manne, S.; Xu, W.; Harmon, S.; Giles, J.R.; Wenz, B.; et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017, 545, 60–65. [Google Scholar] [CrossRef]
- Spitzer, M.H.; Carmi, Y.; Reticker-Flynn, N.E.; Kwek, S.S.; Madhireddy, D.; Martins, M.M.; Gherardini, P.F.; Prestwood, T.R.; Chabon, J.; Bendall, S.C.; et al. Systemic Immunity Is Required for Effective Cancer Immunotherapy. Cell 2017, 168, 487–502.e15. [Google Scholar] [CrossRef]
- Walker, R.; Poleszczuk, J.; Pilon-Thomas, S.; Kim, S.; Anderson, A.A.R.A.; Czerniecki, B.J.; Harrison, L.B.; Moros, E.G.; Enderling, H. Immune interconnectivity of anatomically distant tumors as a potential mediator of systemic responses to local therapy. Sci. Rep. 2018, 8, 9474. [Google Scholar] [CrossRef]
- Iwahori, K.; Shintani, Y.; Funaki, S.; Yamamoto, Y.; Matsumoto, M.; Yoshida, T.; Morimoto-Okazawa, A.; Kawashima, A.; Sato, E.; Gottschalk, S.; et al. Peripheral T cell cytotoxicity predicts T cell function in the tumor microenvironment. Sci. Rep. 2019, 9, 2636. [Google Scholar] [CrossRef]
- Zuazo, M.; Arasanz, H.; Fernández-Hinojal, G.; García-Granda, M.J.; Gato, M.; Bocanegra, A.; Martínez, M.; Hernández, B.; Teijeira, L.; Morilla, I.; et al. Functional systemic CD4 immunity is required for clinical responses to PD-L1/PD-1 blockade therapy. EMBO Mol. Med. 2019, 11, e10293. [Google Scholar] [CrossRef] [PubMed]
- Kagamu, H.; Kitano, S.; Yamaguchi, O.; Yoshimura, K.; Horimoto, K.; Kitazawa, M.; Fukui, K.; Shiono, A.; Mouri, A.; Nishihara, F.; et al. CD4+ T-cell Immunity in the Peripheral Blood Correlates with Response to Anti-PD-1 Therapy. Cancer Immunol. Res. 2020, 8, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Brunsvig, P.F.; Kyte, J.A.; Kersten, C.; Sundstrøm, S.; Møller, M.; Nyakas, M.; Hansen, G.L.; Gaudernack, G.; Aamdal, S. Telomerase peptide vaccination in NSCLC: A phase II trial in stage III patients vaccinated after chemoradiotherapy and an 8-year update on a phase I/II trial. Clin. Cancer Res. 2011, 17, 6847–6857. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, H.S.; Grindey, G.B. Adriamycin and daunorubicin: A comparison of antitumor activities and tissue uptake in mice following immunosuppression. Cancer Res. 1973, 33, 1837–1844. [Google Scholar] [PubMed]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Godet, Y.; Dosset, M.; Borg, C.; Adotevi, O. Is preexisting antitumor CD4 T cell response indispensable for the chemotherapy induced immune regression of cancer? Oncoimmunology 2012, 1, 1617–1619. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Galaine, J.; Kellermann, G.; Guillaume, Y.; Boidot, R.; Picard, E.; Loyon, R.; Queiroz, L.; Boullerot, L.; Beziaud, L.; Jary, M.; et al. Heparan Sulfate Proteoglycans Promote Telomerase Internalization and MHC Class II Presentation on Dendritic Cells. J. Immunol. 2016, 197, 1597–1608. [Google Scholar] [CrossRef]
- Voutsas, I.F.; Anastasopoulou, E.A.; Tzonis, P.; Papamichail, M.; Perez, S.A.; Baxevanis, C.N. Unraveling the role of preexisting immunity in prostate cancer patients vaccinated with a HER-2/neu hybrid peptide. J. Immunother. Cancer 2016, 4, 75. [Google Scholar] [CrossRef]
- Arakawa, A.; Vollmer, S.; Tietze, J.; Galinski, A.; Heppt, M.V.; Bürdek, M.; Berking, C.; Prinz, J.C. Clonality of CD4+ Blood T Cells Predicts Longer Survival With CTLA4 or PD-1 Checkpoint Inhibition in Advanced Melanoma. Front. Immunol. 2019, 10, 1336. [Google Scholar] [CrossRef]
- Lauret Marie Joseph, E.; Laheurte, C.; Jary, M.; Boullerot, L.; Asgarov, K.; Gravelin, E.; Bouard, A.; Rangan, L.; Dosset, M.; Borg, C.; et al. Immunoregulation and Clinical Implications of ANGPT2/TIE2+ M-MDSC Signature in Non-Small Cell Lung Cancer. Cancer Immunol. Res. 2020, 8, 268–279. [Google Scholar] [CrossRef]
- Yost, K.E.; Satpathy, A.T.; Wells, D.K.; Qi, Y.; Wang, C.; Kageyama, R.; McNamara, K.L.; Granja, J.M.; Sarin, K.Y.; Brown, R.A.; et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 2019, 25, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Orillard, E.; Boullerot, L.; Laheurte, C.; Martin, A.; Jacquin, M.; Berthod, D.; Ramseyer, M.; Jacoulet, P.; Lahourcade, J.; Gainet Brun, M.; et al. Association of reinvigoration of circulating anti-telomerase CD4 Th1 response in cancer patients with anti-PD-1 response. JCO 2020, 38, 3044–3044. [Google Scholar] [CrossRef]
- Schlapbach, C.; Yerly, D.; Daubner, B.; Yawalkar, N.; Hunger, R.E. Telomerase-specific GV1001 peptide vaccination fails to induce objective tumor response in patients with cutaneous T cell lymphoma. J. Dermatol. Sci. 2011, 62, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, S.L.; Gjertsen, M.K.; Trachsel, S.; Møller, M.; Eriksen, J.A.; Meo, M.; Buanes, T.; Gaudernack, G. Telomerase peptide vaccination of patients with non-resectable pancreatic cancer: A dose escalating phase I/II study. Br. J. Cancer 2006, 95, 1474–1482. [Google Scholar] [CrossRef] [PubMed]
- Staff, C.; Mozaffari, F.; Frödin, J.-E.; Mellstedt, H.; Liljefors, M. Telomerase (GV1001) vaccination together with gemcitabine in advanced pancreatic cancer patients. Int. J. Oncol. 2014, 45, 1293–1303. [Google Scholar] [CrossRef]
- Thalmensi, J.; Pliquet, E.; Liard, C.; Escande, M.; Bestetti, T.; Julithe, M.; Kostrzak, A.; Pailhes-Jimenez, A.-S.; Bourges, E.; Loustau, M.; et al. Anticancer DNA vaccine based on human telomerase reverse transcriptase generates a strong and specific T cell immune response. Oncoimmunology 2016, 5, e1083670. [Google Scholar] [CrossRef]
- Teixeira, L.; Medioni, J.; Garibal, J.; Adotevi, O.; Doucet, L.; Durey, M.-A.D.; Ghrieb, Z.; Kiladjian, J.-J.; Brizard, M.; Laheurte, C.; et al. A First-in-Human Phase I Study of INVAC-1, an Optimized Human Telomerase DNA Vaccine in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2020, 26, 588–597. [Google Scholar] [CrossRef]
- Slingluff, C.L. Building on the Promise of Cancer Vaccines for Solid Tumors. Clin. Cancer Res. 2020, 26, 529–531. [Google Scholar] [CrossRef]
- Fenoglio, D.; Parodi, A.; Lavieri, R.; Kalli, F.; Ferrera, F.; Tagliamacco, A.; Guastalla, A.; Lamperti, M.G.; Giacomini, M.; Filaci, G. Immunogenicity of GX301 cancer vaccine: Four (telomerase peptides) are better than one. Hum. Vaccines Immunother. 2015, 11, 838–850. [Google Scholar] [CrossRef]
- Gattinoni, L.; Lugli, E.; Ji, Y.; Pos, Z.; Paulos, C.M.; Quigley, M.F.; Almeida, J.R.; Gostick, E.; Yu, Z.; Carpenito, C.; et al. A human memory T cell subset with stem cell-like properties. Nat. Med. 2011, 17, 1290–1297. [Google Scholar] [CrossRef]
- Gattinoni, L.; Speiser, D.E.; Lichterfeld, M.; Bonini, C. T memory stem cells in health and disease. Nat. Med. 2017, 23, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Nizard, M.; Roussel, H.; Diniz, M.O.; Karaki, S.; Tran, T.; Voron, T.; Dransart, E.; Sandoval, F.; Riquet, M.; Rance, B.; et al. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat. Commun. 2017, 8, 15221. [Google Scholar] [CrossRef] [PubMed]
- Gálvez-Cancino, F.; López, E.; Menares, E.; Díaz, X.; Flores, C.; Cáceres, P.; Hidalgo, S.; Chovar, O.; Alcántara-Hernández, M.; Borgna, V.; et al. Vaccination-induced skin-resident memory CD8+ T cells mediate strong protection against cutaneous melanoma. Oncoimmunology 2018, 7, e1442163. [Google Scholar] [CrossRef]
- Edwards, J.; Wilmott, J.S.; Madore, J.; Gide, T.N.; Quek, C.; Tasker, A.; Ferguson, A.; Chen, J.; Hewavisenti, R.; Hersey, P.; et al. CD103+ Tumor-Resident CD8+ T Cells Are Associated with Improved Survival in Immunotherapy-Naïve Melanoma Patients and Expand Significantly During Anti-PD-1 Treatment. Clin. Cancer Res. 2018, 24, 3036–3045. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, I.; Schaeuble, K.; Chennupati, V.; Fuertes Marraco, S.A.; Calderon-Copete, S.; Pais Ferreira, D.; Carmona, S.J.; Scarpellino, L.; Gfeller, D.; Pradervand, S.; et al. Intratumoral Tcf1+PD-1+CD8+ T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 2019, 50, 195–211.e10. [Google Scholar] [CrossRef]
- Kurtulus, S.; Madi, A.; Escobar, G.; Klapholz, M.; Nyman, J.; Christian, E.; Pawlak, M.; Dionne, D.; Xia, J.; Rozenblatt-Rosen, O.; et al. Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1-CD8+ Tumor-Infiltrating T Cells. Immunity 2019, 50, 181–194.e6. [Google Scholar] [CrossRef] [PubMed]
- Gide, T.N.; Quek, C.; Menzies, A.M.; Tasker, A.T.; Shang, P.; Holst, J.; Madore, J.; Lim, S.Y.; Velickovic, R.; Wongchenko, M.; et al. Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell 2019, 35, 238–255.e6. [Google Scholar] [CrossRef]
- Jensen, K.K.; Andreatta, M.; Marcatili, P.; Buus, S.; Greenbaum, J.A.; Yan, Z.; Sette, A.; Peters, B.; Nielsen, M. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 2018, 154, 394–406. [Google Scholar] [CrossRef]
- Wong, M.S.; Wright, W.E.; Shay, J.W. Alternative splicing regulation of telomerase: A new paradigm? Trends Genet. 2014, 30, 430–438. [Google Scholar] [CrossRef]
- Liu, X.; Wang, Y.; Chang, G.; Wang, F.; Wang, F.; Geng, X. Alternative Splicing of hTERT Pre-mRNA: A Potential Strategy for the Regulation of Telomerase Activity. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Position | Sequence | Main HLA Restriction | Year | Ref. |
|---|---|---|---|---|---|
| p68 | TERT68-82 | APSFRQVSCLKELVA | HLA-DR | 2018 | [108] |
| p911 1 | TERT911-927 | DEALGGTAFVQMPAH | HLA-DP4 | 2016 | [107] |
| UCP1 | TERT44-58 | PAAFRALVAQCLVCV | HLA-DR | 2012 | [105,113] |
| UCP2 | TERT578-592 | KSVWSKLQSIGIRQH | HLA-DR | ||
| UCP3 1 | TERT916-930 | GTAFVQMPAHGLFPW | HLA-DR | ||
| UCP4 | TERT1041-1055 | SLCYSILKAKNAGMS | HLA-DR | ||
| p541 | TERT541-555 | LAKFLHWLMSVYVVE | HLA-DP4 | 2011 | [102,103] |
| p573 | TERT573-587 | LFFYRKSVWSKLQSI | HLA-DP4 | ||
| p613 2 | TERT613-627 | RPALLTSRLRFIPKP | HLA-DP4 | ||
| p386 | TERT386-400 | YWQMRPLFLELLGNH | HLA-DP4 | 2011 | [104] |
| p660 3 | TERT660-689 | ALFSVLNYERARRPGLLGASVLGLDDIHRA | HLA-DR | ||
| p6633 | TERT663-677 | SVLNYERARRPGLLG | HLA-DR | ||
| p673 3 | TERT673-687 | PGLLGASVLGLDDIH | HLA-DR | ||
| GV1001 2 | TERT611–626 | EARPALLTSRLRFIPK | HLA-DP4 | 2006 | [101] |
| p766 | TERT766-780 | LTDLQPYMRQFVAHL | HLA-DR | 2003 | [100] |
| p6723 | TERT672-686 | RPGLLGASVLGLDDI | HLA-DR | 2002 | [99] |
| Cancer type | Treatment | Responders | Overall Survival and anti-TERT CD4 T Cell Response at: | Ref. | ||
|---|---|---|---|---|---|---|
| Baseline | Post-Treatment | Baseline | Post-Treatment | |||
| Metastatic non-small cell lung cancer (NSCLC) | Platinum-based chemo therapies | 38% (32/84) | ND | Patients with CD: median OS 53 months in TERT responders vs. 40 months in non-responders (p = 0.049) | - | [105] |
| Non-small cell lung cancer (NSCLC) | Platinum-based chemo therapies | 45% (39/87) of localized 24% (20/83) of metastatic | ND | Two-year OS rate of 59% in anti-TERT Th1highvs. 22% in anti-TERT Th1low (p = 0.006). Similar significant differences in localized and metastatic disease analyzed separately | - | [110] |
| Metastatic Renal cell carcinoma (mRCC) | Rapalog everolimus | 48% (11/23) | 74% (17/23) two months after treatment | ND | Better PFS achieved in patients with increased anti-TERT Th1 immunity and reduced Treg | [112] |
| Metastatic anal squamous cell carcinoma | Docetaxel, cisplatin and fluorouracil (DCF) | 27% (17/64) | 32% (16/50) one month after the last DCF cycle | Median PFS p = 0.059) | One-year PFS rate of 62.5% in TERT responders vs. 23.5 % in non-responders, (p = 0.017) | [111] |
| TERT MHC-II Based Therapy | Cancer | Phase | Estimated Enrollment | Status | ID |
|---|---|---|---|---|---|
| UCPVax: pool UCPs peptides | Metastatic NSCLC | I/II | 54 | Recruiting | NCT02818426 |
| UCPVax -Glio: pool UCPs peptides | Glioblastoma | I/II | 28 | Recruiting | NCT04280848 |
| Optim-UCPVax: pool UCPs + Nivolumab (anti-PD-1) | Advanced NSCLC | II | 111 | Not yet recruiting | NCT04263051 |
| VolATIL: pool UCPs (UCPVax) + Atezolizumab (anti-PD-L1) | Squamous Cell Carcinoma Cervical cancer Advanced Anal Carcinoma | II | 47 | Not yet recruiting | NCT03946358 |
| GV1001 + Gemcitabine + Capecitabine | Pancreatic cancer | III | 148 | Unknown | NCT02854072 |
| INVAC-1: modified TERT DNA plasmid | Chronic Lymphocytic Leukemia | II | 90 | Recruiting | NCT03265717 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dosset, M.; Castro, A.; Carter, H.; Zanetti, M. Telomerase and CD4 T Cell Immunity in Cancer. Cancers 2020, 12, 1687. https://doi.org/10.3390/cancers12061687
Dosset M, Castro A, Carter H, Zanetti M. Telomerase and CD4 T Cell Immunity in Cancer. Cancers. 2020; 12(6):1687. https://doi.org/10.3390/cancers12061687
Chicago/Turabian StyleDosset, Magalie, Andrea Castro, Hannah Carter, and Maurizio Zanetti. 2020. "Telomerase and CD4 T Cell Immunity in Cancer" Cancers 12, no. 6: 1687. https://doi.org/10.3390/cancers12061687
APA StyleDosset, M., Castro, A., Carter, H., & Zanetti, M. (2020). Telomerase and CD4 T Cell Immunity in Cancer. Cancers, 12(6), 1687. https://doi.org/10.3390/cancers12061687