Exploring the Therapeutic Potential of Membrane Transport Proteins: Focus on Cancer and Chemoresistance



Abstract
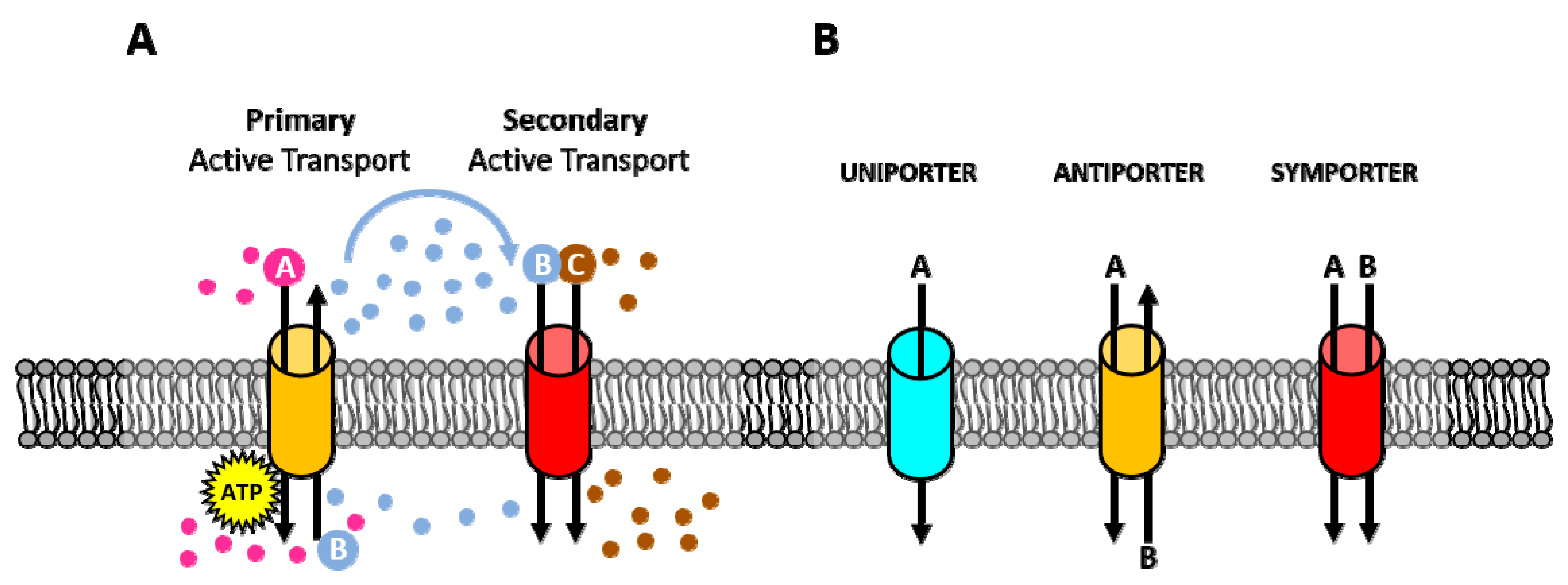
1. Introduction
2. Membrane Pumps
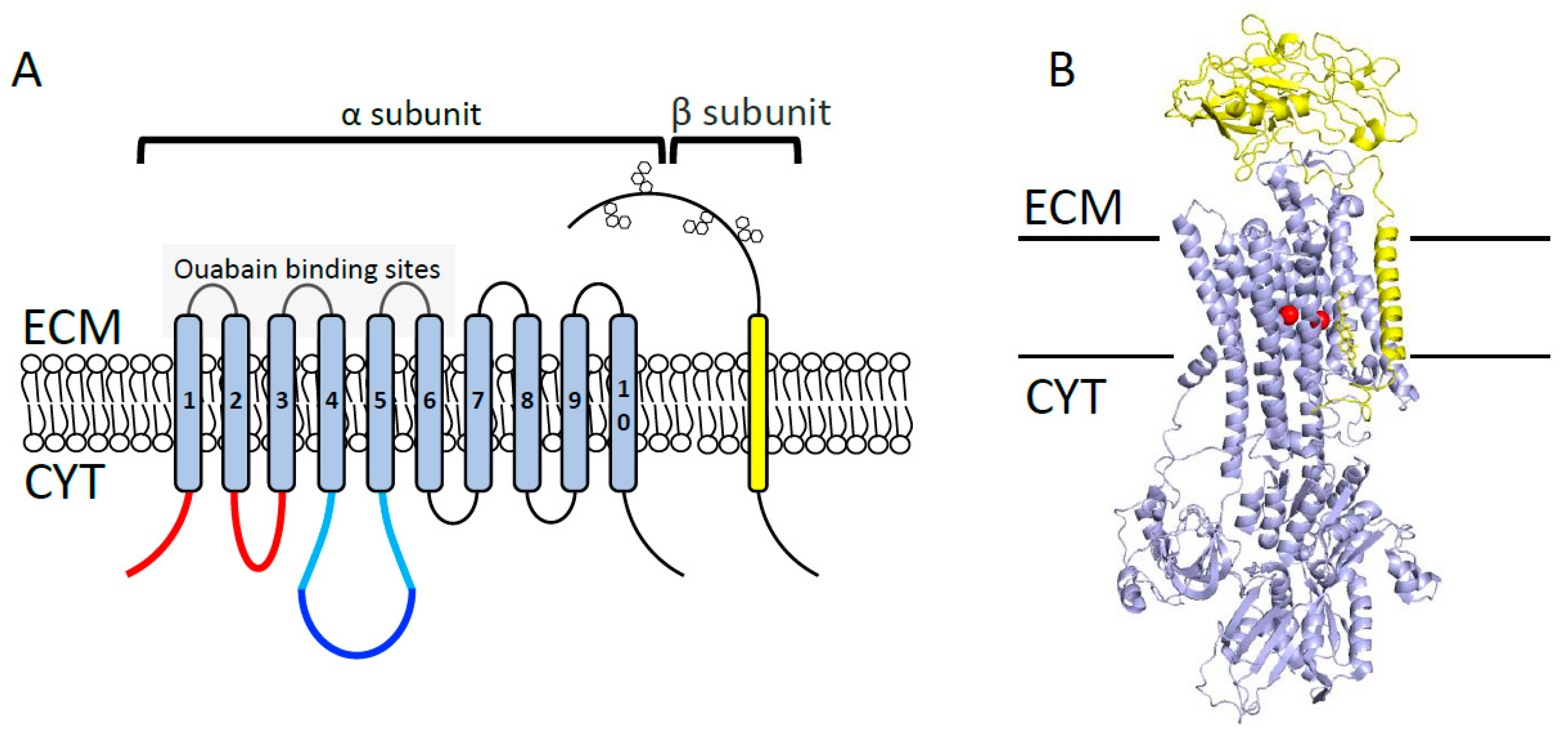
2.1. Na+/K+-ATPase
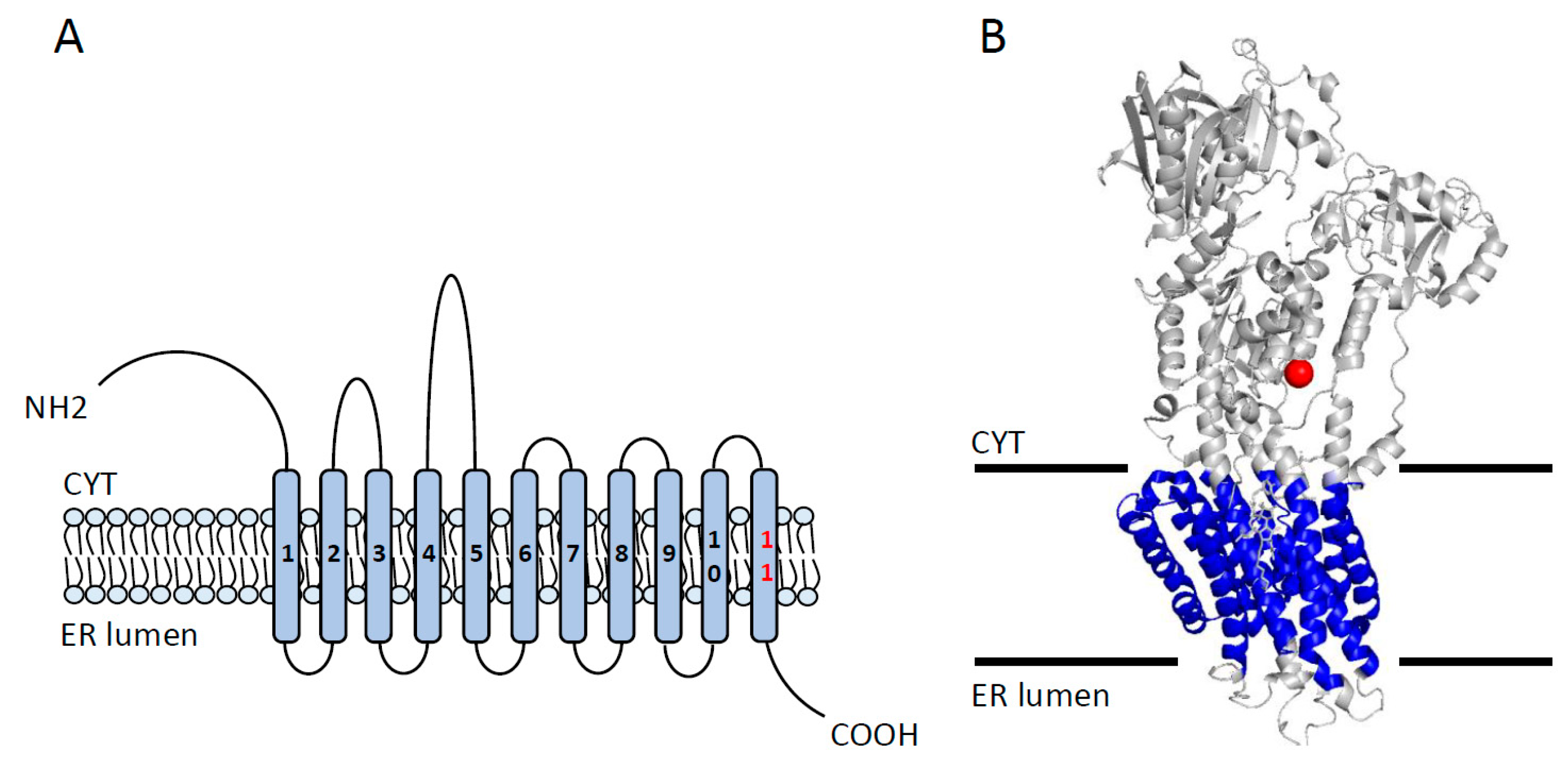
2.2. SERCA
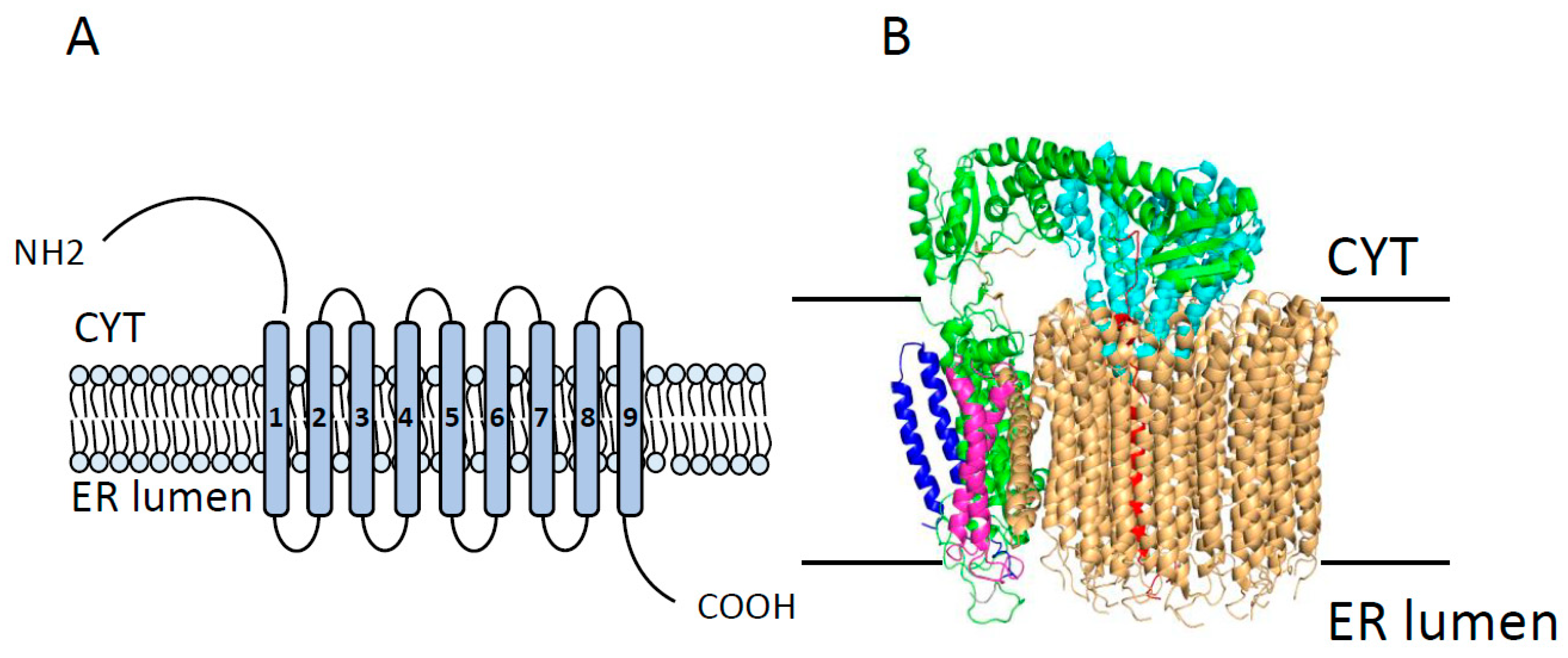
2.3. Vacuolar ATPase (V-ATPase)
3. Ion Channels
3.1. Ca2+ Channels
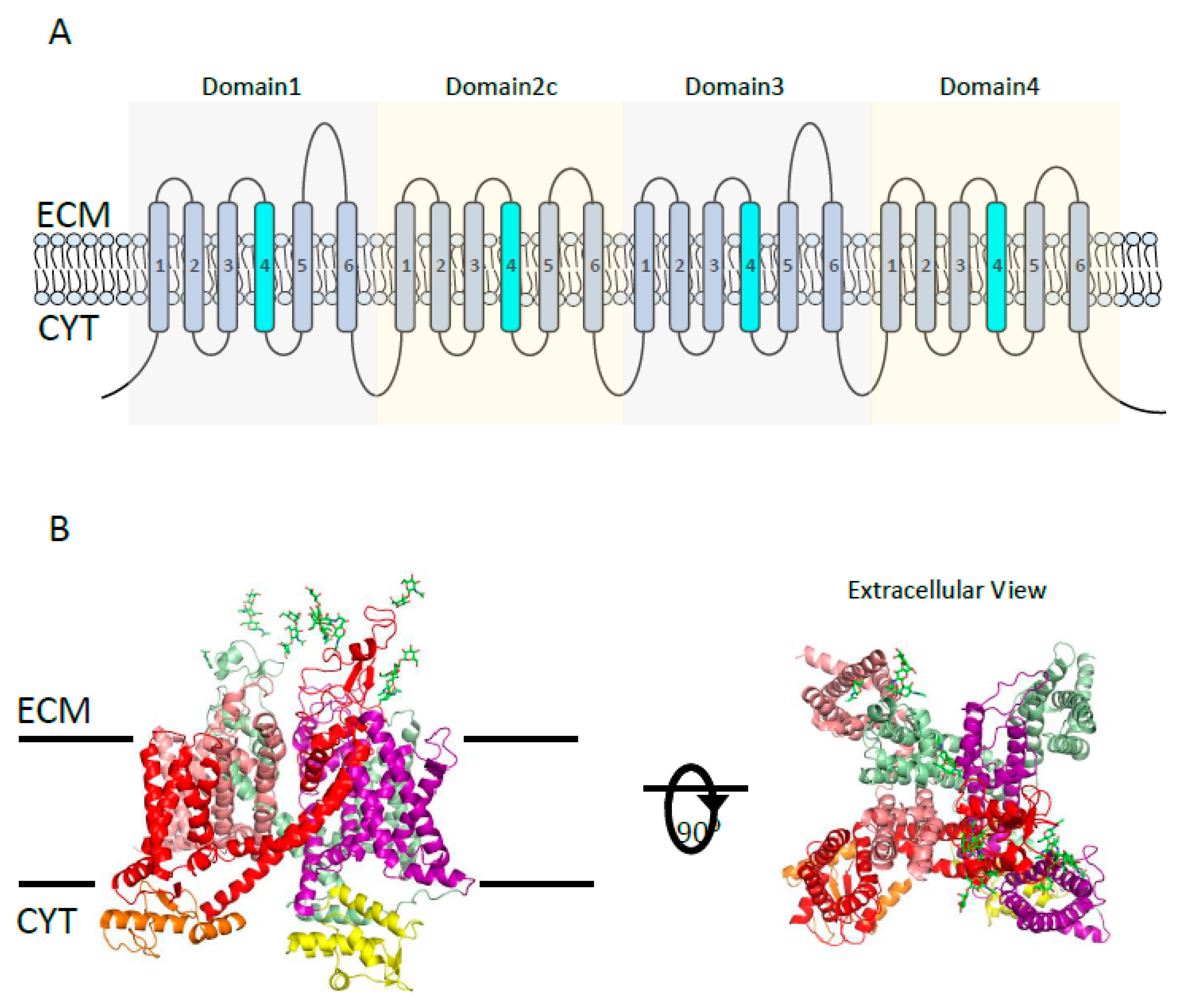
3.1.1. Voltage-Gated Ca2+ Channels (VGCC)
3.1.2. Orai-Mediated Store-Operated Ca2+ Entry
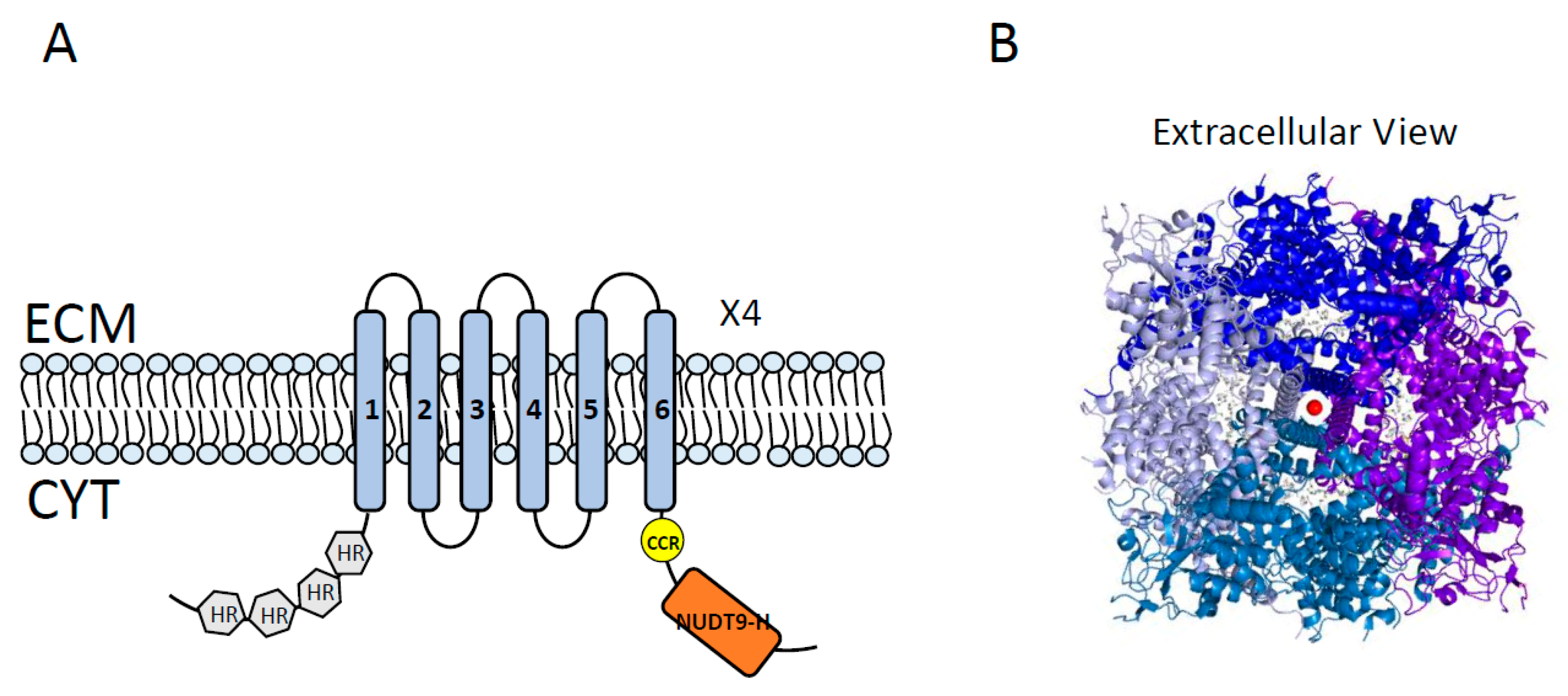
3.1.3. TRP-Mediated Ca2+ Transport
TRPC
TRPM
TRPV
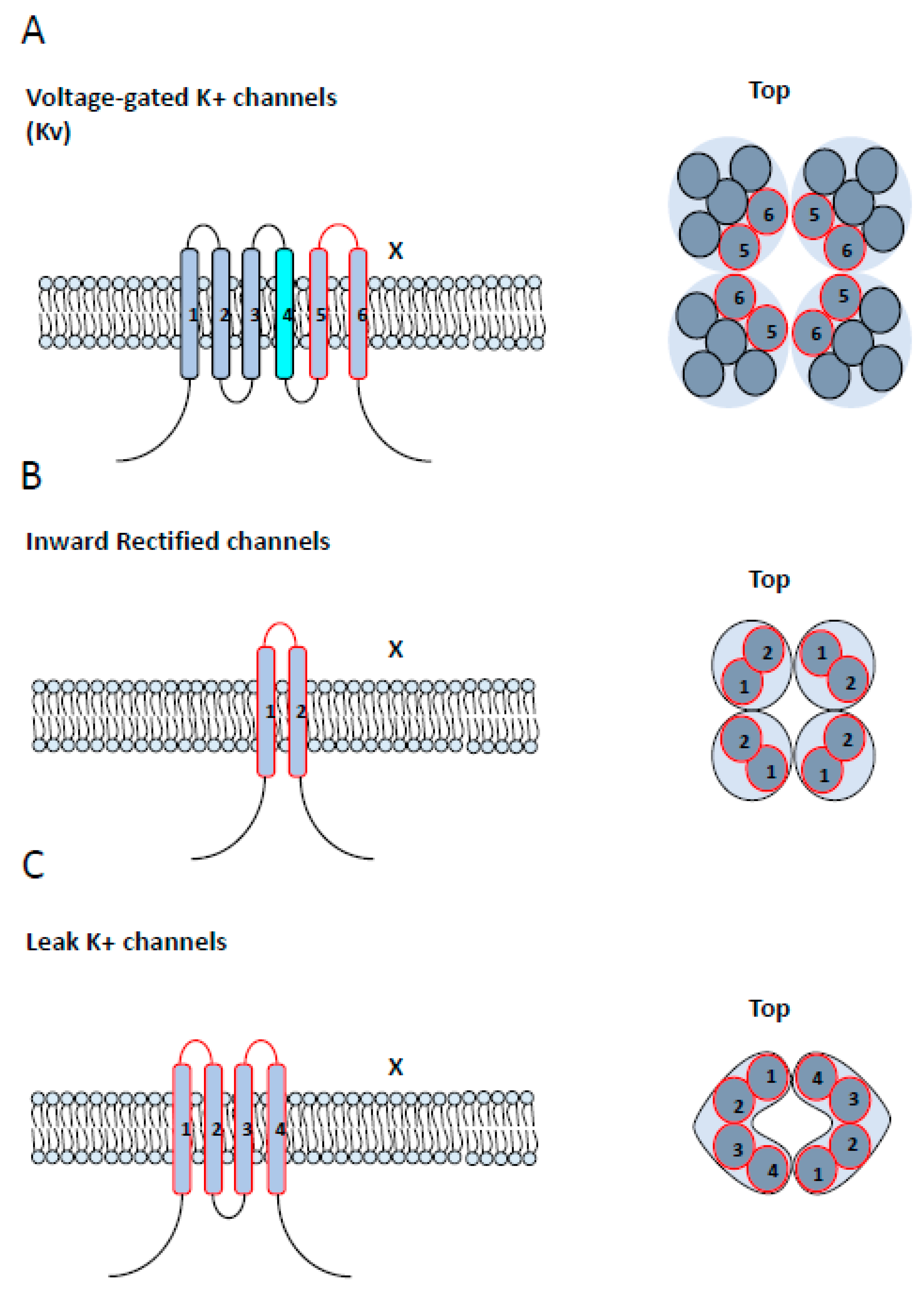
3.2. K+ Channels
3.3. Na+ Channels
3.3.1. VGSCs (NaV Channels)
3.3.2. LGSCs (NaL Channels)
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Membrane Transport Protein | Type of Cancer | Cell Lines | Level of Expression or Activity | Chemoresistance |
|---|---|---|---|---|
| Na+/K+ ATPase | Ovarian | 2000, CB10 | Low | Cisplatin [39], oxaliplatin [45] |
| Leukemia | NIH/3T3 | Cisplatin [40] | ||
| Lung | PC-14, SBC-1 | Cisplatin [41,42] | ||
| Prostate | LNCaP | Cisplatin [43] | ||
| Squamous | BHY | Cisplatin [45] | ||
| SERCA | Ovarian | MDAH-2774 | Low | Cisplatin [67] |
| Lung | H1339 | Cisplatin [68] | ||
| V-ATPase | Epidermoid | KB/PC4 | Hight | Cisplatin [104] |
| Prostate | P/CDP5 | Cisplatin [104] | ||
| Leukemia | HL-60 | Cisplatin, vincristine [105] | ||
| Melanoma | Mel P, Mel M | Cisplatin, vinblastine, & 5-FU [109] | ||
| Colon | Colo1, Colo2 | |||
| Breast | SKRB3, JIMT-1 | Trastuzumab [110] | ||
| CaV3 | Ovarian | A2780 | Hight | Carboplatin [133] |
| Prostate | LNCaP, PC3 | Thapsigargin, Paclitaxel [290] | ||
| Orai1 | Ovarian | A2780 | Hight | Cisplatin [143] |
| Hepatocellular | HepG2 | 5-FU [144] | ||
| Pancreas | Panc1 | 5-FU, gemcitabine [145] | ||
| Orai3 | Breast cancer | T47D | Hight | Cisplatin, 5-FU, paclitaxel [146] |
| TRPC1 | Ovarian | A2780, SKOV3 | Low | Cisplatin, Carboplatin [169] |
| TRPC5 | Colorectal | HCT-8, LoVo | High | 5-FU [170] |
| Breast | MCF7, T47D, MDA231 | Doxorubicin [171] | ||
| TRPC6 | Hepatocellular | Huh7 and HepG2 | High | Doxorubicin [173] |
| TRPM2 | Breast | MCF7, MDA231 | High | Doxorubicin, paclitaxel [195] |
| Neuroblastoma | SH-SY5Y | Doxorubicin [179] | ||
| Gastric | AGS, MKN45 | Doxorubicin, paclitaxel [178] | ||
| TRPM7 | Colon | LoVo | Low | Doxorubicin [196] |
| TRPM8 | Prostate | LNCaP | High | Paclitaxel [197] |
| Bone | MG-63, U2OS | Epirubicin [198] | ||
| TRPV1 | Breast | MCF7 | High | Cisplatin [225], Doxorubicin [226], 5-FU [224] |
| Bladder | 5637, T24 | Pirarubicin [227] | ||
| TRPV2 | Glioma | U87MG, MZC | Low | Carmustine, temozolomide, doxorubicin [230] |
| Melanoma | RPMI and U166 | Bortezomib [231] | ||
| TRPV6 | Prostate | LNCaP | High | Cisplatin, thapsigargin [232] |
| KCa1.1 | Ovarian | A2780 | Low | Cisplatin [250] |
| KCa3.1 | Epidermoid | KB, KCP-4 | Low | Cisplatin [252] |
| Kv1.5 | Gastric | SGC7901 | Low | Doxorubicin [253] |
| Kv10.1 | Breast | MDA-MB-435S | High | Doxorubicin, paclitaxel [254] |
| Kv11.1 | Colorectal | HCT116 | High | Cisplatin [255] |
| NaV | Breast cancer | MCF7, MDA231, MDA468, 4T1 | High | Cisplatin [274,275], Taxol [277] |
| Hepatocellular | HepG2 | Cisplatin [276] | ||
| H+-NaL – ASIC1a | Hepatocellular | BEL-7402, HepG2 | High | Doxorubicin, 5-FU [285] |
References
- Kulbacka, J.; Choromanska, A.; Rossowska, J.; Wezgowiec, J.; Saczko, J.; Rols, M.P. Cell membrane transport mechanisms: Ion channels and electrical properties of cell membranes. Adv. Anat. Embryol. Cell Biol. 2017, 227, 39–58. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion channels in cancer: Are cancer hallmarks oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef]
- Yamashita, N.; Hamada, H.; Tsuruo, T.; Ogata, E. Enhancement of voltage-gated Na+ channel current associated with multidrug resistance in human leukemia cells. Cancer Res. 1987, 47, 3736–3741. [Google Scholar]
- Lee, S.C.; Deutsch, C.; Beck, W.T. Comparison of ion channels in multidrug-resistant and -sensitive human leukemic cells. Proc. Natl. Acad. Sci. USA 1988, 85, 2019–2023. [Google Scholar] [CrossRef]
- Huang, Y.; Sadee, W. Membrane transporters and channels in chemoresistance and -sensitivity of tumor cells. Cancer Lett. 2006, 239, 168–182. [Google Scholar] [CrossRef]
- Huang, Y.; Anderle, P.; Bussey, K.J.; Barbacioru, C.; Shankavaram, U.; Dai, Z.; Reinhold, W.C.; Papp, A.; Weinstein, J.N.; Sadee, W. Membrane transporters and channels: Role of the transportome in cancer chemosensitivity and chemoresistance. Cancer Res. 2004, 64, 4294–4301. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion channels and the hallmarks of cancer. Trends Mol. Med. 2010, 16, 107–121. [Google Scholar] [CrossRef]
- Litan, A.; Langhans, S.A. Cancer as a channelopathy: Ion channels and pumps in tumor development and progression. Front. Cell. Neurosci. 2015, 9, 86. [Google Scholar] [CrossRef]
- Gadsby, D.C. Ion channels versus ion pumps: The principal difference, in principle. Nat. Rev. Mol. Cell Biol. 2009, 10, 344–352. [Google Scholar] [CrossRef]
- Konig, J.; Muller, F.; Fromm, M.F. Transporters and drug-drug interactions: Important determinants of drug disposition and effects. Pharm. Rev. 2013, 65, 944–966. [Google Scholar] [CrossRef]
- Poguntke, M.; Hazai, E.; Fromm, M.F.; Zolk, O. Drug transport by breast cancer resistance protein. Expert. Opin. Drug Metab. Toxicol. 2010, 6, 1363–1384. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T. Drug transporters as targets for cancer chemotherapy. Cancer Genom. Proteom. 2007, 4, 241–254. [Google Scholar]
- Bosch, T.M.; Meijerman, I.; Beijnen, J.H.; Schellens, J.H. Genetic polymorphisms of drug-metabolising enzymes and drug transporters in the chemotherapeutic treatment of cancer. Clin. Pharm. 2006, 45, 253–285. [Google Scholar] [CrossRef] [PubMed]
- Krisnamurti, D.G.; Louisa, M.; Anggraeni, E.; Wanandi, S.I. Drug efflux transporters are overexpressed in short-term tamoxifen-induced MCF7 breast cancer cells. Adv. Pharm. Sci. 2016, 2016, 6702424. [Google Scholar] [CrossRef] [PubMed]
- Vadlapatla, R.K.; Vadlapudi, A.D.; Pal, D.; Mitra, A.K. Mechanisms of drug resistance in cancer chemotherapy: Coordinated role and regulation of efflux transporters and metabolizing enzymes. Curr. Pharm. Des. 2013, 19, 7126–7140. [Google Scholar] [CrossRef]
- Shinoda, T.; Ogawa, H.; Cornelius, F.; Toyoshima, C. Crystal structure of the sodium-potassium pump at 2.4 A resolution. Nature 2009, 459, 446–450. [Google Scholar] [CrossRef]
- Morth, J.P.; Pedersen, B.P.; Toustrup-Jensen, M.S.; Sorensen, T.L.; Petersen, J.; Andersen, J.P.; Vilsen, B.; Nissen, P. Crystal structure of the sodium-potassium pump. Nature 2007, 450, 1043–1049. [Google Scholar] [CrossRef]
- Blanco, G. Na,K-ATPase subunit heterogeneity as a mechanism for tissue-specific ion regulation. Semin. Nephrol. 2005, 25, 292–303. [Google Scholar] [CrossRef]
- Castillo, J.P.; Rui, H.; Basilio, D.; Das, A.; Roux, B.; Latorre, R.; Bezanilla, F.; Holmgren, M. Mechanism of potassium ion uptake by the Na(+)/K(+)-ATPase. Nat. Commun. 2015, 6, 7622. [Google Scholar] [CrossRef]
- Skou, J.C.; Esmann, M. The Na,K-ATPase. J. Bioenerg. Biomembr. 1992, 24, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, H.; Shinoda, T.; Cornelius, F.; Toyoshima, C. Crystal structure of the sodium-potassium pump (Na+,K+-ATPase) with bound potassium and ouabain. Proc. Natl. Acad. Sci. USA 2009, 106, 13742–13747. [Google Scholar] [CrossRef]
- Lingrel, J.B.; Arguello, J.M.; Van Huysse, J.; Kuntzweiler, T.A. Cation and cardiac glycoside binding sites of the Na,K-ATPase. Ann. N. Y. Acad. Sci. 1997, 834, 194–206. [Google Scholar] [CrossRef]
- Rajasekaran, S.A.; Huynh, T.P.; Wolle, D.G.; Espineda, C.E.; Inge, L.J.; Skay, A.; Lassman, C.; Nicholas, S.B.; Harper, J.F.; Reeves, A.E.; et al. Na,K-ATPase subunits as markers for epithelial-mesenchymal transition in cancer and fibrosis. Mol. Cancer 2010, 9, 1515–1524. [Google Scholar] [CrossRef]
- Mijatovic, T.; Dufrasne, F.; Kiss, R. Na+/K+-ATPase and cancer. Pharm Pat. Anal. 2012, 1, 91–106. [Google Scholar] [CrossRef]
- Espineda, C.; Seligson, D.B.; James Ball, W., Jr.; Rao, J.; Palotie, A.; Horvath, S.; Huang, Y.; Shi, T.; Rajasekaran, A.K. Analysis of the Na,K-ATPase alpha- and beta-subunit expression profiles of bladder cancer using tissue microarrays. Cancer 2003, 97, 1859–1868. [Google Scholar] [CrossRef]
- Sakai, H.; Suzuki, T.; Maeda, M.; Takahashi, Y.; Horikawa, N.; Minamimura, T.; Tsukada, K.; Takeguchi, N. Up-regulation of Na(+),K(+)-ATPase alpha 3-isoform and down-regulation of the alpha1-isoform in human colorectal cancer. FEBS Lett. 2004, 563, 151–154. [Google Scholar] [CrossRef]
- Wang, Q.; Li, S.B.; Zhao, Y.Y.; Dai, D.N.; Du, H.; Lin, Y.Z.; Ye, J.C.; Zhao, J.; Xiao, W.; Mei, Y.; et al. Corrigendum to “Identification of a sodium pump Na+/K+ ATPase alpha1-targeted peptide for PET imaging of breast cancer” [Journal of Controlled Release 281C (2018) 178–188]. J. Control. Release 2019, 311, 324–325. [Google Scholar] [CrossRef]
- Lefranc, F.; Mijatovic, T.; Kondo, Y.; Sauvage, S.; Roland, I.; Debeir, O.; Krstic, D.; Vasic, V.; Gailly, P.; Kondo, S.; et al. Targeting the alpha 1 subunit of the sodium pump to combat glioblastoma cells. Neurosurgery 2008, 62, 211–221. [Google Scholar] [CrossRef]
- Lan, Y.L.; Zou, Y.J.; Lou, J.C.; Xing, J.S.; Wang, X.; Zou, S.; Ma, B.B.; Ding, Y.; Zhang, B. The sodium pump alpha1 subunit regulates bufalin sensitivity of human glioblastoma cells through the p53 signaling pathway. Cell Biol. Toxicol. 2019, 35, 521–539. [Google Scholar] [CrossRef]
- Lan, Y.L.; Yu, Z.L.; Lou, J.C.; Ma, X.C.; Zhang, B. Update on the effects of the sodium pump alpha1 subunit on human glioblastoma: From the laboratory to the clinic. Expert Opin. Investig. Drugs 2018, 27, 753–763. [Google Scholar] [CrossRef]
- Huang, X.; Lei, Z.; Li, X.P.; El-Mallakh, R.S. Response of sodium pump to ouabain challenge in human glioblastoma cells in culture. World J. Biol. Psychiatry 2009, 10, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Mobasheri, A.; Fox, R.; Evans, I.; Cullingham, F.; Martin-Vasallo, P.; Foster, C.S. Epithelial Na, K-ATPase expression is down-regulated in canine prostate cancer; a possible consequence of metabolic transformation in the process of prostate malignancy. Cancer Cell Int. 2003, 3, 8. [Google Scholar] [CrossRef]
- Mijatovic, T.; Roland, I.; Van Quaquebeke, E.; Nilsson, B.; Mathieu, A.; Van Vynckt, F.; Darro, F.; Blanco, G.; Facchini, V.; Kiss, R. The alpha1 subunit of the sodium pump could represent a novel target to combat non-small cell lung cancers. J. Pathol. 2007, 212, 170–179. [Google Scholar] [CrossRef]
- Rajasekaran, S.A.; Ball, W.J., Jr.; Bander, N.H.; Liu, H.; Pardee, J.D.; Rajasekaran, A.K. Reduced expression of beta-subunit of Na,K-ATPase in human clear-cell renal cell carcinoma. J. Urol. 1999, 162, 574–580. [Google Scholar] [CrossRef]
- Rajasekaran, S.A.; Palmer, L.G.; Quan, K.; Harper, J.F.; Ball, W.J., Jr.; Bander, N.H.; Peralta Soler, A.; Rajasekaran, A.K. Na,K-ATPase beta-subunit is required for epithelial polarization, suppression of invasion, and cell motility. Mol. Biol. Cell 2001, 12, 279–295. [Google Scholar] [CrossRef]
- Inge, L.J.; Rajasekaran, S.A.; Yoshimoto, K.; Mischel, P.S.; McBride, W.; Landaw, E.; Rajasekaran, A.K. Evidence for a potential tumor suppressor role for the Na,K-ATPase beta1-subunit. Histol. Histopathol. 2008, 23, 459–467. [Google Scholar] [CrossRef]
- Espineda, C.E.; Chang, J.H.; Twiss, J.; Rajasekaran, S.A.; Rajasekaran, A.K. Repression of Na,K-ATPase beta1-subunit by the transcription factor snail in carcinoma. Mol. Biol. Cell 2004, 15, 1364–1373. [Google Scholar] [CrossRef]
- Mijatovic, T.; Jungwirth, U.; Heffeter, P.; Hoda, M.A.; Dornetshuber, R.; Kiss, R.; Berger, W. The Na+/K+-ATPase is the Achilles heel of multi-drug-resistant cancer cells. Cancer Lett. 2009, 282, 30–34. [Google Scholar] [CrossRef]
- Andrews, P.A.; Mann, S.C.; Huynh, H.H.; Albright, K.D. Role of the Na+, K(+)-adenosine triphosphatase in the accumulation of cis-diamminedichloroplatinum(II) in human ovarian carcinoma cells. Cancer Res. 1991, 51, 3677–3681. [Google Scholar]
- Shinohara, N.; Ogiso, Y.; Arai, T.; Takami, S.; Nonomura, K.; Koyanagi, T.; Kuzumaki, N. Differential Na+,K(+)-ATPase activity and cisplatin sensitivity between transformants induced by H-ras and those induced by K-ras. Int. J. Cancer 1994, 58, 672–677. [Google Scholar] [CrossRef]
- Ohmori, T.; Nishio, K.; Ohta, S.; Kubota, N.; Adachi, M.; Komiya, K.; Saijo, N. Ouabain-resistant non-small-cell lung-cancer cell line shows collateral sensitivity to cis-diamminedichloroplatinum(II) (CDDP). Int. J. Cancer 1994, 57, 111–116. [Google Scholar] [CrossRef]
- Bando, T.; Fujimura, M.; Kasahara, K.; Matsuda, T. Significance of Na+,K(+)-ATPase on intracellular accumulation of cis-diamminedichloroplatinum(II) in human non-small-cell but not in small-cell lung cancer cell lines. Anticancer Res. 1998, 18, 1085–1089. [Google Scholar] [PubMed]
- Blok, L.J.; Chang, G.T.; Steenbeek-Slotboom, M.; van Weerden, W.M.; Swarts, H.G.; De Pont, J.J.; van Steenbrugge, G.J.; Brinkmann, A.O. Regulation of expression of Na+,K+-ATPase in androgen-dependent and androgen-independent prostate cancer. Br. J. Cancer 1999, 81, 28–36. [Google Scholar] [CrossRef]
- Ahmed, Z.; Deyama, Y.; Yoshimura, Y.; Suzuki, K. Cisplatin sensitivity of oral squamous carcinoma cells is regulated by Na+,K+-ATPase activity rather than copper-transporting P-type ATPases, ATP7A and ATP7B. Cancer Chemother. Pharm. 2009, 63, 643–650. [Google Scholar] [CrossRef]
- Tummala, R.; Wolle, D.; Barwe, S.P.; Sampson, V.B.; Rajasekaran, A.K.; Pendyala, L. Expression of Na,K-ATPase-beta(1) subunit increases uptake and sensitizes carcinoma cells to oxaliplatin. Cancer Chemother. Pharm. 2009, 64, 1187–1194. [Google Scholar] [CrossRef]
- Toyoshima, C.; Nakasako, M.; Nomura, H.; Ogawa, H. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 A resolution. Nature 2000, 405, 647–655. [Google Scholar] [CrossRef]
- Altshuler, I.; Vaillant, J.J.; Xu, S.; Cristescu, M.E. The evolutionary history of sarco(endo)plasmic calcium ATPase (SERCA). PLoS ONE 2012, 7, e52617. [Google Scholar] [CrossRef]
- Dally, S.; Corvazier, E.; Bredoux, R.; Bobe, R.; Enouf, J. Multiple and diverse coexpression, location, and regulation of additional SERCA2 and SERCA3 isoforms in nonfailing and failing human heart. J. Mol. Cell. Cardiol. 2010, 48, 633–644. [Google Scholar] [CrossRef]
- Inoue, M.; Sakuta, N.; Watanabe, S.; Zhang, Y.; Yoshikaie, K.; Tanaka, Y.; Ushioda, R.; Kato, Y.; Takagi, J.; Tsukazaki, T.; et al. Structural basis of sarco/endoplasmic reticulum Ca(2+)-ATPase 2b regulation via transmembrane helix interplay. Cell Rep. 2019, 27, 1221–1230. [Google Scholar] [CrossRef]
- Periasamy, M.; Kalyanasundaram, A. SERCA pump isoforms: Their role in calcium transport and disease. Muscle Nerve 2007, 35, 430–442. [Google Scholar] [CrossRef]
- Higgins, E.R.; Cannell, M.B.; Sneyd, J. A buffering SERCA pump in models of calcium dynamics. Biophys. J. 2006, 91, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Luo, J.; Wang, H.; Chen, J. SERCA regulates collective cell migration by maintaining cytoplasmic Ca(2+) homeostasis. J. Genet. Genom. 2019, 46, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Periasamy, M.; Huke, S. SERCA pump level is a critical determinant of Ca(2+)homeostasis and cardiac contractility. J. Mol. Cell. Cardiol. 2001, 33, 1053–1063. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef]
- Papp, B.; Brouland, J.P.; Arbabian, A.; Gelebart, P.; Kovacs, T.; Bobe, R.; Enouf, J.; Varin-Blank, N.; Apati, A. Endoplasmic reticulum calcium pumps and cancer cell differentiation. Biomolecules 2012, 2, 165–186. [Google Scholar] [CrossRef]
- Prasad, V.; Boivin, G.P.; Miller, M.L.; Liu, L.H.; Erwin, C.R.; Warner, B.W.; Shull, G.E. Haploinsufficiency of Atp2a2, encoding the sarco(endo)plasmic reticulum Ca2+-ATPase isoform 2 Ca2+ pump, predisposes mice to squamous cell tumors via a novel mode of cancer susceptibility. Cancer Res. 2005, 65, 8655–8661. [Google Scholar] [CrossRef]
- Monteith, G.R.; McAndrew, D.; Faddy, H.M.; Roberts-Thomson, S.J. Calcium and cancer: Targeting Ca2+ transport. Nat. Rev. Cancer 2007, 7, 519–530. [Google Scholar] [CrossRef]
- Parkash, J.; Asotra, K. Calcium wave signaling in cancer cells. Life Sci. 2010, 87, 587–595. [Google Scholar] [CrossRef]
- Korosec, B.; Glavac, D.; Rott, T.; Ravnik-Glavac, M. Alterations in the ATP2A2 gene in correlation with colon and lung cancer. Cancer Genet. Cytogenet. 2006, 171, 105–111. [Google Scholar] [CrossRef]
- Pacifico, F.; Ulianich, L.; De Micheli, S.; Treglia, S.; Leonardi, A.; Vito, P.; Formisano, S.; Consiglio, E.; Di Jeso, B. The expression of the sarco/endoplasmic reticulum Ca2+-ATPases in thyroid and its down-regulation following neoplastic transformation. J. Mol. Endocrinol. 2003, 30, 399–409. [Google Scholar] [CrossRef]
- Chung, F.Y.; Lin, S.R.; Lu, C.Y.; Yeh, C.S.; Chen, F.M.; Hsieh, J.S.; Huang, T.J.; Wang, J.Y. Sarco/endoplasmic reticulum calcium-ATPase 2 expression as a tumor marker in colorectal cancer. Am. J. Surg. Pathol. 2006, 30, 969–974. [Google Scholar] [CrossRef]
- Xu, X.Y.; Gou, W.F.; Yang, X.; Wang, G.L.; Takahashi, H.; Yu, M.; Mao, X.Y.; Takano, Y.; Zheng, H.C. Aberrant SERCA3 expression is closely linked to pathogenesis, invasion, metastasis, and prognosis of gastric carcinomas. Tumour Biol. 2012, 33, 1845–1854. [Google Scholar] [CrossRef]
- Gelebart, P.; Kovacs, T.; Brouland, J.P.; van Gorp, R.; Grossmann, J.; Rivard, N.; Panis, Y.; Martin, V.; Bredoux, R.; Enouf, J.; et al. Expression of endomembrane calcium pumps in colon and gastric cancer cells. Induction of SERCA3 expression during differentiation. J. Biol. Chem. 2002, 277, 26310–26320. [Google Scholar] [CrossRef]
- Azeez, J.M.; Vini, R.; Remadevi, V.; Surendran, A.; Jaleel, A.; Santhosh Kumar, T.R.; Sreeja, S. VDAC1 and SERCA3 Mediate Progesterone-Triggered Ca2(+) Signaling in Breast Cancer Cells. J. Proteome Res. 2018, 17, 698–709. [Google Scholar] [CrossRef]
- Papp, B.; Brouland, J.P. Altered endoplasmic reticulum calcium pump expression during breast tumorigenesis. Breast Cancer Basic Clin. Res. 2011, 5, 163–174. [Google Scholar] [CrossRef]
- Launay, S.; Gianni, M.; Kovacs, T.; Bredoux, R.; Bruel, A.; Gelebart, P.; Zassadowski, F.; Chomienne, C.; Enouf, J.; Papp, B. Lineage-specific modulation of calcium pump expression during myeloid differentiation. Blood 1999, 93, 4395–4405. [Google Scholar] [CrossRef]
- Kucukkaya, B.; Basoglu, H.; Erdag, D.; Akbas, F.; Susgun, S.; Yalcintepe, L. Calcium homeostasis in cisplatin resistant epithelial ovarian cancer. Gen. Physiol. Biophys. 2019, 38, 353–363. [Google Scholar] [CrossRef]
- Schrodl, K.; Oelmez, H.; Edelmann, M.; Huber, R.M.; Bergner, A. Altered Ca2+-homeostasis of cisplatin-treated and low level resistant non-small-cell and small-cell lung cancer cells. Cell. Oncol. 2009, 31, 301–315. [Google Scholar] [CrossRef]
- Chemaly, E.R.; Troncone, L.; Lebeche, D. SERCA control of cell death and survival. Cell Calcium 2018, 69, 46–61. [Google Scholar] [CrossRef]
- Casemore, D.; Xing, C. SERCA as a target for cancer therapies. Integr. Cancer Sci. 2015, 2, 100–103. [Google Scholar]
- Izquierdo-Torres, E.; Hernandez-Oliveras, A.; Meneses-Morales, I.; Rodriguez, G.; Fuentes-Garcia, G.; Zarain-Herzberg, A. Resveratrol up-regulates ATP2A3 gene expression in breast cancer cell lines through epigenetic mechanisms. Int. J. Biochem. Cell Biol. 2019, 113, 37–47. [Google Scholar] [CrossRef]
- Fan, L.; Li, A.; Li, W.; Cai, P.; Yang, B.; Zhang, M.; Gu, Y.; Shu, Y.; Sun, Y.; Shen, Y.; et al. Novel role of Sarco/endoplasmic reticulum calcium ATPase 2 in development of colorectal cancer and its regulation by F36, a curcumin analog. Biomed. Pharm. 2014, 68, 1141–1148. [Google Scholar] [CrossRef]
- Aridoss, G.; Zhou, B.; Hermanson, D.L.; Bleeker, N.P.; Xing, C. Structure-activity relationship (SAR) study of ethyl 2-amino-6-(3,5-dimethoxyphenyl)-4-(2-ethoxy-2-oxoethyl)-4H-chromene-3-carboxylate (CXL017) and the potential of the lead against multidrug resistance in cancer treatment. J. Med. Chem. 2012, 55, 5566–5581. [Google Scholar] [CrossRef]
- Bleeker, N.P.; Cornea, R.L.; Thomas, D.D.; Xing, C. A novel SERCA inhibitor demonstrates synergy with classic SERCA inhibitors and targets multidrug-resistant AML. Mol. Pharm. 2013, 10, 4358–4366. [Google Scholar] [CrossRef][Green Version]
- Das, S.G.; Hermanson, D.L.; Bleeker, N.; Lowman, X.; Li, Y.; Kelekar, A.; Xing, C. Ethyl 2-Amino-6-(3, 5-dimethoxyphenyl)-4-(2-ethoxy-2-oxoethyl)-4 H-chromene-3-carboxylate (CXL017): A Novel Scaffold That Resensitizes Multidrug Resistant Leukemia Cells to Chemotherapy. ACS Chem. Biol. 2013, 8, 327–335. [Google Scholar] [CrossRef]
- Maxson, M.E.; Grinstein, S. The vacuolar-type H(+)-ATPase at a glance-more than a proton pump. J. Cell Sci. 2014, 127, 4987–4993. [Google Scholar] [CrossRef] [PubMed]
- Gruber, G.; Marshansky, V. New insights into structure-function relationships between archeal ATP synthase (A1A0) and vacuolar type ATPase (V1V0). Bioessays 2008, 30, 1096–1109. [Google Scholar] [CrossRef]
- Forgac, M. Structure and function of vacuolar class of ATP-driven proton pumps. Physiol. Rev. 1989, 69, 765–796. [Google Scholar] [CrossRef]
- Manolson, M.F.; Proteau, D.; Jones, E.W. Evidence for a conserved 95–120 kDa subunit associated with and essential for activity of V-ATPases. J. Exp. Biol. 1992, 172, 105–112. [Google Scholar]
- Manolson, M.F.; Proteau, D.; Preston, R.A.; Stenbit, A.; Roberts, B.T.; Hoyt, M.A.; Preuss, D.; Mulholland, J.; Botstein, D.; Jones, E.W. The VPH1 gene encodes a 95-kDa integral membrane polypeptide required for in vivo assembly and activity of the yeast vacuolar H(+)-ATPase. J. Biol. Chem. 1992, 267, 14294–14303. [Google Scholar]
- Cross, R.L.; Muller, V. The evolution of A-, F-, and V-type ATP synthases and ATPases: Reversals in function and changes in the H+/ATP coupling ratio. FEBS Lett. 2004, 576, 1–4. [Google Scholar] [CrossRef]
- Kettner, C.; Bertl, A.; Obermeyer, G.; Slayman, C.; Bihler, H. Electrophysiological analysis of the yeast V-type proton pump: Variable coupling ratio and proton shunt. Biophys. J. 2003, 85, 3730–3738. [Google Scholar] [CrossRef]
- Stransky, L.; Cotter, K.; Forgac, M. The function of v-ATPases in cancer. Physiol. Rev. 2016, 96, 1071–1091. [Google Scholar] [CrossRef]
- Sun-Wada, G.H.; Wada, Y. Role of vacuolar-type proton ATPase in signal transduction. Biochim. Biophys. Acta 2015, 1847, 1166–1172. [Google Scholar] [CrossRef]
- Pamarthy, S.; Kulshrestha, A.; Katara, G.K.; Beaman, K.D. The curious case of vacuolar ATPase: Regulation of signaling pathways. Mol. Cancer 2018, 17, 41. [Google Scholar] [CrossRef]
- Hinton, A.; Bond, S.; Forgac, M. V-ATPase functions in normal and disease processes. Pflügers Arch. Eur. J. Physiol. 2009, 457, 589–598. [Google Scholar] [CrossRef]
- Sennoune, S.R.; Martinez-Zaguilan, R. Vacuolar H(+)-ATPase signaling pathway in cancer. Curr. Protein Pept. Sci. 2012, 13, 152–163. [Google Scholar] [CrossRef]
- Song, T.; Jeon, H.K.; Hong, J.E.; Choi, J.J.; Kim, T.J.; Choi, C.H.; Bae, D.S.; Kim, B.G.; Lee, J.W. Proton pump inhibition enhances the cytotoxicity of paclitaxel in cervical cancer. Cancer Res. Treat. J. Korean Cancer Assoc. 2017, 49, 595–606. [Google Scholar] [CrossRef]
- Liu, P.; Chen, H.; Han, L.; Zou, X.; Shen, W. Expression and role of V1A subunit of V-ATPases in gastric cancer cells. Int. J. Clin. Oncol. 2015, 20, 725–735. [Google Scholar] [CrossRef]
- Cotter, K.; Liberman, R.; Sun-Wada, G.; Wada, Y.; Sgroi, D.; Naber, S.; Brown, D.; Breton, S.; Forgac, M. The a3 isoform of subunit a of the vacuolar ATPase localizes to the plasma membrane of invasive breast tumor cells and is overexpressed in human breast cancer. Oncotarget 2016, 7, 46142–46157. [Google Scholar] [CrossRef]
- Katara, G.K.; Kulshrestha, A.; Jaiswal, M.K.; Pamarthy, S.; Gilman-Sachs, A.; Beaman, K.D. Inhibition of vacuolar ATPase subunit in tumor cells delays tumor growth by decreasing the essential macrophage population in the tumor microenvironment. Oncogene 2016, 35, 1058–1065. [Google Scholar] [CrossRef]
- Katara, G.K.; Jaiswal, M.K.; Kulshrestha, A.; Kolli, B.; Gilman-Sachs, A.; Beaman, K.D. Tumor-associated vacuolar ATPase subunit promotes tumorigenic characteristics in macrophages. Oncogene 2014, 33, 5649–5654. [Google Scholar] [CrossRef]
- Chueca, E.; Apostolova, N.; Esplugues, J.V.; Garcia-Gonzalez, M.A.; Lanas, A.; Piazuelo, E. Proton pump inhibitors display antitumor effects in barrett’s adenocarcinoma cells. Front. Pharm. 2016, 7, 452. [Google Scholar] [CrossRef]
- Son, S.W.; Kim, S.H.; Moon, E.Y.; Kim, D.H.; Pyo, S.; Um, S.H. Prognostic significance and function of the vacuolar H+-ATPase subunit V1E1 in esophageal squamous cell carcinoma. Oncotarget 2016, 7, 49334–49348. [Google Scholar] [CrossRef]
- Ohta, T.; Numata, M.; Yagishita, H.; Futagami, F.; Tsukioka, Y.; Kitagawa, H.; Kayahara, M.; Nagakawa, T.; Miyazaki, I.; Yamamoto, M.; et al. Expression of 16 kDa proteolipid of vacuolar-type H(+)-ATPase in human pancreatic cancer. Br. J. Cancer 1996, 73, 1511–1517. [Google Scholar] [CrossRef]
- Capecci, J.; Forgac, M. The function of vacuolar ATPase (V-ATPase) a subunit isoforms in invasiveness of MCF10a and MCF10CA1a human breast cancer cells. J. Biol. Chem. 2013, 288, 32731–32741. [Google Scholar] [CrossRef]
- Cotter, K.; Capecci, J.; Sennoune, S.; Huss, M.; Maier, M.; Martinez-Zaguilan, R.; Forgac, M. Activity of plasma membrane V-ATPases is critical for the invasion of MDA-MB231 breast cancer cells. J. Biol. Chem. 2015, 290, 3680–3692. [Google Scholar] [CrossRef]
- Michel, V.; Licon-Munoz, Y.; Trujillo, K.; Bisoffi, M.; Parra, K.J. Inhibitors of vacuolar ATPase proton pumps inhibit human prostate cancer cell invasion and prostate-specific antigen expression and secretion. Int. J. Cancer 2013, 132, E1–E10. [Google Scholar] [CrossRef]
- Nishisho, T.; Hata, K.; Nakanishi, M.; Morita, Y.; Sun-Wada, G.H.; Wada, Y.; Yasui, N.; Yoneda, T. The a3 isoform vacuolar type H(+)-ATPase promotes distant metastasis in the mouse B16 melanoma cells. Mol. Cancer Res. 2011, 9, 845–855. [Google Scholar] [CrossRef]
- Kubisch, R.; Frohlich, T.; Arnold, G.J.; Schreiner, L.; von Schwarzenberg, K.; Roidl, A.; Vollmar, A.M.; Wagner, E. V-ATPase inhibition by archazolid leads to lysosomal dysfunction resulting in impaired cathepsin B activation in vivo. Int. J. Cancer 2014, 134, 2478–2488. [Google Scholar] [CrossRef]
- Hendrix, A.; Sormunen, R.; Westbroek, W.; Lambein, K.; Denys, H.; Sys, G.; Braems, G.; Van den Broecke, R.; Cocquyt, V.; Gespach, C.; et al. Vacuolar H+ ATPase expression and activity is required for Rab27B-dependent invasive growth and metastasis of breast cancer. Int. J. Cancer 2013, 133, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Whitton, B.; Okamoto, H.; Packham, G.; Crabb, S.J. Vacuolar ATPase as a potential therapeutic target and mediator of treatment resistance in cancer. Cancer Med. 2018, 7, 3800–3811. [Google Scholar] [CrossRef] [PubMed]
- Torigoe, T.; Izumi, H.; Ishiguchi, H.; Uramoto, H.; Murakami, T.; Ise, T.; Yoshida, Y.; Tanabe, M.; Nomoto, M.; Itoh, H.; et al. Enhanced expression of the human vacuolar H+-ATPase c subunit gene (ATP6L) in response to anticancer agents. J. Biol. Chem. 2002, 277, 36534–36543. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Shibuya, I.; Ise, T.; Chen, Z.S.; Akiyama, S.; Nakagawa, M.; Izumi, H.; Nakamura, T.; Matsuo, K.; Yamada, Y.; et al. Elevated expression of vacuolar proton pump genes and cellular PH in cisplatin resistance. Int. J. Cancer 2001, 93, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Center, M.S. The gene encoding vacuolar H+-ATPase subunit C is overexpressed in multidrug resistant HL60 cells. Biochem. Biophys. Res. Commun. 1992, 182, 675–681. [Google Scholar] [CrossRef]
- Ouar, Z.; Bens, M.; Vignes, C.; Paulais, M.; Pringel, C.; Fleury, J.; Cluzeaud, F.; Lacave, R.; Vandewalle, A. Inhibitors of vacuolar H+-ATPase impair the preferential accumulation of daunomycin in lysosomes and reverse the resistance to anthracyclines in drug-resistant renal epithelial cells. Biochem. J. 2003, 370, 185–193. [Google Scholar] [CrossRef]
- Supino, R.; Scovassi, A.I.; Croce, A.C.; Dal Bo, L.; Favini, E.; Corbelli, A.; Farina, C.; Misiano, P.; Zunino, F. Biological effects of a new vacuolar-H,-ATPase inhibitor in colon carcinoma cell lines. Ann. N. Y. Acad. Sci. 2009, 1171, 606–616. [Google Scholar] [CrossRef]
- Petrangolini, G.; Supino, R.; Pratesi, G.; Dal Bo, L.; Tortoreto, M.; Croce, A.C.; Misiano, P.; Belfiore, P.; Farina, C.; Zunino, F. Effect of a novel vacuolar-H+-ATPase inhibitor on cell and tumor response to camptothecins. J. Pharm. Exp. 2006, 318, 939–946. [Google Scholar] [CrossRef]
- Luciani, F.; Spada, M.; De Milito, A.; Molinari, A.; Rivoltini, L.; Montinaro, A.; Marra, M.; Lugini, L.; Logozzi, M.; Lozupone, F.; et al. Effect of proton pump inhibitor pretreatment on resistance of solid tumors to cytotoxic drugs. J. Natl. Cancer Inst. 2004, 96, 1702–1713. [Google Scholar] [CrossRef]
- von Schwarzenberg, K.; Lajtos, T.; Simon, L.; Müller, R.; Vereb, G.; Vollmar, A.M. V-ATPase inhibition overcomes trastuzumab resistance in breast cancer. Mol. Oncol. 2014, 8, 9–19. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Ion channels and the electrical properties of membranes. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- North, R.A. Ligand-and Voltage-Gated Ion Channels; CRC Press: Boca Raton, FL, USA, 1995. [Google Scholar]
- Bates, E. Ion channels in development and cancer. Annu. Rev. Cell Dev. Biol. 2015, 31, 231–247. [Google Scholar] [CrossRef] [PubMed]
- Leanza, L.; Biasutto, L.; Managò, A.; Gulbins, E.; Zoratti, M.; Szabò, I. Intracellular ion channels and cancer. Front. Physiol. 2013, 4, 227. [Google Scholar] [CrossRef] [PubMed]
- Peruzzo, R.; Biasutto, L.; Szabò, I.; Leanza, L. Impact of intracellular ion channels on cancer development and progression. Eur. Biophys. J. 2016, 45, 685–707. [Google Scholar] [CrossRef] [PubMed]
- Sterea, A.M.; Almasi, S.; El Hiani, Y. The hidden potential of lysosomal ion channels: A new era of oncogenes. Cell Calcium 2018, 72, 91–103. [Google Scholar] [CrossRef]
- Sterea, A.M.; El Hiani, Y. The role of mitochondrial calcium signaling in the pathophysiology of cancer cells. In Calcium Signaling; Springer: Berlin, Germany, 2020; pp. 747–770. [Google Scholar]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Kim, J. Emerging role of transient receptor potential (TRP) channels in cancer progression. BMB Rep. 2020, 53, 125–132. [Google Scholar] [CrossRef]
- Chalmers, S.B.; Monteith, G.R. ORAI channels and cancer. Cell Calcium 2018, 74, 160–167. [Google Scholar] [CrossRef]
- Catterall, W.A. Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947. [Google Scholar] [CrossRef]
- Fiske, J.L.; Fomin, V.P.; Brown, M.L.; Duncan, R.L.; Sikes, R.A. Voltage-sensitive ion channels and cancer. Cancer Metastasis Rev. 2006, 25, 493–500. [Google Scholar] [CrossRef]
- Rao, V.R.; Perez-Neut, M.; Kaja, S.; Gentile, S. Voltage-gated ion channels in cancer cell proliferation. Cancers 2015, 7, 849–875. [Google Scholar] [CrossRef]
- Xiao, X.; Li, B.X.; Mitton, B.; Ikeda, A.; Sakamoto, K.M. Targeting CREB for cancer therapy: Friend or foe. Curr. Cancer Drug Targets 2010, 10, 384–391. [Google Scholar] [CrossRef]
- Buchanan, P.J.; McCloskey, K.D. Ca V channels and cancer: Canonical functions indicate benefits of repurposed drugs as cancer therapeutics. Eur. Biophys. J. 2016, 45, 621–633. [Google Scholar] [CrossRef]
- Shi, Z.-Z.; Shang, L.; Jiang, Y.-Y.; Shi, F.; Xu, X.; Wang, M.-R.; Hao, J.-J. Identification of genomic biomarkers associated with the clinicopathological parameters and prognosis of esophageal squamous cell carcinoma. Cancer Biomark. 2015, 15, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Bao, X.; Jin, B.; Wang, X.; Mao, Z.; Li, X.; Wei, L.; Shen, D.; Wang, J.-L. Ca2+ channel subunit α 1D promotes proliferation and migration of endometrial cancer cells mediated by 17β-estradiol via the G protein-coupled estrogen receptor. FASEB J. 2015, 29, 2883–2893. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zeng, X.; Zhang, R.; Huang, J.; Kuang, X.; Yang, J.; Liu, J.; Tawfik, O.; Thrasher, J.B.; Li, B. Cav1. 3 channel α1D protein is overexpressed and modulates androgen receptor transactivation in prostate cancers. Urol. Oncol. 2014, 32, 524–536. [Google Scholar] [CrossRef]
- Natrajan, R.; Little, S.E.; Reis-Filho, J.S.; Hing, L.; Messahel, B.; Grundy, P.E.; Dome, J.S.; Schneider, T.; Vujanic, G.M.; Pritchard-Jones, K. Amplification and overexpression of CACNA1E correlates with relapse in favorable histology Wilms’ tumors. Clin. Cancer Res. 2006, 12, 7284–7293. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Wang, P.; Ma, H.; Zhang, G.; Yulin, Z.; Lu, B.; Wang, H.; Dong, M. Suppression of T-type Ca2+ channels inhibited human laryngeal squamous cell carcinoma cell proliferation running title: Roles of T-type Ca2+ channels in LSCC cell proliferation. Clin. Lab. 2014, 60, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Valerie, N.C.; Dziegielewska, B.; Hosing, A.S.; Augustin, E.; Gray, L.S.; Brautigan, D.L.; Larner, J.M.; Dziegielewski, J. Inhibition of T-type calcium channels disrupts Akt signaling and promotes apoptosis in glioblastoma cells. Biochem. Pharmacol. 2013, 85, 888–897. [Google Scholar] [CrossRef]
- Nie, C.; Qin, X.; Li, X.; Tian, B.; Zhao, Y.; Jin, Y.; Li, Y.; Wang, Q.; Zeng, D.; Hong, A. CACNA2D3 enhances the chemosensitivity of esophageal squamous cell carcinoma to cisplatin via inducing ca2+-mediated apoptosis and suppressing PI3K/Akt pathways. Front. Oncol. 2019, 9, 185. [Google Scholar] [CrossRef]
- Dziegielewska, B.; Casarez, E.V.; Yang, W.Z.; Gray, L.S.; Dziegielewski, J.; Slack-Davis, J.K. T-type Ca2+ channel inhibition sensitizes ovarian cancer to carboplatin. Mol. Cancer Ther. 2016, 15, 460–470. [Google Scholar] [CrossRef]
- Hou, X.; Pedi, L.; Diver, M.M.; Long, S.B. Crystal structure of the calcium release–activated calcium channel Orai. Science 2012, 338, 1308–1313. [Google Scholar] [CrossRef] [PubMed]
- Shuba, Y.M. Ca(2+) channel-forming ORAI proteins: Cancer foes or cancer allies? Exp. Oncol. 2019, 41, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Fiorio Pla, A.; Kondratska, K.; Prevarskaya, N. STIM and ORAI proteins: Crucial roles in hallmarks of cancer. Am. J. Physiol. Physiol. 2016, 310, C509–C519. [Google Scholar] [CrossRef] [PubMed]
- Dubois, C.; Vanden Abeele, F.; Lehen’kyi, V.; Gkika, D.; Guarmit, B.; Lepage, G.; Slomianny, C.; Borowiec, A.S.; Bidaux, G.; Benahmed, M.; et al. Remodeling of channel-forming ORAI proteins determines an oncogenic switch in prostate cancer. Cancer Cell 2014, 26, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hao, J.; Zhang, Y.; Yang, Z.; Cao, Y.; Lu, W.; Shu, Y.; Jiang, L.; Hu, Y.; Lv, W. Orai1 mediates tumor-promoting store-operated Ca2+ entry in human gastrointestinal stromal tumors via c-KIT and the extracellular signal–regulated kinase pathway. Tumor Biol. 2017, 39, 1010428317691426. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, H.; Jin, F.; Fang, M.; Huang, M.; Yang, C.S.; Chen, T.; Fu, L.; Pan, Z. Elevated Orai1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma. Oncotarget 2014, 5, 3455. [Google Scholar] [CrossRef]
- Faouzi, M.; Hague, F.; Potier, M.; Ahidouch, A.; Sevestre, H.; Ouadid-Ahidouch, H. Down-regulation of Orai3 arrests cell-cycle progression and induces apoptosis in breast cancer cells but not in normal breast epithelial cells. J. Cell. Physiol. 2011, 226, 542–551. [Google Scholar] [CrossRef]
- Ay, A.-S.; Benzerdjerb, N.; Sevestre, H.; Ahidouch, A.; Ouadid-Ahidouch, H. Orai3 constitutes a native store-operated calcium entry that regulates non small cell lung adenocarcinoma cell proliferation. PLoS ONE 2013, 8, e72889. [Google Scholar] [CrossRef]
- Kischel, P.; Girault, A.; Rodat-Despoix, L.; Chamlali, M.; Radoslavova, S.; Abou Daya, H.; Lefebvre, T.; Foulon, A.; Rybarczyk, P.; Hague, F.; et al. Ion channels: New actors playing in chemotherapeutic resistance. Cancers 2019, 11. [Google Scholar] [CrossRef]
- Schmidt, S.; Liu, G.; Liu, G.; Yang, W.; Honisch, S.; Pantelakos, S.; Stournaras, C.; Hönig, A.; Lang, F. Enhanced Orai1 and STIM1 expression as well as store operated Ca2+ entry in therapy resistant ovary carcinoma cells. Oncotarget 2014, 5, 4799. [Google Scholar] [CrossRef]
- Tang, B.D.; Xia, X.; Lv, X.F.; Yu, B.X.; Yuan, J.N.; Mai, X.Y.; Shang, J.Y.; Zhou, J.G.; Liang, S.J.; Pang, R.P. Inhibition of Orai1-mediated Ca2+ entry enhances chemosensitivity of HepG2 hepatocarcinoma cells to 5-fluorouracil. J. Cell. Mol. Med. 2017, 21, 904–915. [Google Scholar] [CrossRef] [PubMed]
- Kondratska, K.; Kondratskyi, A.; Yassine, M.; Lemonnier, L.; Lepage, G.; Morabito, A.; Skryma, R.; Prevarskaya, N. Orai1 and STIM1 mediate SOCE and contribute to apoptotic resistance of pancreatic adenocarcinoma. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 2263–2269. [Google Scholar] [CrossRef] [PubMed]
- Hasna, J.; Hague, F.; Rodat-Despoix, L.; Geerts, D.; Leroy, C.; Tulasne, D.; Ouadid-Ahidouch, H.; Kischel, P. Orai3 calcium channel and resistance to chemotherapy in breast cancer cells: The p53 connection. Cell Death Differ. 2018, 25, 691. [Google Scholar] [CrossRef]
- Li, H. TRP channel classification. In Transient Receptor Potential Canonical Channels and Brain Diseases; Springer: Berlin, Germany, 2017; pp. 1–8. [Google Scholar]
- Owsianik, G.; D’hoedt, D.; Voets, T.; Nilius, B. Structure-function relationship of the TRP channel superfamily. Rev. Physiol. Biochem. Pharmacol. 2006, 156, 61–90. [Google Scholar] [PubMed]
- Clapham, D.E. TRP channels as cellular sensors. Nature 2003, 426, 517–524. [Google Scholar] [CrossRef]
- Gees, M.; Colsoul, B.; Nilius, B. The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a003962. [Google Scholar] [CrossRef]
- Minke, B. TRP channels and Ca2+ signaling. Cell Calcium 2006, 40, 261–275. [Google Scholar] [CrossRef]
- Canales, J.; Morales, D.; Blanco, C.; Rivas, J.; Diaz, N.; Angelopoulos, I.; Cerda, O. A TR(i)P to cell migration: New roles of trp channels in mechanotransduction and cancer. Front. Physiol. 2019, 10, 757. [Google Scholar] [CrossRef]
- Shapovalov, G.; Ritaine, A.; Skryma, R.; Prevarskaya, N. Role of TRP ion channels in cancer and tumorigenesis. Semin. Immunopathol. 2016, 38, 357–369. [Google Scholar] [CrossRef]
- Chen, J.; Luan, Y.; Yu, R.; Zhang, Z.; Zhang, J.; Wang, W. Transient receptor potential (TRP) channels, promising potential diagnostic and therapeutic tools for cancer. Biosci. Trends 2014, 8, 1–10. [Google Scholar] [CrossRef]
- Lehen’kyi, V.; Prevarskaya, N. Study of TRP Channels in Cancer Cells. In TRP Channels; Zhu, M.X., Ed.; CRC Press: Boca Raton, FL, USA, 2011. [Google Scholar]
- Santoni, G.; Maggi, F.; Morelli, M.B.; Santoni, M.; Marinelli, O. Transient receptor potential cation channels in cancer therapy. Med. Sci. 2019, 7, 108. [Google Scholar] [CrossRef] [PubMed]
- Birnbaumer, L. The TRPC class of ion channels: A critical review of their roles in slow, sustained increases in intracellular Ca2+ concentrations. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 395–426. [Google Scholar] [CrossRef] [PubMed]
- Dhennin-Duthille, I.; Gautier, M.; Faouzi, M.; Guilbert, A.; Brevet, M.; Vaudry, D.; Ahidouch, A.; Sevestre, H.; Ouadid-Ahidouch, H. High expression of transient receptor potential channels in human breast cancer epithelial cells and tissues: Correlation with pathological parameters. Cell. Physiol. Biochem. 2011, 28, 813–822. [Google Scholar] [CrossRef] [PubMed]
- El Hiani, Y.; Ahidouch, A.; Lehen’kyi, V.; Hague, F.; Gouilleux, F.; Mentaverri, R.; Kamel, S.; Lassoued, K.; Brule, G.; Ouadid-Ahidouch, H. Extracellular signal-regulated kinases 1 and 2 and TRPC1 channels are required for calcium-sensing receptor-stimulated MCF-7 breast cancer cell proliferation. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2009, 23, 335–346. [Google Scholar] [CrossRef] [PubMed]
- El Hiani, Y.; Lehen’kyi, V.; Ouadid-Ahidouch, H.; Ahidouch, A. Activation of the calcium-sensing receptor by high calcium induced breast cancer cell proliferation and TRPC1 cation channel over-expression potentially through EGFR pathways. Arch. Biochem. Biophys. 2009, 486, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Elzamzamy, O.M.; Penner, R.; Hazlehurst, L.A. The role of TRPC1 in modulating cancer progression. Cells 2020, 9, 388. [Google Scholar] [CrossRef]
- Yang, S.; Cao, Q.; Zhou, K.; Feng, Y.; Wang, Y. Transient receptor potential channel C3 contributes to the progression of human ovarian cancer. Oncogene 2009, 28, 1320–1328. [Google Scholar] [CrossRef]
- Tiapko, O.; Groschner, K. TRPC3 as a target of novel therapeutic interventions. Cells 2018, 7, 83. [Google Scholar] [CrossRef]
- Ding, X.; He, Z.; Zhou, K.; Cheng, J.; Yao, H.; Lu, D.; Cai, R.; Jin, Y.; Dong, B.; Xu, Y. Essential role of TRPC6 channels in G2/M phase transition and development of human glioma. J. Natl. Cancer Inst. 2010, 102, 1052–1068. [Google Scholar] [CrossRef]
- Cai, R.; Ding, X.; Zhou, K.; Shi, Y.; Ge, R.; Ren, G.; Jin, Y.; Wang, Y. Blockade of TRPC6 channels induced G2/M phase arrest and suppressed growth in human gastric cancer cells. Int. J. Cancer 2009, 125, 2281–2287. [Google Scholar] [CrossRef]
- Aydar, E.; Yeo, S.; Djamgoz, M.; Palmer, C. Abnormal expression, localization and interaction of canonical transient receptor potential ion channels in human breast cancer cell lines and tissues: A potential target for breast cancer diagnosis and therapy. Cancer Cell Int. 2009, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Veliceasa, D.; Ivanovic, M.; Hoepfner, F.T.S.; Thumbikat, P.; Volpert, O.V.; Smith, N.D. Transient potential receptor channel 4 controls thrombospondin-1 secretion and angiogenesis in renal cell carcinoma. FEBS J. 2007, 274, 6365–6377. [Google Scholar] [CrossRef] [PubMed]
- Carson, C.; Raman, P.; Tullai, J.; Xu, L.; Henault, M.; Thomas, E.; Yeola, S.; Lao, J.; McPate, M.; Verkuyl, J.M. Englerin A agonizes the TRPC4/C5 cation channels to inhibit tumor cell line proliferation. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zou, J.; Su, J.; Lu, Y.; Zhang, J.; Li, L.; Yin, F. Down regulation of transient receptor potential cation channel, subfamily C, member 1 contributes to drug resistance and high histological grade in ovarian cancer. Int. J. Oncol. 2016, 48, 243–252. [Google Scholar] [CrossRef]
- Wang, T.; Chen, Z.; Zhu, Y.; Pan, Q.; Liu, Y.; Qi, X.; Jin, L.; Jin, J.; Ma, X.; Hua, D. Inhibition of transient receptor potential channel 5 reverses 5-Fluorouracil resistance in human colorectal cancer cells. J. Biol. Chem. 2015, 290, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Liu, X.; Li, H.; Chen, Z.; Yao, X.; Jin, J.; Ma, X. TRPC5-induced autophagy promotes drug resistance in breast carcinoma via CaMKKβ/AMPKα/mTOR pathway. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Ning, K.; Lu, T.X.; Sun, X.; Jin, L.; Qi, X.; Jin, J.; Hua, D. Increasing circulating exosomes-carrying TRPC 5 predicts chemoresistance in metastatic breast cancer patients. Cancer Sci. 2017, 108, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Liang, C.; Chen, E.; Chen, W.; Liang, F.; Zhi, X.; Wei, T.; Xue, F.; Li, G.; Yang, Q. Regulation of Multi-drug Resistance in hepatocellular carcinoma cells is TRPC6/Calcium Dependent. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Fliegert, R.; Guse, A.H.; Lü, W.; Du, J. A Structural overview of the ion channels of the TRPM family. Cell Calcium 2019, 85, 102111. [Google Scholar] [CrossRef]
- Duncan, L.M.; Deeds, J.; Cronin, F.E.; Donovan, M.; Sober, A.J.; Kauffman, M.; McCarthy, J.J. Melastatin expression and prognosis in cutaneous malignant melanoma. J. Clin. Oncol. 2001, 19, 568–576. [Google Scholar] [CrossRef]
- Deeds, J.; Cronin, F.; Duncan, L.M. Patterns of melastatin mRNA expression in melanocytic tumors. Hum. Pathol. 2000, 31, 1346–1356. [Google Scholar] [CrossRef]
- Duncan, L.M.; Deeds, J.; Hunter, J.; Shao, J.; Holmgren, L.M.; Woolf, E.A.; Tepper, R.I.; Shyjan, A.W. Down-regulation of the novel gene melastatin correlates with potential for melanoma metastasis. Cancer Res. 1998, 58, 1515–1520. [Google Scholar] [PubMed]
- Almasi, S.; Kennedy, B.E.; El-Aghil, M.; Sterea, A.M.; Gujar, S.; Partida-Sanchez, S.; El Hiani, Y. TRPM2 channel-mediated regulation of autophagy maintains mitochondrial function and promotes gastric cancer cell survival via the JNK-signaling pathway. J. Biol. Chem. 2018, 293, 3637–3650. [Google Scholar] [CrossRef]
- Bao, L.; Chen, S.J.; Conrad, K.; Keefer, K.; Abraham, T.; Lee, J.P.; Wang, J.; Zhang, X.Q.; Hirschler-Laszkiewicz, I.; Wang, H.G.; et al. Depletion of the human ion channel TRPM2 in neuroblastoma demonstrates its key role in cell survival through modulation of mitochondrial reactive oxygen species and bioenergetics. J. Biol. Chem. 2016, 291, 24449–24464. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.A. TRPM2 in cancer. Cell Calcium 2019, 80, 8–17. [Google Scholar] [CrossRef]
- Almasi, S.; Long, C.Y.; Sterea, A.; Clements, D.R.; Gujar, S.; El Hiani, Y. TRPM2 silencing causes G2/M arrest and apoptosis in lung cancer cells via increasing intracellular ROS and RNS levels and activating the JNK pathway. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2019, 52, 742–757. [Google Scholar] [CrossRef]
- Almasi, S.; Sterea, A.M.; Fernando, W.; Clements, D.R.; Marcato, P.; Hoskin, D.W.; Gujar, S.; El Hiani, Y. TRPM2 ion channel promotes gastric cancer migration, invasion and tumor growth through the AKT signaling pathway. Sci. Rep. 2019, 9, 4182. [Google Scholar] [CrossRef]
- Hall, D.P.; Cost, N.G.; Hegde, S.; Kellner, E.; Mikhaylova, O.; Stratton, Y.; Ehmer, B.; Abplanalp, W.A.; Pandey, R.; Biesiada, J. TRPM3 and miR-204 establish a regulatory circuit that controls oncogenic autophagy in clear cell renal cell carcinoma. Cancer Cell 2014, 26, 738–753. [Google Scholar] [CrossRef]
- Yee, N.S. Role of TRPM7 in cancer: Potential as molecular biomarker and therapeutic target. Pharmaceuticals 2017, 10, 39. [Google Scholar] [CrossRef]
- Rybarczyk, P.; Gautier, M.; Hague, F.; Dhennin-Duthille, I.; Chatelain, D.; Kerr-Conte, J.; Pattou, F.; Regimbeau, J.M.; Sevestre, H.; Ouadid-Ahidouch, H. Transient receptor potential melastatin-related 7 channel is overexpressed in human pancreatic ductal adenocarcinomas and regulates human pancreatic cancer cell migration. Int. J. Cancer 2012, 131, E851–E861. [Google Scholar] [CrossRef]
- Yee, N.S.; Kazi, A.A.; Li, Q.; Yang, Z.; Berg, A.; Yee, R.K. Aberrant over-expression of TRPM7 ion channels in pancreatic cancer: Required for cancer cell invasion and implicated in tumor growth and metastasis. Biol. Open 2015, 4, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xiao, L.; Luo, C.-H.; Zhou, H.; Hu, J.; Tang, Y.-X.; Fang, K.-N.; Zhang, Y. Overexpression of TRPM7 is associated with poor prognosis in human ovarian carcinoma. Asian Pac. J. Cancer Prev. 2014, 15, 3955–3958. [Google Scholar] [CrossRef]
- Gao, S.-L.; Kong, C.-Z.; Zhang, Z.; Li, Z.-L.; Bi, J.-B.; Liu, X.-K. TRPM7 is overexpressed in bladder cancer and promotes proliferation, migration, invasion and tumor growth. Oncol. Rep. 2017, 38, 1967–1976. [Google Scholar] [CrossRef]
- Liu, J.; Chen, Y.; Shuai, S.; Ding, D.; Li, R.; Luo, R. TRPM8 promotes aggressiveness of breast cancer cells by regulating EMT via activating AKT/GSK-3β pathway. Tumor Biol. 2014, 35, 8969–8977. [Google Scholar] [CrossRef]
- Yee, N.S.; Zhou, W.; Lee, M. Transient receptor potential channel TRPM8 is over-expressed and required for cellular proliferation in pancreatic adenocarcinoma. Cancer Lett. 2010, 297, 49–55. [Google Scholar] [CrossRef]
- Yee, N.S.; Li, Q.; Kazi, A.A.; Yang, Z.; Berg, A.; Yee, R.K. Aberrantly over-expressed TRPM8 channels in pancreatic adenocarcinoma: Correlation with tumor size/stage and requirement for cancer cells invasion. Cells 2014, 3, 500–516. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Wang, Z.; Yang, Z.; Tao, L.; Liu, Q.; Yi, L.; Wang, X. Overexpression of short TRPM8 variant α promotes cell migration and invasion, and decreases starvation-induced apoptosis in prostate cancer LNCaP cells. Oncol. Lett. 2015, 10, 1378–1384. [Google Scholar] [CrossRef]
- Fuessel, S.; Sickert, D.; Meye, A.; Klenk, U.; Schmidt, U.; Schmitz, M.; Rost, A.-K.; Weigle, B.; Kiessling, A.; Wirth, M.P. Multiple tumor marker analyses (PSA, hK2, PSCA, trp-p8) in primary prostate cancers using quantitative RT-PCR. Int. J. Oncol. 2003, 23, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, H.; Ugawa, S.; Ueda, T.; Morita, A.; Shimada, S. TRPM8 activation suppresses cellular viability in human melanoma. Am. J. Physiol. Cell Physiol. 2008, 295, C296–C301. [Google Scholar] [CrossRef]
- Koh, D.W.; Powell, D.P.; Blake, S.D.; Hoffman, J.L.; Hopkins, M.M.; Feng, X. Enhanced cytotoxicity in triple-negative and estrogen receptor-positive breast adenocarcinoma cells due to inhibition of the transient receptor potential melastatin-2 channel. Oncol. Rep. 2015, 34, 1589–1598. [Google Scholar] [CrossRef]
- Castiglioni, S.; Cazzaniga, A.; Trapani, V.; Cappadone, C.; Farruggia, G.; Merolle, L.; Wolf, F.I.; Iotti, S.; Maier, J.A. Magnesium homeostasis in colon carcinoma LoVo cells sensitive or resistant to doxorubicin. Sci. Rep. 2015, 5, 16538. [Google Scholar] [CrossRef]
- Yu, S.; Xu, Z.; Zou, C.; Wu, D.; Wang, Y.; Yao, X.; Ng, C.F.; Chan, F.L. Ion channel TRPM8 promotes hypoxic growth of prostate cancer cells via an O2-independent and RACK1-mediated mechanism of HIF-1α stabilization. J. Pathol. 2014, 234, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, Z.; Meng, Z.; Cao, H.; Zhu, G.; Liu, T.; Wang, X. Knockdown of TRPM8 suppresses cancer malignancy and enhances epirubicin-induced apoptosis in human osteosarcoma cells. Int. J. Biol. Sci. 2014, 10, 90. [Google Scholar] [CrossRef]
- Santoni, G.; Farfariello, V.; Amantini, C. TRPV channels in tumor growth and progression. In Transient Receptor Potential Channels; Springer: Berlin, Germany, 2011; pp. 947–967. [Google Scholar]
- Yang, Y.; Guo, W.; Ma, J.; Xu, P.; Zhang, W.; Guo, S.; Liu, L.; Ma, J.; Shi, Q.; Jian, Z. Downregulated TRPV1 expression contributes to melanoma growth via the calcineurin-ATF3-p53 pathway. J. Investig. Dermatol. 2018, 138, 2205–2215. [Google Scholar] [CrossRef] [PubMed]
- Hou, N.; He, X.; Yang, Y.; Fu, J.; Zhang, W.; Guo, Z.; Hu, Y.; Liang, L.; Xie, W.; Xiong, H. TRPV1 induced apoptosis of colorectal cancer cells by activating calcineurin-NFAT2-p53 signaling pathway. BioMed Res. Int. 2019, 2019. [Google Scholar] [CrossRef]
- Wu, Y.-Y.; Liu, X.-Y.; Zhuo, D.-X.; Huang, H.-B.; Zhang, F.-B.; Liao, S.-F. Decreased expression of TRPV1 in renal cell carcinoma: Association with tumor Fuhrman grades and histopathological subtypes. Cancer Manag. Res. 2018, 10, 1647. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Wang, G.; Tao, H.; Yang, Z.; Wang, Y.; Meng, Z.; Cao, R.; Xiao, Y.; Wang, X.; Zhou, J. Capsaicin mediates caspases activation and induces apoptosis through P38 and JNK MAPK pathways in human renal carcinoma. BMC Cancer 2016, 16, 790. [Google Scholar] [CrossRef]
- de Jong, P.R.; Takahashi, N.; Harris, A.R.; Lee, J.; Bertin, S.; Jeffries, J.; Jung, M.; Duong, J.; Triano, A.I.; Lee, J. Ion channel TRPV1-dependent activation of PTP1B suppresses EGFR-associated intestinal tumorigenesis. J. Clin. Investig. 2014, 124, 3793–3806. [Google Scholar] [CrossRef]
- Morelli, M.B.; Amantini, C.; Nabissi, M.; Liberati, S.; Cardinali, C.; Farfariello, V.; Tomassoni, D.; Quaglia, W.; Piergentili, A.; Bonifazi, A. Cross-talk between alpha 1D-adrenoceptors and transient receptor potential vanilloid type 1 triggers prostate cancer cell proliferation. BMC Cancer 2014, 14, 921. [Google Scholar] [CrossRef]
- Weber, L.V.; Al-Refae, K.; Wölk, G.; Bonatz, G.; Altmüller, J.; Becker, C.; Gisselmann, G.; Hatt, H. Expression and functionality of TRPV1 in breast cancer cells. Breast Cancer Targets Ther. 2016, 8, 243. [Google Scholar] [CrossRef]
- Pecze, L.; Jósvay, K.; Blum, W.; Petrovics, G.; Vizler, C.; Oláh, Z.; Schwaller, B. Activation of endogenous TRPV1 fails to induce overstimulation-based cytotoxicity in breast and prostate cancer cells but not in pain-sensing neurons. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 2054–2064. [Google Scholar] [CrossRef] [PubMed]
- Elbaz, M.; Ahirwar, D.; Xiaoli, Z.; Zhou, X.; Lustberg, M.; Nasser, M.W.; Shilo, K.; Ganju, R.K. TRPV2 is a novel biomarker and therapeutic target in triple negative breast cancer. Oncotarget 2018, 9, 33459. [Google Scholar] [CrossRef] [PubMed]
- Caprodossi, S.; Lucciarini, R.; Amantini, C.; Nabissi, M.; Canesin, G.; Ballarini, P.; Di Spilimbergo, A.; Cardarelli, M.A.; Servi, L.; Mammana, G. Transient receptor potential vanilloid type 2 (TRPV2) expression in normal urothelium and in urothelial carcinoma of human bladder: Correlation with the pathologic stage. Eur. Urol. 2008, 54, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Zhang, S.-S.; Yan, Y.; Zhao, S. Overexpression of transient receptor potential vanilloid 2 is associated with poor prognosis in patients with esophageal squamous cell carcinoma. Med. Oncol. 2014, 31, 17. [Google Scholar] [CrossRef]
- Santoni, G.; Amantini, C.; Maggi, F.; Marinelli, O.; Santoni, M.; Nabissi, M.; Morelli, M.B. The TRPV2 cation channels: From urothelial cancer invasiveness to glioblastoma multiforme interactome signature. Lab. Investig. 2019, 100, 1–13. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Q.; Fan, K.; Li, B.; Li, H.; Qi, H.; Guo, J.; Cao, Y.; Sun, H. Overexpression of TRPV3 correlates with tumor progression in non-small cell lung cancer. Int. J. Mol. Sci. 2016, 17, 437. [Google Scholar] [CrossRef]
- Hoeft, B.; Linseisen, J.; Beckmann, L.; Müller-Decker, K.; Canzian, F.; Hüsing, A.; Kaaks, R.; Vogel, U.; Jakobsen, M.U.; Overvad, K. Polymorphisms in fatty acid metabolism-related genes are associated with colorectal cancer risk. Carcinogenesis 2010, 31, 466–472. [Google Scholar] [CrossRef]
- Lee, W.H.; Choong, L.Y.; Mon, N.N.; Lu, S.; Lin, Q.; Pang, B.; Yan, B.; Krishna, V.S.R.; Singh, H.; Tan, T.Z. TRPV4 regulates breast cancer cell extravasation, stiffness and actin cortex. Sci. Rep. 2016, 6, 27903. [Google Scholar] [CrossRef]
- Fang, Y.; Liu, G.; Xie, C.; Qian, K.; Lei, X.; Liu, Q.; Liu, G.; Cao, Z.; Fu, J.; Du, H. Pharmacological inhibition of TRPV4 channel suppresses malignant biological behavior of hepatocellular carcinoma via modulation of ERK signaling pathway. Biomed. Pharmacother. 2018, 101, 910–919. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, P.; Xie, C.; Sham, K.W.; Ng, S.S.; Chen, Y.; Cheng, C.H. Activation of PTEN by inhibition of TRPV4 suppresses colon cancer development. Cell Death Dis. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Adapala, R.K.; Thoppil, R.J.; Ghosh, K.; Cappelli, H.; Dudley, A.C.; Paruchuri, S.; Keshamouni, V.; Klagsbrun, M.; Meszaros, J.G.; Chilian, W.M. Activation of mechanosensitive ion channel TRPV4 normalizes tumor vasculature and improves cancer therapy. Oncogene 2016, 35, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Huang, S.; Ding, Y.; Wang, W.; Wang, A.; Lu, Y. Transient receptor potential ion-channel subfamily V member 4: A potential target for cancer treatment. Cell Death Dis. 2019, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Shen, Y.-X.; Yuan, Y.-F. Expression and prognostic roles of TRPV5 and TRPV6 in non-small cell lung cancer after curative resection. Asian Pac. J. Cancer Prev. 2014, 15, 2559–2563. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Miyamoto, T.; Li, K.; Nakagomi, H.; Sawada, N.; Kira, S.; Kobayashi, H.; Zakohji, H.; Tsuchida, T.; Fukazawa, M. Decreased expression of the epithelial Ca2+ channel TRPV5 and TRPV6 in human renal cell carcinoma associated with vitamin D receptor. J. Urol. 2011, 186, 2419–2425. [Google Scholar] [CrossRef]
- Lehen’kyi, V.Y.; Raphaël, M.; Prevarskaya, N. The role of the TRPV6 channel in cancer. J. Physiol. 2012, 590, 1369–1376. [Google Scholar] [CrossRef]
- Stewart, J.M. TRPV6 as a target for cancer therapy. J. Cancer 2020, 11, 374. [Google Scholar] [CrossRef]
- Zhang, S.-S.; Xie, X.; Wen, J.; Luo, K.-J.; Liu, Q.-W.; Yang, H.; Hu, Y.; Fu, J.-H. TRPV6 plays a new role in predicting survival of patients with esophageal squamous cell carcinoma. Diagn. Pathol. 2016, 11, 14. [Google Scholar] [CrossRef]
- Deveci, H.A.; Nazıroğlu, M.; Nur, G. 5-Fluorouracil-induced mitochondrial oxidative cytotoxicity and apoptosis are increased in MCF-7 human breast cancer cells by TRPV1 channel activation but not Hypericum perforatum treatment. Mol. Cell. Biochem. 2018, 439, 189–198. [Google Scholar] [CrossRef]
- Nur, G.; Nazıroğlu, M.; Deveci, H.A. Synergic prooxidant, apoptotic and TRPV1 channel activator effects of alpha-lipoic acid and cisplatin in MCF-7 breast cancer cells. J. Recept. Signal. Transduct. 2017, 37, 569–577. [Google Scholar] [CrossRef]
- Koşar, P.A.; Nazıroğlu, M.; Övey, İ.S.; Çiğ, B. Synergic effects of doxorubicin and melatonin on apoptosis and mitochondrial oxidative stress in MCF-7 breast cancer cells: Involvement of TRPV1 channels. J. Membr. Biol. 2016, 249, 129–140. [Google Scholar] [CrossRef]
- Zheng, L.; Chen, J.; Ma, Z.; Liu, W.; Yang, F.; Yang, Z.; Wang, K.; Wang, X.; He, D.; Li, L. Capsaicin enhances anti-proliferation efficacy of pirarubicin via activating TRPV1 and inhibiting PCNA nuclear translocation in 5637 cells. Mol. Med. Rep. 2016, 13, 881–887. [Google Scholar] [CrossRef]
- Ortega-Guerrero, A.; Espinosa-Duran, J.M.; Velasco-Medina, J. TRPV1 channel as a target for cancer therapy using CNT-based drug delivery systems. Eur. Biophys. J. 2016, 45, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Nabissi, M.; Morelli, M.B.; Amantini, C.; Farfariello, V.; Ricci-Vitiani, L.; Caprodossi, S.; Arcella, A.; Santoni, M.; Giangaspero, F.; De Maria, R. TRPV2 channel negatively controls glioma cell proliferation and resistance to Fas-induced apoptosis in ERK-dependent manner. Carcinogenesis 2010, 31, 794–803. [Google Scholar] [CrossRef] [PubMed]
- Nabissi, M.; Morelli, M.B.; Santoni, M.; Santoni, G. Triggering of the TRPV2 channel by cannabidiol sensitizes glioblastoma cells to cytotoxic chemotherapeutic agents. Carcinogenesis 2013, 34, 48–57. [Google Scholar] [CrossRef]
- Morelli, M.B.; Offidani, M.; Alesiani, F.; Discepoli, G.; Liberati, S.; Olivieri, A.; Santoni, M.; Santoni, G.; Leoni, P.; Nabissi, M. The effects of cannabidiol and its synergism with bortezomib in multiple myeloma cell lines. A role for transient receptor potential vanilloid type-2. Int. J. Cancer 2014, 134, 2534–2546. [Google Scholar] [CrossRef] [PubMed]
- Raphaël, M.; Lehen’kyi, V.Y.; Vandenberghe, M.; Beck, B.; Khalimonchyk, S.; Abeele, F.V.; Farsetti, L.; Germain, E.; Bokhobza, A.; Mihalache, A. TRPV6 calcium channel translocates to the plasma membrane via Orai1-mediated mechanism and controls cancer cell survival. Proc. Natl. Acad. Sci. USA 2014, 111, E3870–E3879. [Google Scholar] [CrossRef]
- Montgomery, D.R.; Buffington, J.M. Channel Classification, Prediction of Channel Response, and Assessment of Channel Condition; University of Washington Seattle: Washington, DC, USA, 1993. [Google Scholar]
- Kim, D.M.; Nimigean, C.M. Voltage-gated potassium channels: A structural examination of selectivity and gating. Cold Spring Harb. Perspect. Biol. 2016, 8, a029231. [Google Scholar] [CrossRef]
- Berkefeld, H.; Fakler, B.; Schulte, U. Ca2+-activated K+ channels: From protein complexes to function. Physiol. Rev. 2010, 90, 1437–1459. [Google Scholar] [CrossRef]
- Hibino, H.; Inanobe, A.; Furutani, K.; Murakami, S.; Findlay, I.; Kurachi, Y. Inwardly rectifying potassium channels: Their structure, function, and physiological roles. Physiol. Rev. 2010, 90, 291–366. [Google Scholar] [CrossRef]
- Kim, D. Physiology and pharmacology of two-pore domain potassium channels. Curr. Pharm. Des. 2005, 11, 2717–2736. [Google Scholar] [CrossRef]
- Kuang, Q.; Purhonen, P.; Hebert, H. Structure of potassium channels. Cell. Mol. Life Sci. 2015, 72, 3677–3693. [Google Scholar] [CrossRef] [PubMed]
- Piechotta, P.L.; Rapedius, M.; Stansfeld, P.J.; Bollepalli, M.K.; Erhlich, G.; Andres-Enguix, I.; Fritzenschaft, H.; Decher, N.; Sansom, M.S.; Tucker, S.J. The pore structure and gating mechanism of K2P channels. EMBO J. 2011, 30, 3607–3619. [Google Scholar] [CrossRef] [PubMed]
- Ouadid-Ahidouch, H.; Ahidouch, A. K+ channel expression in human breast cancer cells: Involvement in cell cycle regulation and carcinogenesis. J. Membr. Biol. 2008, 221, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Comes, N.; Serrano-Albarras, A.; Capera, J.; Serrano-Novillo, C.; Condom, E.; y Cajal, S.R.; Ferreres, J.C.; Felipe, A. Involvement of potassium channels in the progression of cancer to a more malignant phenotype. Biochim. Biophys. Acta Biomembr. 2015, 1848, 2477–2492. [Google Scholar] [CrossRef]
- Huang, X.; Dubuc, A.M.; Hashizume, R.; Berg, J.; He, Y.; Wang, J.; Chiang, C.; Cooper, M.K.; Northcott, P.A.; Taylor, M.D. Voltage-gated potassium channel EAG2 controls mitotic entry and tumor growth in medulloblastoma via regulating cell volume dynamics. Genes Dev. 2012, 26, 1780–1796. [Google Scholar] [CrossRef]
- Song, P.; Du, Y.; Song, W.; Chen, H.; Xuan, Z.; Zhao, L.; Chen, J.; Chen, J.; Guo, D.; Jin, C. KCa3. 1 as an effective target for inhibition of growth and progression of intrahepatic cholangiocarcinoma. J. Cancer 2017, 8, 1568. [Google Scholar] [CrossRef]
- Haren, N.; Khorsi, H.; Faouzi, M.; Ahidouch, A.; Sevestre, H.; Ouadid-Ahidouch, H. Intermediate conductance Ca2+ activated K+ channels are expressed and functional in breast adenocarcinomas: Correlation with tumour grade and metastasis status. Histol. Histopathol. 2010, 25, 1247–1255. [Google Scholar]
- Rabjerg, M.; Oliván-Viguera, A.; Hansen, L.K.; Jensen, L.; Sevelsted-Møller, L.; Walter, S.; Jensen, B.L.; Marcussen, N.; Köhler, R. High expression of KCa3. 1 in patients with clear cell renal carcinoma predicts high metastatic risk and poor survival. PLoS ONE 2015, 10, e0122992. [Google Scholar] [CrossRef]
- Ji, C.-D.; Wang, Y.-X.; Xiang, D.-F.; Liu, Q.; Zhou, Z.-H.; Qian, F.; Yang, L.; Ren, Y.; Cui, W.; Xu, S.-L. Kir2. 1 Interaction with Stk38 Promotes Invasion and Metastasis of Human Gastric Cancer by Enhancing MEKK2–MEK1/2–ERK1/2 Signaling. Cancer Res. 2018, 78, 3041–3053. [Google Scholar] [CrossRef]
- Zhang, G.-M.; Wan, F.-N.; Qin, X.-J.; Cao, D.-L.; Zhang, H.-L.; Zhu, Y.; Dai, B.; Shi, G.-H.; Ye, D.-W. Prognostic significance of the TREK-1 K2P potassium channels in prostate cancer. Oncotarget 2015, 6, 18460. [Google Scholar] [CrossRef][Green Version]
- Chen, S.-Z.; Jiang, M.; Zhen, Y.-S. HERG K+ channel expression-related chemosensitivity in cancer cells and its modulation by erythromycin. Cancer Chemother. Pharmacol. 2005, 56, 212–220. [Google Scholar] [CrossRef]
- Laniado, M.E.; Fraser, S.P.; Djamgoz, M.B. Voltage-gated K+ channel activity in human prostate cancer cell lines of markedly different metastatic potential: Distinguishing characteristics of PC-3 and LNCaP cells. Prostate 2001, 46, 262–274. [Google Scholar] [CrossRef]
- Samuel, P.; Pink, R.C.; Caley, D.P.; Currie, J.M.S.; Brooks, S.A.; Carter, D.R.F. Over-expression of miR-31 or loss of KCNMA1 leads to increased cisplatin resistance in ovarian cancer cells. Tumor Biol. 2016, 37, 2565–2573. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wei, L.; Zhao, B.; Cai, X.; Dong, C.; Yin, F. Low expression of KCNN3 may affect drug resistance in ovarian cancer. Mol. Med. Rep. 2018, 18, 1377–1386. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.L.; Hasegawa, Y.; Shimizu, T.; Okada, Y. IK1 channel activity contributes to cisplatin sensitivity of human epidermoid cancer cells. Am. J. Physiol. Cell Physiol. 2008, 294, C1398–C1406. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Shi, Y.; Han, Z.; Sun, L.; Fan, D. Detection of potassium currents and regulation of multidrug resistance by potassium channels in human gastric cancer cells. Cell Biol. Int. 2007, 31, 741–747. [Google Scholar] [CrossRef]
- Hartung, F.; Pardo, L.A. Guiding TRAIL to cancer cells through Kv10. 1 potassium channel overcomes resistance to doxorubicin. Eur. Biophys. J. 2016, 45, 709–719. [Google Scholar] [CrossRef]
- Fortunato, A. The role of hERG1 ion channels in epithelial-mesenchymal transition and the capacity of riluzole to reduce cisplatin resistance in colorectal cancer cells. Cell. Oncol. 2017, 40, 367–378. [Google Scholar] [CrossRef]
- Catterall, W.A. From ionic currents to molecular mechanisms: The structure and function of voltage-gated sodium channels. Neuron 2000, 26, 13–25. [Google Scholar] [CrossRef]
- Diss, J.; Fraser, S.; Djamgoz, M. Voltage-gated Na+ channels: Multiplicity of expression, plasticity, functional implications and pathophysiological aspects. Eur. Biophys. J. 2004, 33, 180–193. [Google Scholar] [CrossRef]
- Black, J.A.; Waxman, S.G. Noncanonical roles of voltage-gated sodium channels. Neuron 2013, 80, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Mao, W.; Zhang, J.; Korner, H.; Jiang, Y.; Ying, S. The emerging role of voltage-gated sodium channels in tumour biology. Front. Oncol. 2019, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Shen, Y.; Cai, J.; Lei, M.; Wang, Z. Expression of voltage-gated sodium channel α subunit in human ovarian cancer. Oncol. Rep. 2010, 23, 1293–1299. [Google Scholar] [PubMed]
- House, C.D.; Vaske, C.J.; Schwartz, A.M.; Obias, V.; Frank, B.; Luu, T.; Sarvazyan, N.; Irby, R.; Strausberg, R.L.; Hales, T.G. Voltage-gated Na+ channel SCN5A is a key regulator of a gene transcriptional network that controls colon cancer invasion. Cancer Res. 2010, 70, 6957–6967. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.; Yang, M.; Millican-Slater, R.; Brackenbury, W.J. Nav1. 5 regulates breast tumor growth and metastatic dissemination in vivo. Oncotarget 2015, 6, 32914. [Google Scholar] [CrossRef]
- Hernandez-Plata, E.; Ortiz, C.S.; Marquina-Castillo, B.; Medina-Martinez, I.; Alfaro, A.; Berumen, J.; Rivera, M.; Gomora, J.C. Overexpression of Nav1. 6 channels is associated with the invasion capacity of human cervical cancer. Int. J. Cancer 2012, 130, 2013–2023. [Google Scholar] [CrossRef]
- Shan, B.; Dong, M.; Tang, H.; Wang, N.; Zhang, J.; Yan, C.; Jiao, X.; Zhang, H.; Wang, C. Voltage-gated sodium channels were differentially expressed in human normal prostate, benign prostatic hyperplasia and prostate cancer cells. Oncol. Lett. 2014, 8, 345–350. [Google Scholar] [CrossRef]
- Xia, J.; Huang, N.; Huang, H.; Sun, L.; Dong, S.; Su, J.; Zhang, J.; Wang, L.; Lin, L.; Shi, M. Voltage-gated sodium channel Nav1. 7 promotes gastric cancer progression through MACC1-mediated upregulation of NHE1. Int. J. Cancer 2016, 139, 2553–2569. [Google Scholar] [CrossRef]
- Liu, J.; Tan, H.; Yang, W.; Yao, S.; Hong, L. The voltage-gated sodium channel Nav1. 7 associated with endometrial cancer. J. Cancer 2019, 10, 4954. [Google Scholar] [CrossRef]
- Chioni, A.-M.; Brackenbury, W.J.; Calhoun, J.D.; Isom, L.L.; Djamgoz, M.B. A novel adhesion molecule in human breast cancer cells: Voltage-gated Na+ channel β1 subunit. Int. J. Biochem. Cell Biol. 2009, 41, 1216–1227. [Google Scholar] [CrossRef]
- Nelson, M.; Millican-Slater, R.; Forrest, L.C.; Brackenbury, W.J. The sodium channel β1 subunit mediates outgrowth of neurite-like processes on breast cancer cells and promotes tumour growth and metastasis. Int. J. Cancer 2014, 135, 2338–2351. [Google Scholar] [CrossRef] [PubMed]
- Jansson, K.H.; Lynch, J.E.; Lepori-Bui, N.; Czymmek, K.J.; Duncan, R.L.; Sikes, R.A. Overexpression of the VSSC-associated CAM, beta-2, enhances LNCaP cell metastasis associated behavior. Prostate 2012, 72, 1080–1092. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Yang, J.; Wu, W.; Liu, F.; Su, A.; Li, Z.; Zhu, J.; Wei, T. Preserved SCN4B expression is an independent indicator of favorable recurrence-free survival in classical papillary thyroid cancer. PLoS ONE 2018, 13, e0197007. [Google Scholar] [CrossRef] [PubMed]
- Bon, E.; Driffort, V.; Gradek, F.; Martinez-Caceres, C.; Anchelin, M.; Pelegrin, P.; Cayuela, M.-L.; Marionneau-Lambot, S.; Oullier, T.; Guibon, R. SCN4B acts as a metastasis-suppressor gene preventing hyperactivation of cell migration in breast cancer. Nat. Commun. 2016, 7, 1–18. [Google Scholar] [CrossRef]
- Sanchez-Sandoval, A.L.; Gomora, J.C. Contribution of voltage-gated sodium channel β-subunits to cervical cancer cells metastatic behavior. Cancer Cell Int. 2019, 19, 35. [Google Scholar] [CrossRef]
- Djamgoz, M.; Fraser, S.P.; Brackenbury, W.J. In vivo evidence for voltage-gated sodium channel expression in carcinomas and potentiation of metastasis. Cancers 2019, 11, 1675. [Google Scholar] [CrossRef]
- Li, K.; Yang, J.; Han, X. Lidocaine sensitizes the cytotoxicity of cisplatin in breast cancer cells via up-regulation of RARβ2 and RASSF1A demethylation. Int. J. Mol. Sci. 2014, 15, 23519–23536. [Google Scholar] [CrossRef]
- Freeman, J.; Crowley, P.D.; Foley, A.G.; Gallagher, H.C.; Iwasaki, M.; Ma, D.; Buggy, D.J. Effect of perioperative lidocaine and cisplatin on metastasis in a murine model of breast cancer surgery. Anticancer Res. 2018, 38, 5599–5606. [Google Scholar] [CrossRef]
- Xing, W.; Chen, D.-T.; Pan, J.-H.; Chen, Y.-H.; Yan, Y.; Li, Q.; Xue, R.-F.; Yuan, Y.-F.; Zeng, W.-A. Lidocaine induces apoptosis and suppresses tumor growth in human hepatocellular carcinoma cells in vitro and in a xenograft model in vivo. Anesthesiol. J. Am. Soc. Anesthesiol. 2017, 126, 868–881. [Google Scholar] [CrossRef]
- Tran, T.-A.; Gillet, L.; Roger, S.; Besson, P.; White, E.; Le Guennec, J.-Y. Non-anti-mitotic concentrations of taxol reduce breast cancer cell invasiveness. Biochem. Biophys. Res. Commun. 2009, 379, 304–308. [Google Scholar] [CrossRef]
- Adachi, K.; Toyota, M.; Sasaki, Y.; Yamashita, T.; Ishida, S.; Ohe-Toyota, M.; Maruyama, R.; Hinoda, Y.; Saito, T.; Imai, K. Identification of SCN3B as a novel p53-inducible proapoptotic gene. Oncogene 2004, 23, 7791–7798. [Google Scholar] [CrossRef] [PubMed]
- Jasti, J.; Furukawa, H.; Gonzales, E.B.; Gouaux, E. Structure of acid-sensing ion channel 1 at 1.9 Å resolution and low pH. Nature 2007, 449, 316–323. [Google Scholar] [CrossRef]
- Wu, Y.; Gao, B.; Xiong, Q.-J.; Wang, Y.-C.; Huang, D.-K.; Wu, W.-N. Acid-sensing ion channels contribute to the effect of extracellular acidosis on proliferation and migration of A549 cells. Tumor Biol. 2017, 39, 1010428317705750. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Zhou, H.-Y.; Deng, S.-C.; Deng, S.-J.; He, C.; Li, X.; Chen, J.-Y.; Jin, Y.; Hu, Z.-L.; Wang, F. ASIC1 and ASIC3 contribute to acidity-induced EMT of pancreatic cancer through activating Ca2+/RhoA pathway. Cell Death Dis. 2017, 8, e2806. [Google Scholar] [CrossRef] [PubMed]
- Berdiev, B.K.; Xia, J.; McLean, L.A.; Markert, J.M.; Gillespie, G.Y.; Mapstone, T.B.; Naren, A.P.; Jovov, B.; Bubien, J.K.; Ji, H.-L. Acid-sensing ion channels in malignant gliomas. J. Biol. Chem. 2003, 278, 15023–15034. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, N.; Bartoszewski, R.; Qadri, Y.J.; Bebok, Z.; Bubien, J.K.; Fuller, C.M.; Benos, D.J. Knockdown of ASIC1 and epithelial sodium channel subunits inhibits glioblastoma whole cell current and cell migration. J. Biol. Chem. 2009, 284, 24526–24541. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Singh, R.; Asters, M.; Liu, J.; Zhang, X.; Pabbidi, M.R.; Watabe, K.; Mo, Y.-Y. Regulation of breast tumorigenesis through acid sensors. Oncogene 2016, 35, 4102–4111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, T.; Wu, C.; Xia, Q.; Xu, D. ASIC1a mediates the drug resistance of human hepatocellular carcinoma via the Ca(2+)/PI3-kinase/AKT signaling pathway. Lab. Investig. 2017, 97, 53–69. [Google Scholar] [CrossRef]
- Holzmann, C.; Kappel, S.; Kilch, T.; Jochum, M.M.; Urban, S.K.; Jung, V.; Stöckle, M.; Rother, K.; Greiner, M.; Peinelt, C. Transient receptor potential melastatin 4 channel contributes to migration of androgen-insensitive prostate cancer cells. Oncotarget 2015, 6, 41783. [Google Scholar] [CrossRef]
- Armisén, R.; Marcelain, K.; Simon, F.; Tapia, J.C.; Toro, J.; Quest, A.F.; Stutzin, A. TRPM4 enhances cell proliferation through up-regulation of the β-catenin signaling pathway. J. Cell. Physiol. 2011, 226, 103–109. [Google Scholar] [CrossRef]
- Narayan, G.; Bourdon, V.; Chaganti, S.; Arias-Pulido, H.; Nandula, S.V.; Rao, P.H.; Gissmann, L.; Dürst, M.; Schneider, A.; Pothuri, B. Gene dosage alterations revealed by cDNA microarray analysis in cervical cancer: Identification of candidate amplified and overexpressed genes. Genes Chromosomes Cancer 2007, 46, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Suzuki, A.; Koga, K.; Miyamoto, C.; Maehata, Y.; Ozawa, S.; Hata, R.-I.; Nagashima, Y.; Nabeshima, K.; Miyazaki, K. TRPM5 mediates acidic extracellular pH signaling and TRPM5 inhibition reduces spontaneous metastasis in mouse B16-BL6 melanoma cells. Oncotarget 2017, 8, 78312. [Google Scholar] [CrossRef] [PubMed]
- Weaver, E.M.; Zamora, F.J.; Puplampu-Dove, Y.A.; Kiessu, E.; Hearne, J.L.; Martin-Caraballo, M. Regulation of T-type calcium channel expression by sodium butyrate in prostate cancer cells. Eur. J. Pharmacol. 2015, 749, 20–31. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almasi, S.; El Hiani, Y. Exploring the Therapeutic Potential of Membrane Transport Proteins: Focus on Cancer and Chemoresistance. Cancers 2020, 12, 1624. https://doi.org/10.3390/cancers12061624
Almasi S, El Hiani Y. Exploring the Therapeutic Potential of Membrane Transport Proteins: Focus on Cancer and Chemoresistance. Cancers. 2020; 12(6):1624. https://doi.org/10.3390/cancers12061624
Chicago/Turabian StyleAlmasi, Shekoufeh, and Yassine El Hiani. 2020. "Exploring the Therapeutic Potential of Membrane Transport Proteins: Focus on Cancer and Chemoresistance" Cancers 12, no. 6: 1624. https://doi.org/10.3390/cancers12061624
APA StyleAlmasi, S., & El Hiani, Y. (2020). Exploring the Therapeutic Potential of Membrane Transport Proteins: Focus on Cancer and Chemoresistance. Cancers, 12(6), 1624. https://doi.org/10.3390/cancers12061624