Genetic Target Modulation Employing CRISPR/Cas9 Identifies Glyoxalase 1 as a Novel Molecular Determinant of Invasion and Metastasis in A375 Human Malignant Melanoma Cells In Vitro and In Vivo



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
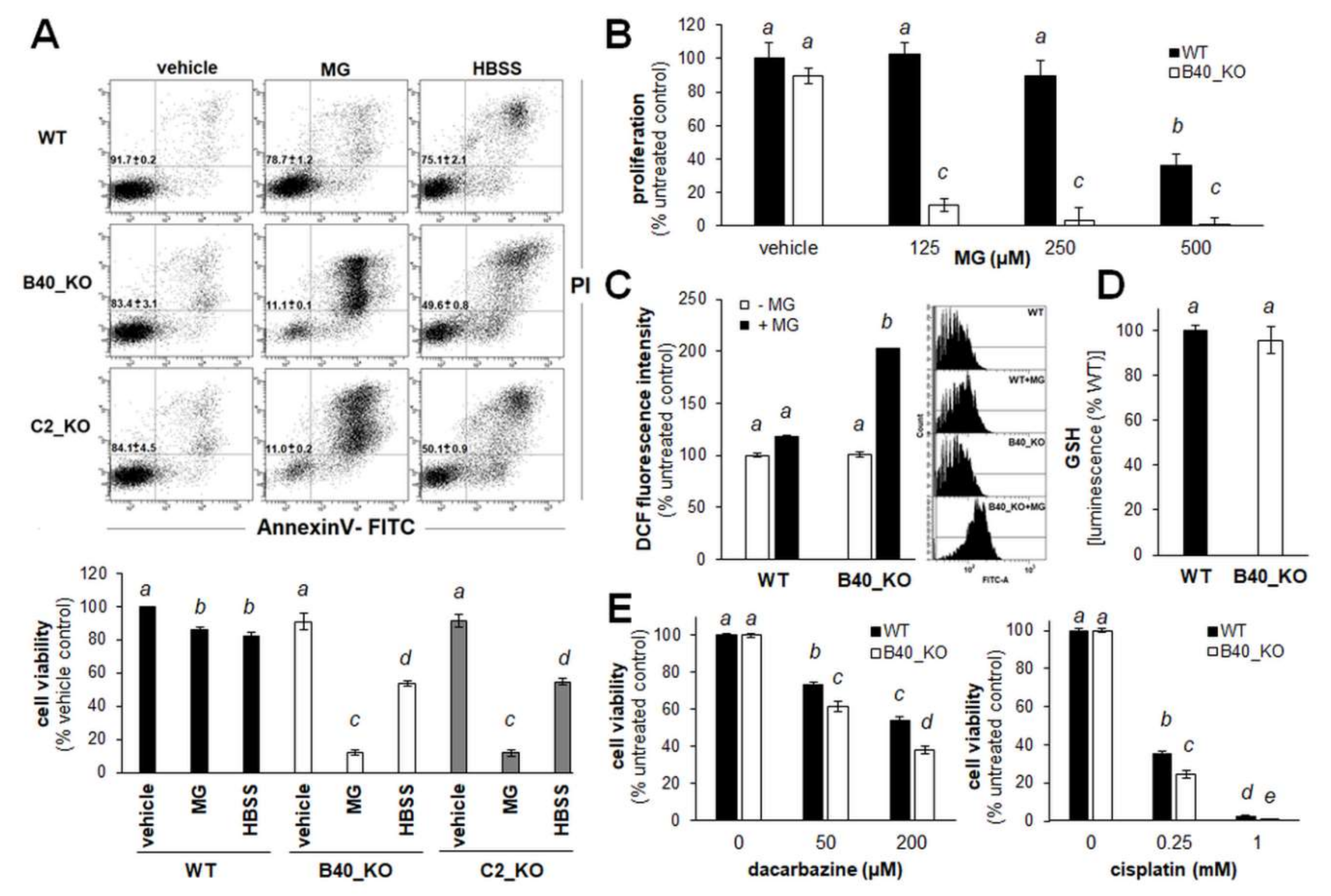
2.1. A375 Human Malignant Melanoma Cells with Genetic GLO1 Deletion (A375-GLO1_KO) Display Sensitization to Methylglyoxal-, Chemotherapy-, and Starvation-Induced Cytotoxic Stress
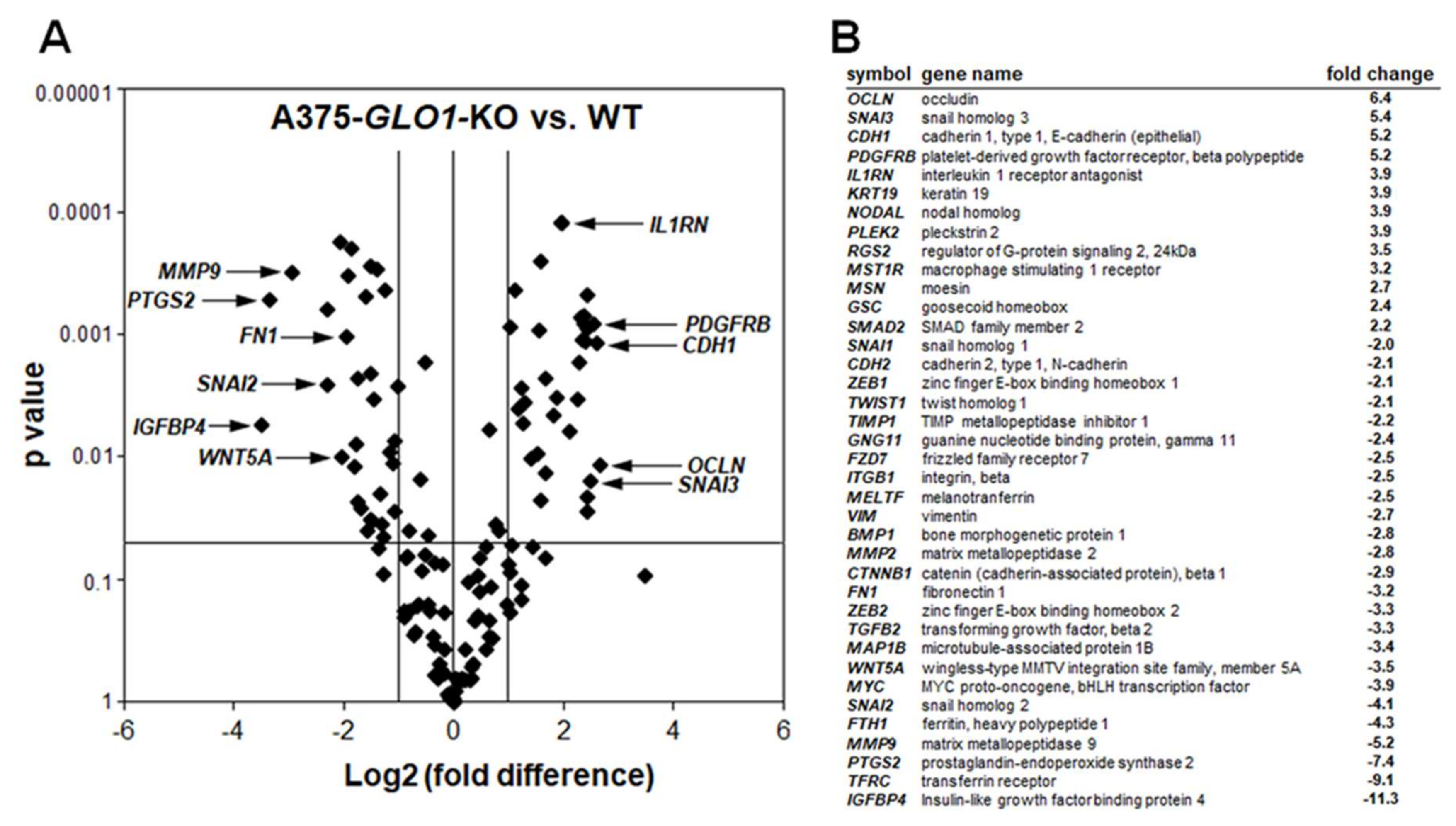
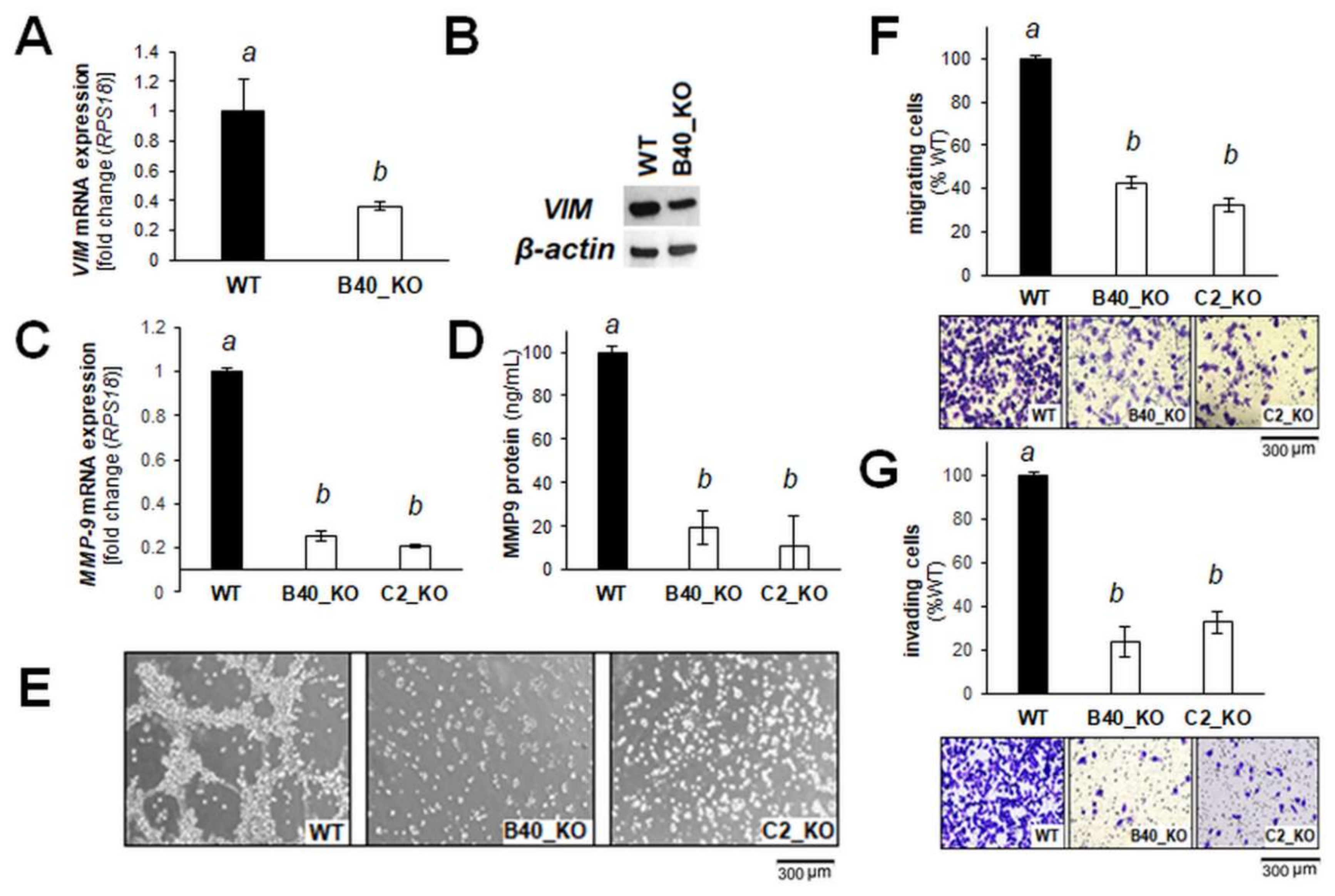
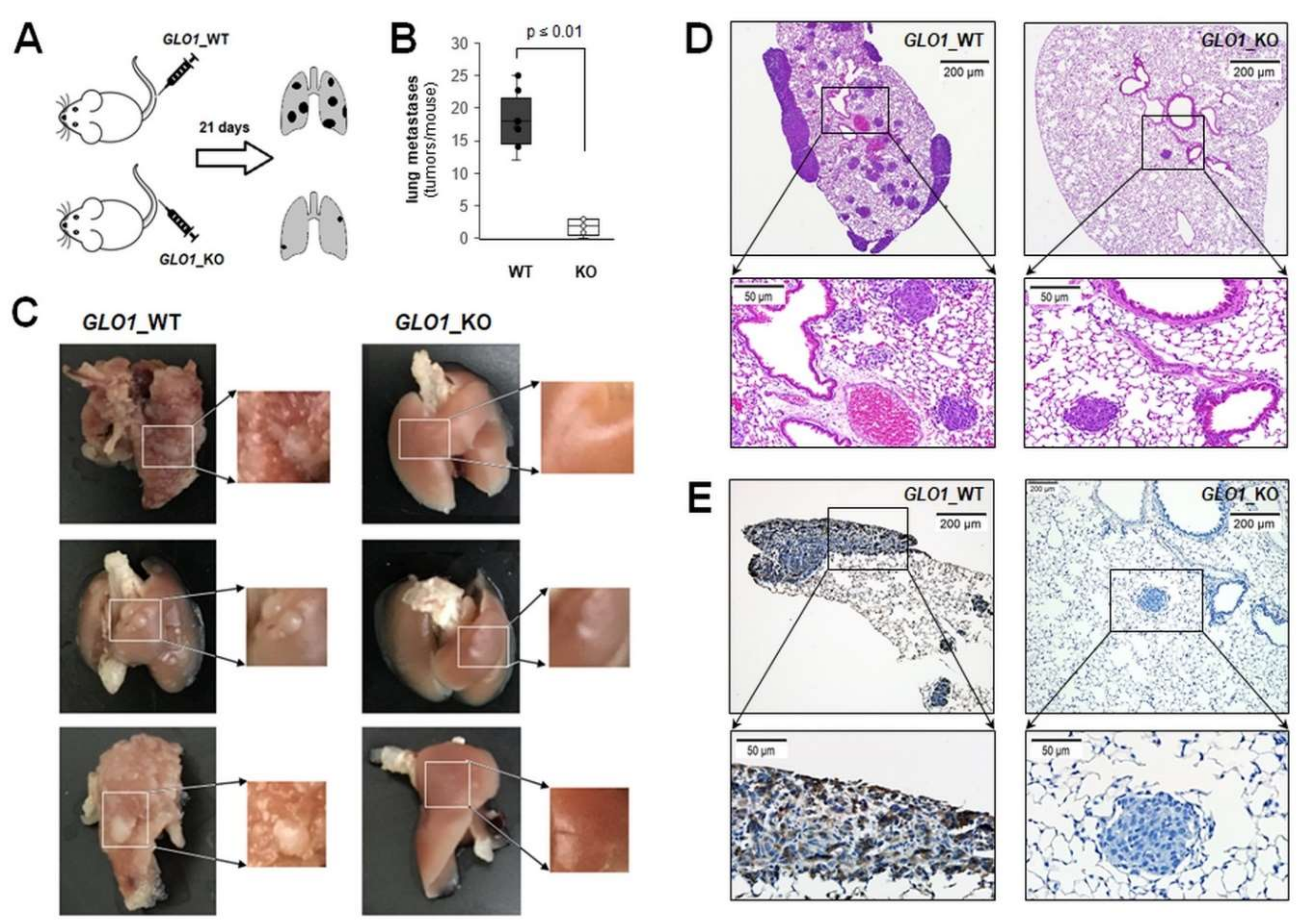
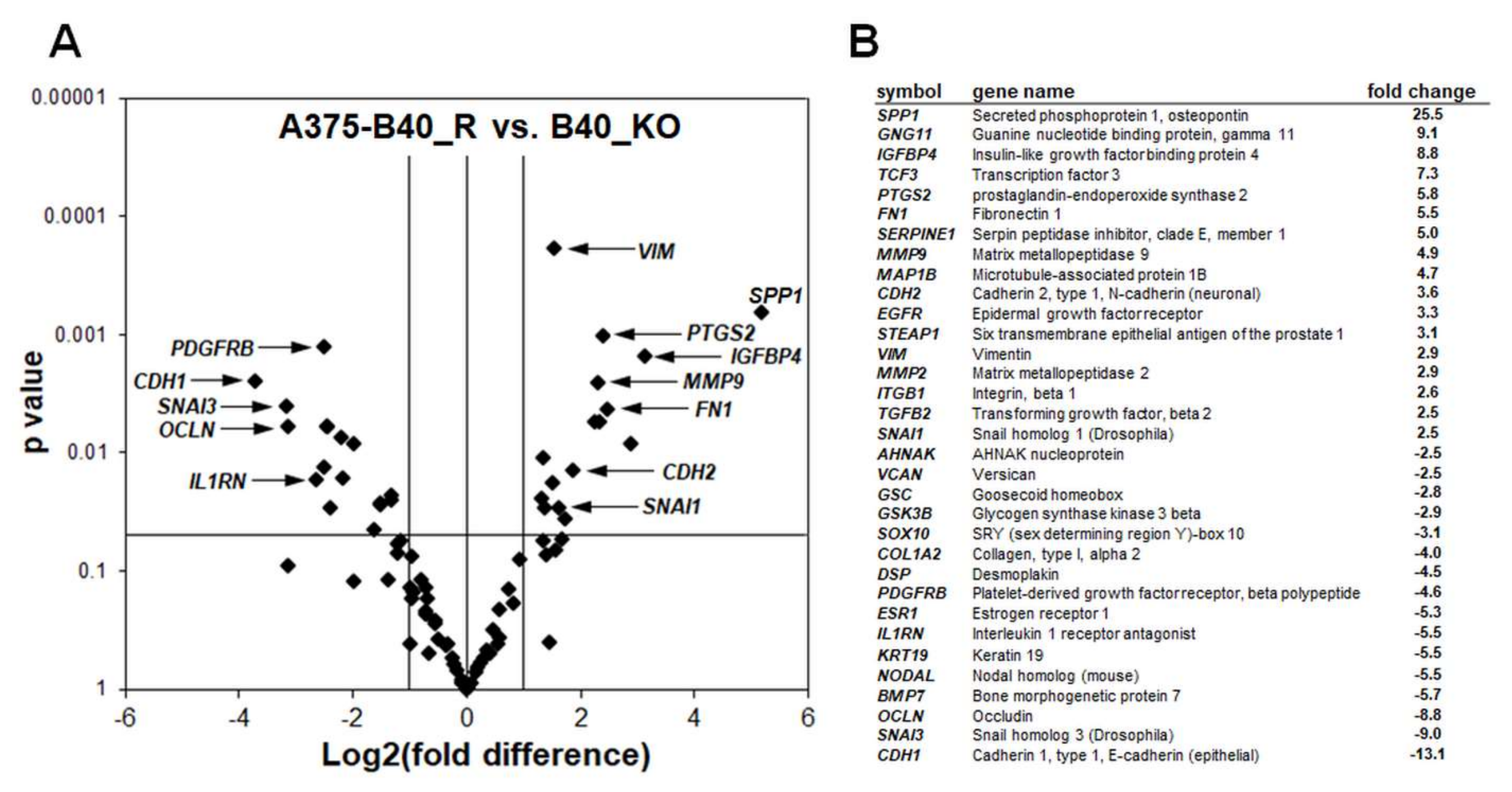
2.2. Differential Array Analysis Reveals Pronounced Modulation of EMT-Related Gene Expression in A375-GLO1_KO Cells that Display Attenuated Migration, Invasiveness, and Metastasis Assessed In Vitro and In Vivo
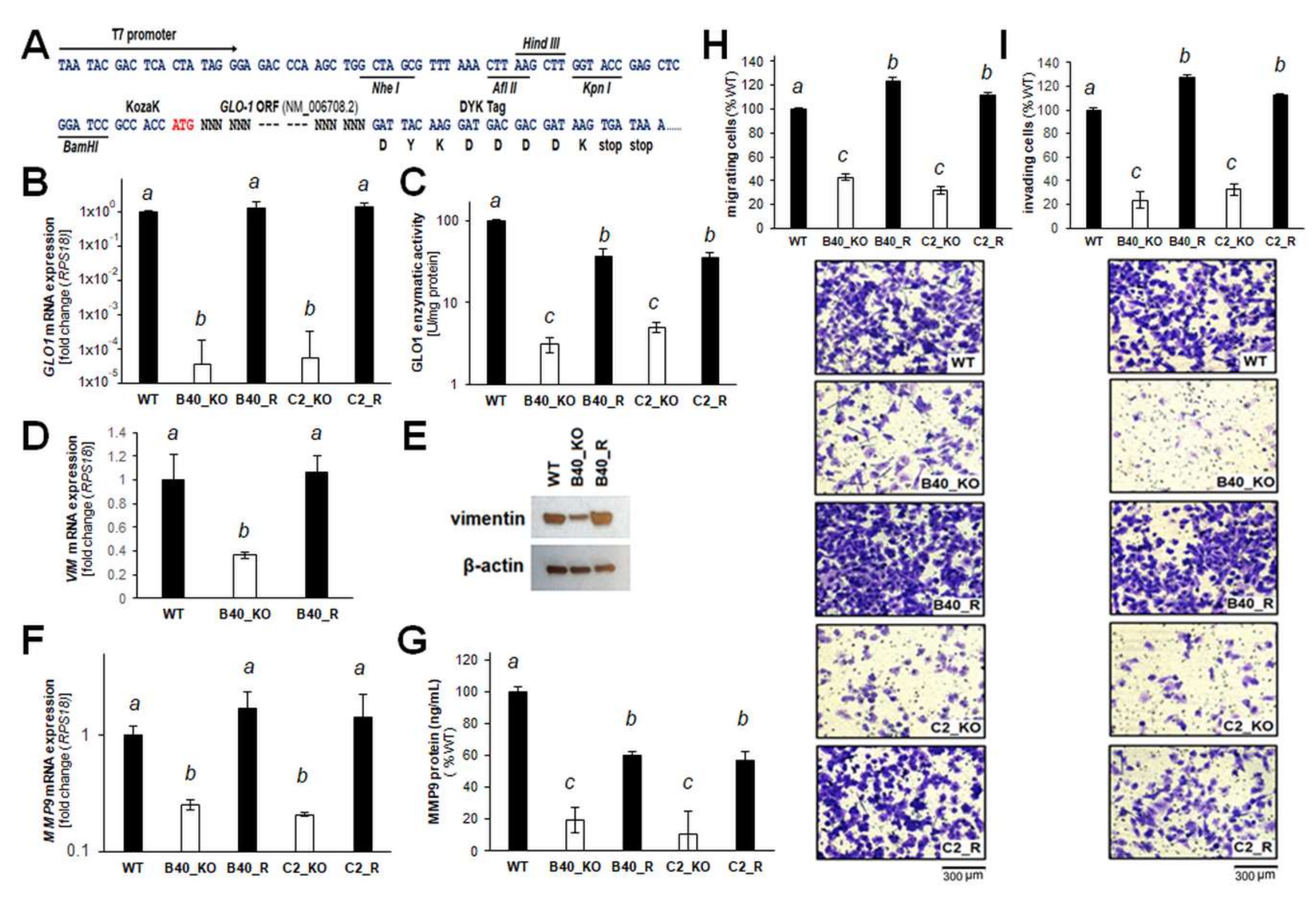
2.3. Re-Expression of GLO1 in A375-GLO1_KO Cells Reverses EMT-Related Gene Expression and Restores Melanoma Cell Migration and Invasiveness In Vitro
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. CRISPR/Cas 9 Genome Editing
4.4. Rescue Expression Construct
4.5. RNA Extraction and Single Reverse Transcrition Quantitative Polymerase Chain Reaction (RT-qPCR)
4.6. Immunoblot Detection
4.7. Glyoxalase 1-Specific Enzymatic Activity
4.8. Flow Cytometric Analysis of Cell Viability
4.9. Cell Glo Viability Assay CellTiter-Glo® Assay
4.10. Cell Proliferation Assay
4.11. Detection of Intracellular Oxidative Stress by Flow Cytometric Analysis
4.12. Determination of Reduced Cellular Glutathione Content
4.13. Human RT2ProfilerTM PCR Expression Array
4.14. Human MMP9 Immunoassay
4.15. Transwell Migration/Invasion Assay
4.16. Matrigel 3D Growth Assay (Branching Morphology Assay)
4.17. Metastasis Model In Vivo
4.18. Immunohistochemistry
4.19. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bollong, M.J.; Lee, G.; Coukos, J.S.; Yun, H.; Zambaldo, C.; Chang, J.W.; Chin, E.N.; Ahmad, I.; Chatterjee, A.K.; Lairson, L.L.; et al. A metabolite-derived protein modification integrates glycolysis with KEAP1-NRF2 signalling. Nature 2018, 562, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Xue, M.; Weickert, M.O.; Thornalley, P.J. Multiple roles of glyoxalase 1-mediated suppression of methylglyoxal glycation in cancer biology-Involvement in tumour suppression, tumour growth, multidrug resistance and target for chemotherapy. Semin. Cancer Biol. 2018, 49, 83–93. [Google Scholar] [CrossRef]
- Antognelli, C.; Mezzasoma, L.; Fettucciari, K.; Talesa, V.N. A novel mechanism of methylglyoxal cytotoxicity in prostate cancer cells. Int. J. Biochem. Cell Biol. 2013, 45, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Chiavarina, B.; Nokin, M.J.; Bellier, J.; Durieux, F.; Bletard, N.; Sherer, F.; Lovinfosse, P.; Peulen, O.; Verset, L.; Dehon, R.; et al. Methylglyoxal-Mediated Stress Correlates with High Metabolic Activity and Promotes Tumor Growth in Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 213. [Google Scholar] [CrossRef]
- Nokin, M.J.; Durieux, F.; Bellier, J.; Peulen, O.; Uchida, K.; Spiegel, D.A.; Cochrane, J.R.; Hutton, C.A.; Castronovo, V.; Bellahcène, A. Hormetic potential of methylglyoxal, a side-product of glycolysis, in switching tumours from growth to death. Sci. Rep. 2017, 7, 11722. [Google Scholar] [CrossRef] [PubMed]
- Shimada, N.; Takasawa, R.; Tanuma, S.I. Interdependence of GLO I and PKM2 in the Metabolic shift to escape apoptosis in GLO I-dependent cancer cells. Arch. Biochem. Biophys. 2018, 638, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Nokin, M.J.; Bellier, J.; Durieux, F.; Peulen, O.; Rademaker, G.; Gabriel, M.; Monseur, C.; Charloteaux, B.; Verbeke, L.; van Laere, S.; et al. Methylglyoxal, a glycolysis metabolite, triggers metastasis through MEK/ERK/SMAD1 pathway activation in breast cancer. Breast Cancer Res. 2019, 21, 11. [Google Scholar] [CrossRef]
- Thornalley, P.J.; Edwards, L.G.; Kang, Y.; Wyatt, C.; Davies, N.; Ladan, M.J.; Double, J. Antitumour activity of S-p-bromobenzylglutathione cyclopentyl diester in vitro and in vivo. Inhibition of glyoxalase I and induction of apoptosis. Biochem. Pharmacol. 1996, 51, 1365–1372. [Google Scholar] [CrossRef]
- Santarius, T.; Bignell, G.R.; Greenman, C.D.; Widaa, S.; Chen, L.; Mahoney, C.L.; Butler, A.; Edkins, S.; Waris, S.; Thornalley, P.J.; et al. GLO1-A novel amplified gene in human cancer. Genes Chromosomes Cancer 2010, 49, 711–725. [Google Scholar] [CrossRef]
- Taniguchi, H.; Horinaka, M.; Yoshida, T.; Yano, K.; Goda, A.E.; Yasuda, S.; Wakada, M.; Sakai, T. Targeting the glyoxalase pathway enhances TRAIL efficacy in cancer cells by downregulating the expression of antiapoptotic molecules. Mol. Cancer Ther. 2012, 11, 2294–2300. [Google Scholar] [CrossRef]
- Zhang, S.; Liang, X.; Zheng, X.; Huang, H.; Chen, X.; Wu, K.; Wang, B.; Ma, S. Glo1 genetic amplification as a potential therapeutic target in hepatocellular carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 2079–2090. [Google Scholar]
- Al-Shar’i, N.A.; Al-Balas, Q.A.; Al-Waqfi, R.A.; Hassan, M.A.; Alkhalifa, A.E.; Ayoub, N.M. Discovery of a nanomolar inhibitor of the human glyoxalase-I enzyme using structure-based poly-pharmacophore modelling and molecular docking. J. Comput. Aided Mol. Des. 2019, 33, 799–815. [Google Scholar] [CrossRef] [PubMed]
- Paluncic, J.; Kovacevic, Z.; Jansson, P.J.; Kalinowski, D.; Merlot, A.M.; Huang, M.L.; Lok, H.C.; Sahni, S.; Lane, D.J.; Richardson, D.R. Roads to melanoma: Key pathways and emerging players in melanoma progression and oncogenic signaling. Biochim. Biophys. Acta 2016, 1863, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef] [PubMed]
- Helgadottir, H.; Rocha Trocoli Drakensjo, I.; Girnita, A. Personalized Medicine in Malignant Melanoma: Towards Patient Tailored Treatment. Front. Oncol. 2018, 8, 202. [Google Scholar] [CrossRef] [PubMed]
- Bair, W.B., 3rd; Cabello, C.M.; Uchida, K.; Bause, A.S.; Wondrak, G.T. GLO1 overexpression in human malignant melanoma. Melanoma Res. 2010, 20, 85–96. [Google Scholar] [CrossRef]
- Hutschenreuther, A.; Bigl, M.; Hemdan, N.Y.; Debebe, T.; Gaunitz, F.; Birkenmeier, G. Modulation of GLO1 Expression Affects Malignant Properties of Cells. Int. J. Mol. Sci. 2016, 17, 2133. [Google Scholar] [CrossRef]
- Antognelli, C.; Cecchetti, R.; Riuzzi, F.; Peirce, M.J.; Talesa, V.N. Glyoxalase 1 sustains the metastatic phenotype of prostate cancer cells via EMT control. J. Cell. Mol. Med. 2018, 22, 2865–2883. [Google Scholar] [CrossRef]
- Antognelli, C.; Moretti, S.; Frosini, R.; Puxeddu, E.; Sidoni, A.; Talesa, V.N. Methylglyoxal Acts as a Tumor-Promoting Factor in Anaplastic Thyroid Cancer. Cells 2019, 8, 547. [Google Scholar] [CrossRef]
- Cheng, W.L.; Tsai, M.M.; Tsai, C.Y.; Huang, Y.H.; Chen, C.Y.; Chi, H.C.; Tseng, Y.H.; Chao, I.W.; Lin, W.C.; Wu, S.M.; et al. Glyoxalase-I is a novel prognosis factor associated with gastric cancer progression. PLoS ONE 2012, 7, e34352. [Google Scholar] [CrossRef]
- Cesi, G.; Walbrecq, G.; Zimmer, A.; Kreis, S.; Haan, C. ROS production induced by BRAF inhibitor treatment rewires metabolic processes affecting cell growth of melanoma cells. Mol. Cancer 2017, 16, 102. [Google Scholar] [CrossRef]
- Ratnikov, B.I.; Scott, D.A.; Osterman, A.L.; Smith, J.W.; Ronai, Z.A. Metabolic rewiring in melanoma. Oncogene 2017, 36, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Qiao, S.; Tao, S.; de la Rojo Vega, M.; Park, S.L.; Vonderfecht, A.A.; Jacobs, S.L.; Zhang, D.D.; Wondrak, G.T. The antimalarial amodiaquine causes autophagic-lysosomal and proliferative blockade sensitizing human melanoma cells to starvation- and chemotherapy-induced cell death. Autophagy 2013, 9, 2087–2102. [Google Scholar] [CrossRef] [PubMed]
- Samulitis, B.K.; Dorr, R.T.; Chow, H.H. Interaction of dacarbazine and imexon, in vitro and in vivo, in human A375 melanoma cells. Anticancer Res. 2011, 31, 2781–2785. [Google Scholar] [PubMed]
- Lo, R.S.; Witte, O.N. Transforming growth factor-beta activation promotes genetic context-dependent invasion of immortalized melanocytes. Cancer Res. 2008, 68, 4248–4257. [Google Scholar] [CrossRef] [PubMed]
- Rolland, Y.; Demeule, M.; Fenart, L.; Béliveau, R. Inhibition of melanoma brain metastasis by targeting melanotransferrin at the cell surface. Pigment Cell Melanoma Res. 2009, 22, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.; Wierinckx, A.; Saldanha, G.; Osborne, J.; Hutchinson, P.; Tse, G.; et al. A switch in the expression of embryonic EMT-inducers drives the development of malignant melanoma. Cancer Cell 2013, 24, 466–480. [Google Scholar] [CrossRef]
- Moro, N.; Mauch, C.; Zigrino, P. Metalloproteinases in melanoma. Eur. J. Cell Biol. 2014, 93, 23–29. [Google Scholar] [CrossRef]
- Li, F.Z.; Dhillon, A.S.; Anderson, R.L.; McArthur, G.; Ferrao, P.T. Phenotype switching in melanoma: Implications for progression and therapy. Front. Oncol. 2015, 5, 31. [Google Scholar] [CrossRef]
- Pearlman, R.L.; de Montes Oca, M.K.; Pal, H.C.; Afaq, F. Potential therapeutic targets of epithelial-mesenchymal transition in melanoma. Cancer Lett. 2017, 391, 125–140. [Google Scholar] [CrossRef]
- Sasaki, K.; Sugai, T.; Ishida, K.; Osakabe, M.; Amano, H.; Kimura, H.; Sakuraba, M.; Kashiwa, K.; Kobayashi, S. Analysis of cancer-associated fibroblasts and the epithelial-mesenchymal transition in cutaneous basal cell carcinoma, squamous cell carcinoma, and malignant melanoma. Hum. Pathol. 2018, 79, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wicklein, D.; Otto, B.; Suling, A.; Elies, E.; Lüers, G.; Lange, T.; Feldhaus, S.; Maar, H.; Schröder-Schwarz, J.; Brunner, G.; et al. CEACAM1 promotes melanoma metastasis and is involved in the regulation of the EMT associated gene network in melanoma cells. Sci. Rep. 2018, 8, 11893. [Google Scholar] [CrossRef] [PubMed]
- Ercolano, G.; De Cicco, P.; Rubino, V.; Terrazzano, G.; Ruggiero, G.; Carriero, R.; Kunderfranco, P.; Ianaro, A. Knockdown of PTGS2 by CRISPR/CAS9 System Designates a New Potential Gene Target for Melanoma Treatment. Front. Pharmacol. 2019, 10, 1456. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.B.; Li, K.Y.; Men, Y.L.; Ding, C.; Du, J.; Liang, T.; Ji, Z.; Chen, L.; Wang, T.; Kang, Q. Protein 4.1B Suppresses Tumor Metastasis by Regulating Epithelial-mesenchymal Transition Progression in Melanoma Cells. Int. J. Med. Sci. 2019, 16, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Prasad, P.; Vasas, A.; Hohmann, J.; Bishayee, A.; Sinha, D. Cirsiliol suppresed epithelial to mesenchymal transition in B16F10 malignant melanoma cells through alteration of the PI3K/Akt/NF-kB signaling pathway. Int. J. Mol. Sci. 2019, 20, 608. [Google Scholar] [CrossRef]
- Falzone, L.; Salemi, R.; Travali, S.; Scalisi, A.; McCubrey, J.A.; Candido, S.; Libra, M. MMP-9 overexpression is associated with intragenic hypermethylation of MMP9 gene in melanoma. Aging 2016, 8, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.Y.; Liu, Y.W.; Xue, M.Z.; Liu, H.; Du, S.; Zhang, L.; Wang, P. Synergistic action of master transcription factors controls epithelial-to-mesenchymal transition. Nucleic Acids Res. 2016, 44, 2514–2527. [Google Scholar] [CrossRef] [PubMed]
- Amicarelli, F.; Bucciarelli, T.; Poma, A.; Aimola, P.; Di Ilio, C.; Ragnelli, A.M.; Miranda, M. Adaptive response of human melanoma cells to methylglyoxal-injury. Carcinogenesis 1998, 19, 519–523. [Google Scholar] [CrossRef]
- Kosmopolou, M.; Giannopoulou, A.F.; Iliou, A.; Benaki, D.; Panagiotakis, A.; Velentzas, A.D.; Konstantakou, E.G.; Papassideri, I.S.; Mikros, E.; Stravopodis, D.J.; et al. Human melanoma-cell metabolic profiling: Identification of novel biomarkers indicating metastasis. Int. J. Mol. Sci. 2020, 17, 2436. [Google Scholar] [CrossRef]
- Rangel, J.; Nosrati, M.; Torabian, S.; Shaikh, L.; Leong, S.P.; Haqq, C.; Miller, J.R., 3rd; Sagebiel, R.W.; Kashani-Sabet, M. Osteopontin as a molecular prognostic marker for melanoma. Cancer 2008, 112, 144–150. [Google Scholar] [CrossRef]
- Antognelli, C.; Gambelunghe, A.; Muzi, G.; Talesa, V.N. Glyoxalase I drives epithelial-to-mesenchymal transition via argpyrimidine-modified Hsp70, miR-21 and SMAD signalling in human bronchial cells BEAS-2B chronically exposed to crystalline silica Min-U-Sil 5: Transformation into a neoplastic-like phenotype. Free Radic. Biol. Med. 2016, 92, 110–125. [Google Scholar] [CrossRef] [PubMed]
- Tamori, S.; Nozaki, Y.; Motomura, H.; Nakane, H.; Katayama, R.; Onaga, C.; Kikuchi, E.; Shimada, N.; Suzuki, Y.; Noike, M.; et al. Glyoxalase 1 gene is highly expressed in basal-like human breast cancers and contributes to survival of ALDH1-positive breast cancer stem cells. Oncotarget 2018, 9, 36515–36529. [Google Scholar] [CrossRef] [PubMed]
- Vickers, T.J.; Greig, N.; Fairlamb, A.H. A trypanothione-dependent glyoxalase I with a prokaryotic ancestry in Leishmania major. Proc. Natl. Acad. Sci. USA 2004, 101, 13186–13191. [Google Scholar] [CrossRef] [PubMed]
- Duggan, M.C.; Stiff, A.R.; Bainazar, M.; Regan, K.; Olaverria Salavaggione, G.N.; Maharry, S.; Blachly, J.S.; Krischak, M.; Walker, C.J.; Latchana, N.; et al. Identification of NRAS isoform 2 overexpression as a mechanism facilitating BRAF inhibitor resistance in malignant melanoma. Proc. Natl. Acad. Sci. USA 2017, 114, 9629–9634. [Google Scholar] [CrossRef]
- Cabello, C.M.; Lamore, S.D.; Bair, W.B., 3rd; Shuxi, Q.; Sara, A.; Jessica, L.L.; Georg, T.W. The redox antimalarial dihydroartemisinin targets human metastatic melanoma cells but not primary melanocytes with induction of NOXA-dependent apoptosis. Investig. New Drugs 2012, 30, 1289–1301. [Google Scholar] [CrossRef]
- Davis, A.L.; Qiao, S.; Lesson, J.L.; Rojo de la Vega, M.; Park, S.L.; Seanez, C.M.; Gokhale, V.; Cabello, C.M.; Wondrak, G.T. The quinone methide aurin is a heat shock response inducer that causes proteotoxic stress and Noxa-dependent apoptosis in malignant melanoma cells. J. Biol. Chem. 2015, 290, 1623–1638. [Google Scholar] [CrossRef]
- Sligh, J.; Janda, J.; Jandova, J. Mutations in BALB mitochondrial DNA induce CCL20 up-regulation promoting tumorigenic phenotypes. Mutat. Res. 2014, 769, 49–58. [Google Scholar] [CrossRef]
- Jandova, J.; Mason, C.J.; Pawar, S.C.; Watts, G.S. Fn14 receptor promotes invasive potential and metastatic capacity of non-small lung adenocarcinoma cells through the up-regulation of integrin alpha6. Neoplasma 2015, 62, 41–52. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jandova, J.; Perer, J.; Hua, A.; Snell, J.A.; Wondrak, G.T. Genetic Target Modulation Employing CRISPR/Cas9 Identifies Glyoxalase 1 as a Novel Molecular Determinant of Invasion and Metastasis in A375 Human Malignant Melanoma Cells In Vitro and In Vivo. Cancers 2020, 12, 1369. https://doi.org/10.3390/cancers12061369
Jandova J, Perer J, Hua A, Snell JA, Wondrak GT. Genetic Target Modulation Employing CRISPR/Cas9 Identifies Glyoxalase 1 as a Novel Molecular Determinant of Invasion and Metastasis in A375 Human Malignant Melanoma Cells In Vitro and In Vivo. Cancers. 2020; 12(6):1369. https://doi.org/10.3390/cancers12061369
Chicago/Turabian StyleJandova, Jana, Jessica Perer, Anh Hua, Jeremy A. Snell, and Georg T. Wondrak. 2020. "Genetic Target Modulation Employing CRISPR/Cas9 Identifies Glyoxalase 1 as a Novel Molecular Determinant of Invasion and Metastasis in A375 Human Malignant Melanoma Cells In Vitro and In Vivo" Cancers 12, no. 6: 1369. https://doi.org/10.3390/cancers12061369
APA StyleJandova, J., Perer, J., Hua, A., Snell, J. A., & Wondrak, G. T. (2020). Genetic Target Modulation Employing CRISPR/Cas9 Identifies Glyoxalase 1 as a Novel Molecular Determinant of Invasion and Metastasis in A375 Human Malignant Melanoma Cells In Vitro and In Vivo. Cancers, 12(6), 1369. https://doi.org/10.3390/cancers12061369