Application of Next-Generation Sequencing for the Genomic Characterization of Patients with Smoldering Myeloma

 , , , ,
, , , ,  and
and 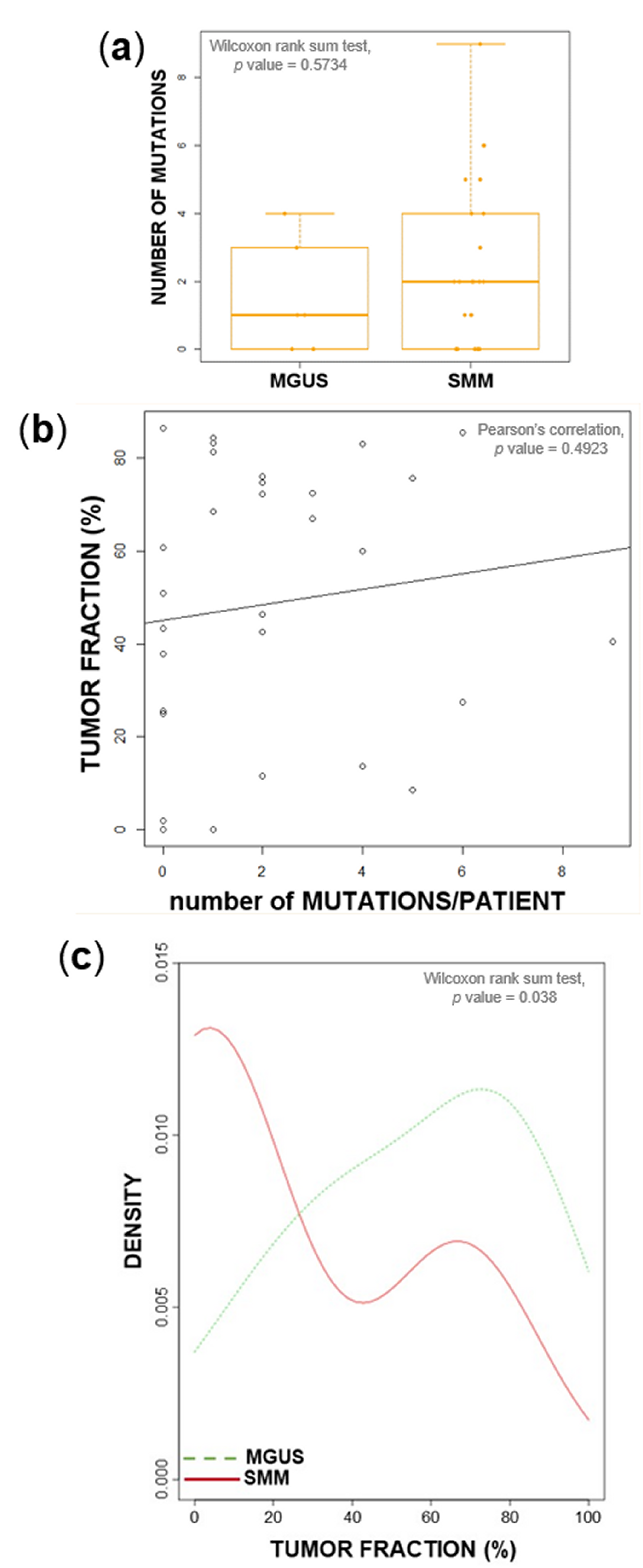{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Tumor Load and Tumor Mutational Burden
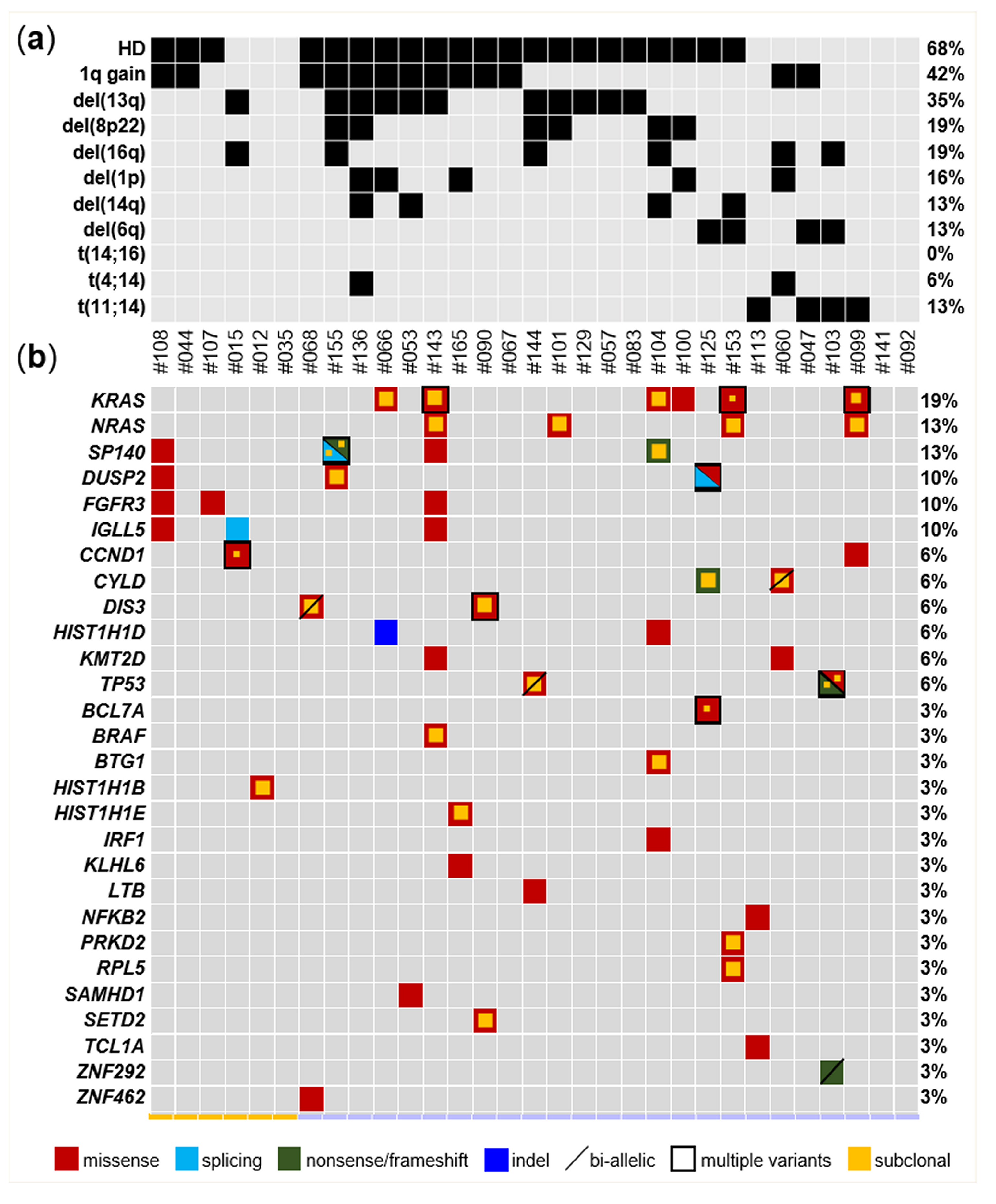
2.2. Mutational Landscape
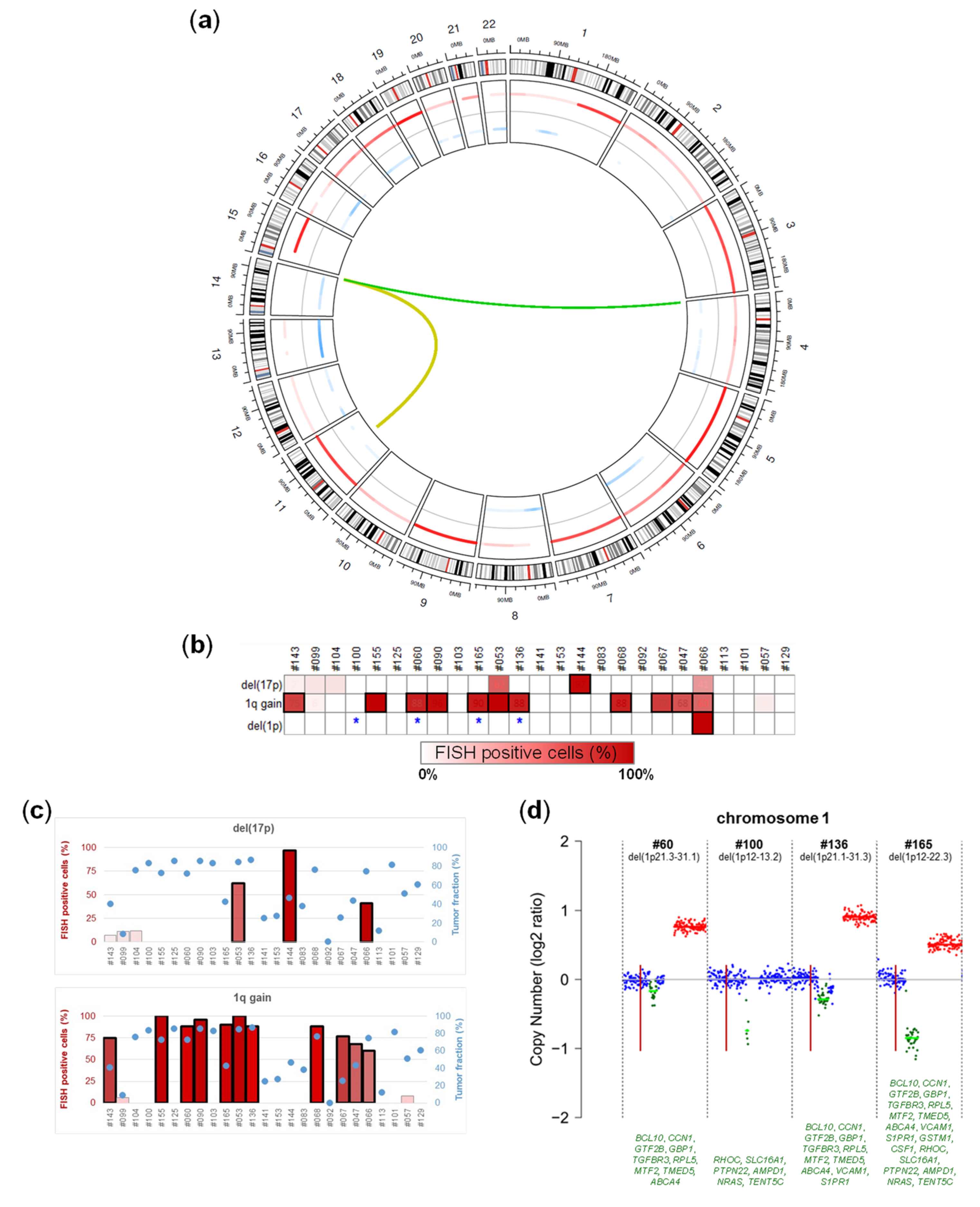
2.3. Chromosomal Alterations
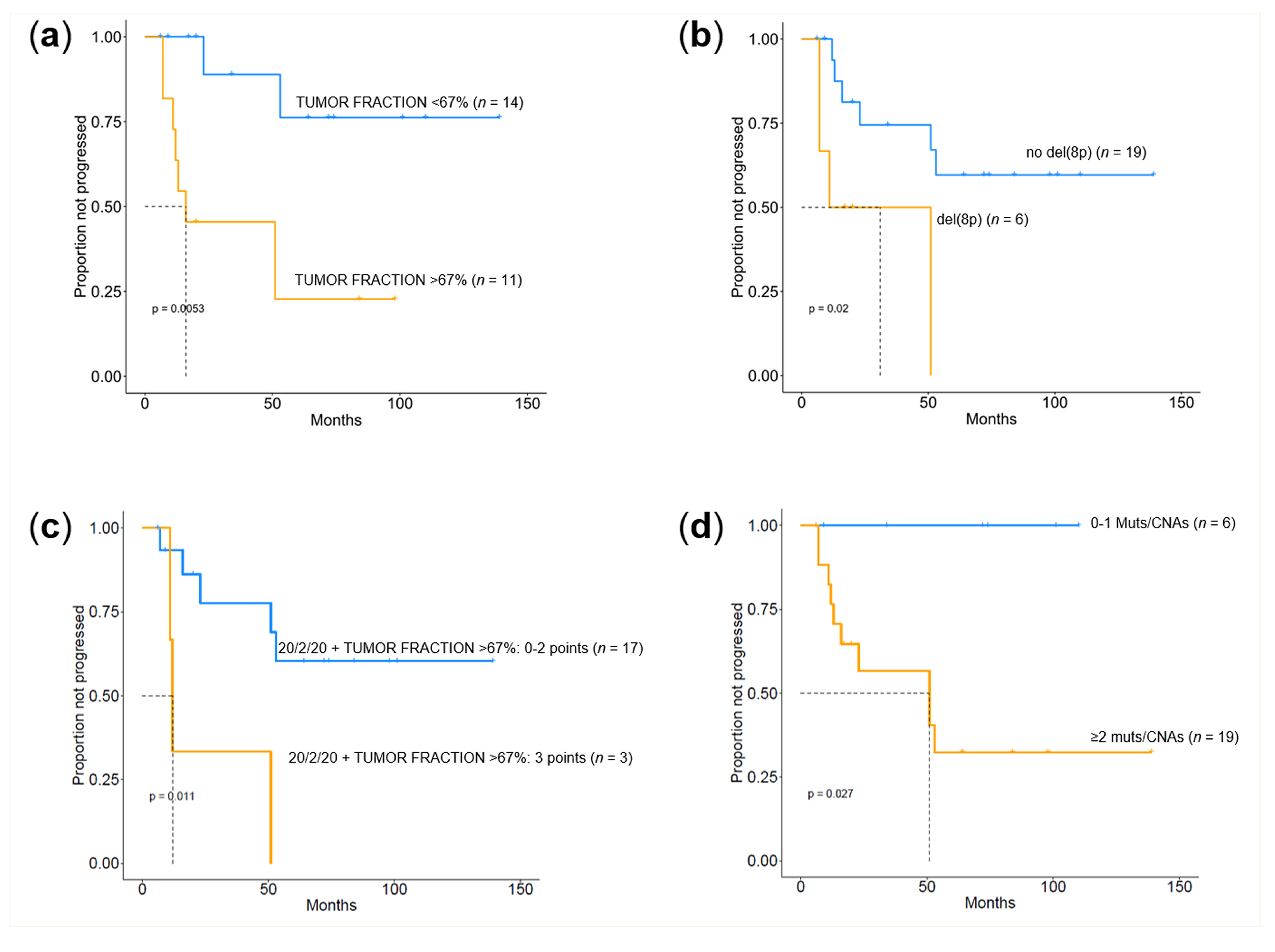
2.4. Prognostic Value of Genomic Findings
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Next-Generation Sequencing
4.3. Fluorescence In Situ Hybridization (FISH) Analysis
4.4. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lionetti, M.; Neri, A. Utilizing next-generation sequencing in the management of multiple myeloma. Expert Rev. Mol. Diagn. 2017, 17, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Bolli, N.; Genuardi, E.; Ziccheddu, B.; Martello, M.; Oliva, S.; Terragna, C. Next-Generation Sequencing for Clinical Management of Multiple Myeloma: Ready for Prime Time? Front. Oncol. 2020, 10, 189. [Google Scholar] [CrossRef] [PubMed]
- Maura, F.; Bolli, N.; Rustad, E.H.; Hultcrantz, M.; Munshi, N.; Landgren, O. Moving From Cancer Burden to Cancer Genomics for Smoldering Myeloma. JAMA Oncol. 2020, 6, 425. [Google Scholar] [CrossRef] [PubMed]
- Smadbeck, J.; Peterson, J.F.; Pearce, K.E.; Pitel, B.A.; Figueroa, A.L.; Timm, M.; Jevremovic, A.D.; Shi, M.; Stewart, A.K.; Braggio, E.; et al. Mate pair sequencing outperforms fluorescence in situ hybridization in the genomic characterization of multiple myeloma. Blood Cancer J. 2019, 9, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Bolli, N.; Maura, F.; Minvielle, S.; Gloznik, D.; Szalat, R.; Fullam, A.; Martincorena, I.; Dawson, K.J.; Samur, M.K.; Zamora, J.; et al. Genomic patterns of progression in smoldering multiple myeloma. Nat. Commun. 2018, 9, 3363. [Google Scholar] [CrossRef]
- Mateos, M.-V.; Hernández, M.-T.; Giraldo, P.; De La Rubia, J.; De Arriba, F.; Corral, L.L.; Rosiñol, L.; Paiva, B.; Palomera, L.; Bargay, J.; et al. Lenalidomide plus dexamethasone versus observation in patients with high-risk smouldering multiple myeloma (QuiRedex): Long-term follow-up of a randomised, controlled, phase 3 trial. Lancet Oncol. 2016, 17, 1127–1136. [Google Scholar] [CrossRef]
- Lonial, S.; Jacobus, S.; Fonseca, R.; Weiss, M.; Kumar, S.; Orlowski, R.Z.; Kaufman, J.L.; Yacoub, A.M.; Buadi, F.K.; O’Brien, T.; et al. Randomized Trial of Lenalidomide Versus Observation in Smoldering Multiple Myeloma. J. Clin. Oncol. 2020, 38, 1126–1137. [Google Scholar] [CrossRef]
- Bolli, N.; Li, Y.; Sathiaseelan, V.; Raine, K.; Jones, D.; Ganly, P.; Cocito, F.; Bignell, G.; Chapman, M.A.; Sperling, A.S.; et al. A DNA target-enrichment approach to detect mutations, copy number changes and immunoglobulin translocations in multiple myeloma. Blood Cancer J. 2016, 6, e467. [Google Scholar] [CrossRef]
- Yellapantula, V.; Hultcrantz, M.; Rustad, E.H.; Wasserman, E.; Londono, D.; Cimera, R.; Ciardiello, A.; Landau, H.; Akhlaghi, T.; Mailankody, S.; et al. Comprehensive detection of recurring genomic abnormalities: A targeted sequencing approach for multiple myeloma. Blood Cancer J. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Jiménez, C.; Jara-Acevedo, M.; Corchete, L.A.; Castillo, D.; Ordoñez, G.R.; Sarasquete, M.E.; Puig, N.; Martínez-López, J.; Prieto-Conde, M.I.; García-Álvarez, M.; et al. A Next-Generation Sequencing Strategy for Evaluating the Most Common Genetic Abnormalities in Multiple Myeloma. J. Mol. Diagn. 2017, 19, 99–106. [Google Scholar] [CrossRef]
- White, B.S.; Lanc, I.; O’Neal, J.; Gupta, H.; Fulton, R.S.; Schmidt, H.; Fronick, C.; Belter, E.A.; Fiala, M.; King, J.; et al. A multiple myeloma-specific capture sequencing platform discovers novel translocations and frequent, risk-associated point mutations in IGLL5. Blood Cancer J. 2018, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Corre, J.; Cleynen, A.; Du Pont, S.R.; Buisson, L.; Bolli, N.; Attal, M.; Munshi, N.; Avet-Loiseau, H. Multiple myeloma clonal evolution in homogeneously treated patients. Leukemia 2018, 32, 2636–2647. [Google Scholar] [CrossRef] [PubMed]
- Chavan, S.S.; He, J.; Tytarenko, R.; Deshpande, S.; Patel, P.; Bailey, M.; Stein, C.K.; Stephens, O.; Weinhold, N.; Petty, N.; et al. Bi-allelic inactivation is more prevalent at relapse in multiple myeloma, identifying RB1 as an independent prognostic marker. Blood Cancer J. 2017, 7, e535. [Google Scholar] [CrossRef] [PubMed]
- Boyd, K.D.; Ross, F.M.; Tapper, W.J.; Chiecchio, L.; Dagrada, G.; Konn, Z.J.; Gonzalez, D.; Walker, B.A.; Hockley, S.L.; Wardell, C.; et al. The clinical impact and molecular biology of del(17p) in multiple myeloma treated with conventional or thalidomide-based therapy. Genes Chromosom. Cancer 2011, 50, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Adalsteinsson, V.; Ha, G.; Freeman, S.S.; Choudhury, A.D.; Stover, D.; Parsons, H.A.; Gydush, G.; Reed, S.C.; Rotem, D.; Rhoades, J.; et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat. Commun. 2017, 8, 1324. [Google Scholar] [CrossRef] [PubMed]
- Lakshman, A.; Rajkumar, S.V.; Buadi, F.K.; Binder, M.; Gertz, M.A.; Lacy, M.Q.; Dispenzieri, A.; Dingli, D.; Fonder, A.L.; Hayman, S.R.; et al. Risk stratification of smoldering multiple myeloma incorporating revised IMWG diagnostic criteria. Blood Cancer J. 2018, 8, 59. [Google Scholar] [CrossRef]
- Kyle, R.A.; Larson, D.R.; Therneau, T.M.; Dispenzieri, A.; Kumar, S.; Cerhan, J.R.; Rajkumar, S.V. Long-Term Follow-up of Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 2018, 378, 241–249. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Landgren, O.; Mateos, M.V. Smoldering multiple myeloma. Blood 2015, 125, 3069–3075. [Google Scholar] [CrossRef]
- Dutta, A.K.; Fink, J.L.; Grady, J.P.; Morgan, G.; Mullighan, C.G.; To, L.B.; Hewett, D.R.; Zannettino, A.C. Subclonal evolution in disease progression from MGUS/SMM to multiple myeloma is characterised by clonal stability. Leukemia 2018, 33, 457–468. [Google Scholar] [CrossRef]
- Drach, J.; Angerler, J.; Schuster, J.; Rothermundt, C.; Thalhammer, R.; Haas, O.; Jager, U.; Fiegl, M.; Geissler, K.; Ludwig, H. Interphase fluorescence in situ hybridization identifies chromosomal abnormalities in plasma cells from patients with monoclonal gammopathy of undetermined significance. Blood 1995, 86, 3915–3921. [Google Scholar] [CrossRef]
- Walker, B.A.; Wardell, C.; Ross, M.; Bentley, D.; Gutiérrez, N.C.; García-Sanz, R.; San-Miguel, J.; Davies, F.E.; Gonzalez, D.; Morgan, G.; et al. Intraclonal heterogeneity is a critical early event in the development of myeloma and precedes the development of clinical symptoms. Leukemia 2013, 28, 384–390. [Google Scholar] [CrossRef] [PubMed]
- López-Corral, L.; Sarasquete, M.E.; Hernández-García, M.T.; Giraldo, P.; Hernández, J.; González, M.; Hernández-Rivas, J.M.; San-Miguel, J.; Gutiérrez, N.C.; Beà, S.; et al. SNP-based mapping arrays reveal high genomic complexity in monoclonal gammopathies, from MGUS to myeloma status. Leukemia 2012, 26, 2521–2529. [Google Scholar] [CrossRef] [PubMed]
- Ziccheddu, B.; Biancon, G.; Bagnoli, F.; De Philippis, C.; Maura, F.; Rustad, E.H.; Dugo, M.; Devecchi, A.; De Cecco, L.; Sensi, M.; et al. Integrative analysis of the genomic and transcriptomic landscape of double-refractory multiple myeloma. Blood Adv. 2020, 4, 830–844. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef]
- Bolli, N.; Biancon, G.; O’Meara, S.; Raine, K.; Teague, J.W.; Butler, A.P.; Carniti, C.; Gerstung, M.; Bagratuni, T.; Kastritis, E.; et al. Analysis of the genomic landscape of multiple myeloma highlights novel prognostic markers and disease subgroups. Leukemia 2018, 32, 2604–2616. [Google Scholar] [CrossRef]
- Lionetti, M.; Barbieri, M.; Todoerti, K.; Agnelli, L.; Marzorati, S.; Fabris, S.; Ciceri, G.; Galletti, S.; Milesi, G.; Manzoni, M.; et al. Molecular spectrum of BRAF, NRAS and KRAS gene mutations in plasma cell dyscrasias: Implication for MEK-ERK pathway activation. Oncotarget 2015, 6, 24205–24217. [Google Scholar] [CrossRef]
- Thakurta, A.; Ortiz, M.; Blecua, P.; Towfic, F.; Corre, J.; Serbina, N.V.; Flynt, E.; Yu, Z.; Yang, Z.; Palumbo, A.; et al. High subclonal fraction of 17p deletion is associated with poor prognosis in multiple myeloma. Blood 2019, 133, 1217–1221. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; A Dimopoulos, M.; Palumbo, A.; Bladé, J.; Merlini, G.; Mateos, M.-V.; Rajkumar, S.V.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Maura, F.; Bolli, N.; Angelopoulos, N.; Dawson, K.J.; Leongamornlert, D.; Martincorena, I.; Mitchell, T.J.; Fullam, A.; Gonzalez, S.; Szalat, R.; et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat. Commun. 2019, 10, 3835. [Google Scholar] [CrossRef]
- Gerber, B.; Manzoni, M.; Spina, V.; Bruscaggin, A.; Lionetti, M.; Fabris, S.; Barbieri, M.; Ciceri, G.; Pompa, A.; Forestieri, G.; et al. Circulating tumor DNA as a liquid biopsy in plasma cell dyscrasias. Haematol. 2018, 103, e245–e248. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.W.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Agnelli, L.; Fabris, S.; Bicciato, S.; Basso, D.; Baldini, L.; Morabito, F.; Verdelli, D.; Todoerti, K.; Lambertenghi-Deliliers, G.; Lombardi, L.; et al. Upregulation of translational machinery and distinct genetic subgroups characterise hyperdiploidy in multiple myeloma. Br. J. Haematol. 2007, 136, 565–573. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manzoni, M.; Marchica, V.; Storti, P.; Ziccheddu, B.; Sammarelli, G.; Todaro, G.; Pelizzoni, F.; Salerio, S.; Notarfranchi, L.; Pompa, A.; et al. Application of Next-Generation Sequencing for the Genomic Characterization of Patients with Smoldering Myeloma. Cancers 2020, 12, 1332. https://doi.org/10.3390/cancers12051332
Manzoni M, Marchica V, Storti P, Ziccheddu B, Sammarelli G, Todaro G, Pelizzoni F, Salerio S, Notarfranchi L, Pompa A, et al. Application of Next-Generation Sequencing for the Genomic Characterization of Patients with Smoldering Myeloma. Cancers. 2020; 12(5):1332. https://doi.org/10.3390/cancers12051332
Chicago/Turabian StyleManzoni, Martina, Valentina Marchica, Paola Storti, Bachisio Ziccheddu, Gabriella Sammarelli, Giannalisa Todaro, Francesca Pelizzoni, Simone Salerio, Laura Notarfranchi, Alessandra Pompa, and et al. 2020. "Application of Next-Generation Sequencing for the Genomic Characterization of Patients with Smoldering Myeloma" Cancers 12, no. 5: 1332. https://doi.org/10.3390/cancers12051332
APA StyleManzoni, M., Marchica, V., Storti, P., Ziccheddu, B., Sammarelli, G., Todaro, G., Pelizzoni, F., Salerio, S., Notarfranchi, L., Pompa, A., Baldini, L., Bolli, N., Neri, A., Giuliani, N., & Lionetti, M. (2020). Application of Next-Generation Sequencing for the Genomic Characterization of Patients with Smoldering Myeloma. Cancers, 12(5), 1332. https://doi.org/10.3390/cancers12051332