Immune Phenotype and Immune Checkpoint Inhibitors for the Treatment of Human Hepatocellular Carcinoma



Abstract
1. Introduction
2. Molecular Classification and Immune Phenotype of HCC
2.1. Oncogenic Signal Activation in HCC
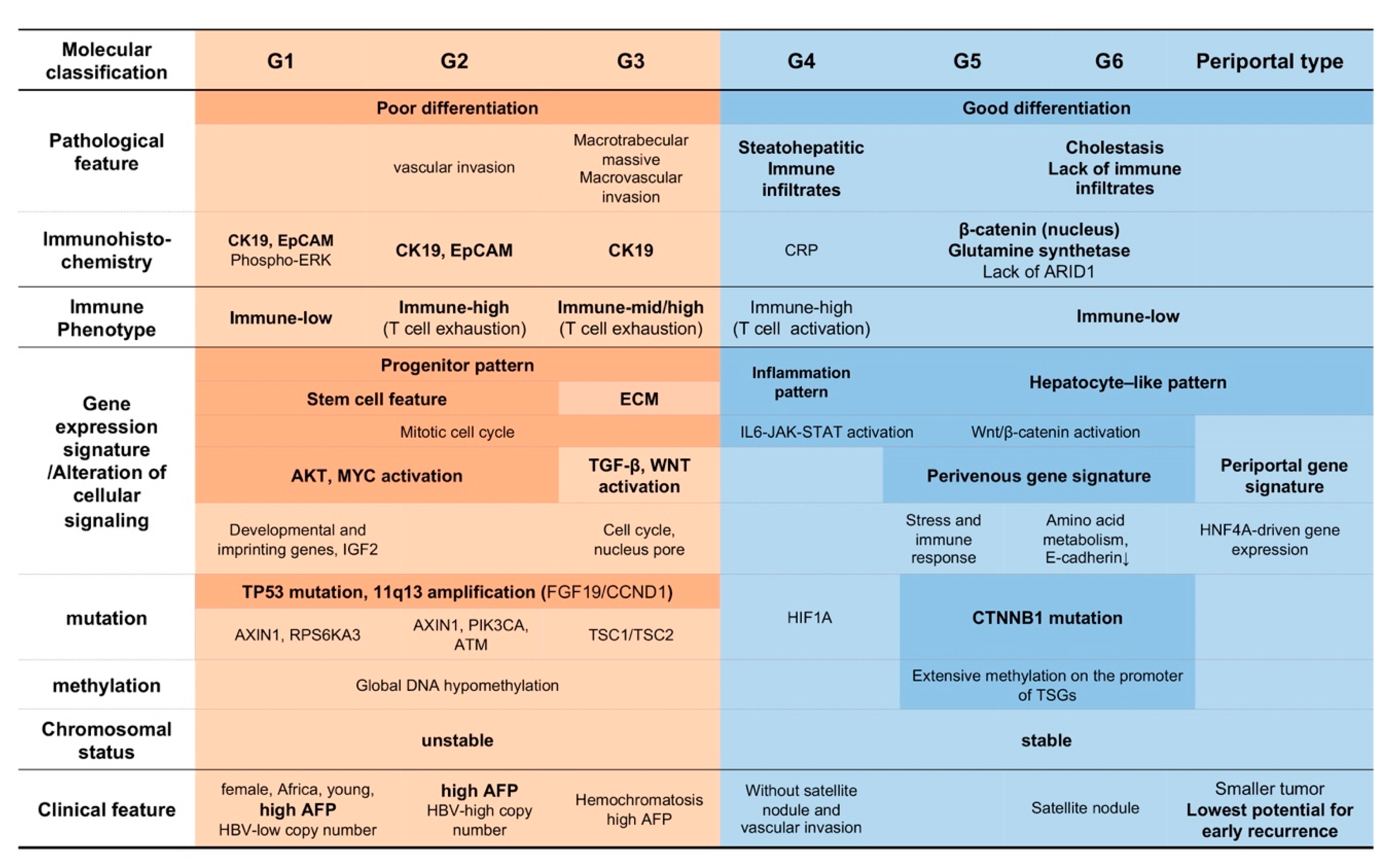
2.2. Molecular Subclass and Tumor Characteristics
2.3. Immune Phenothype of HCC
2.3.1. Classification of HCC Based on the Gene Expression Pattern and Immune Milieu
2.3.2. Characteristics of Inflammatory Infiltrates in HCC Tissues
3. Effective Application of Immune Checkpoint Inhibitors for HCC Cases
3.1. HCC Response to Immune Checkpoint Inhibitors
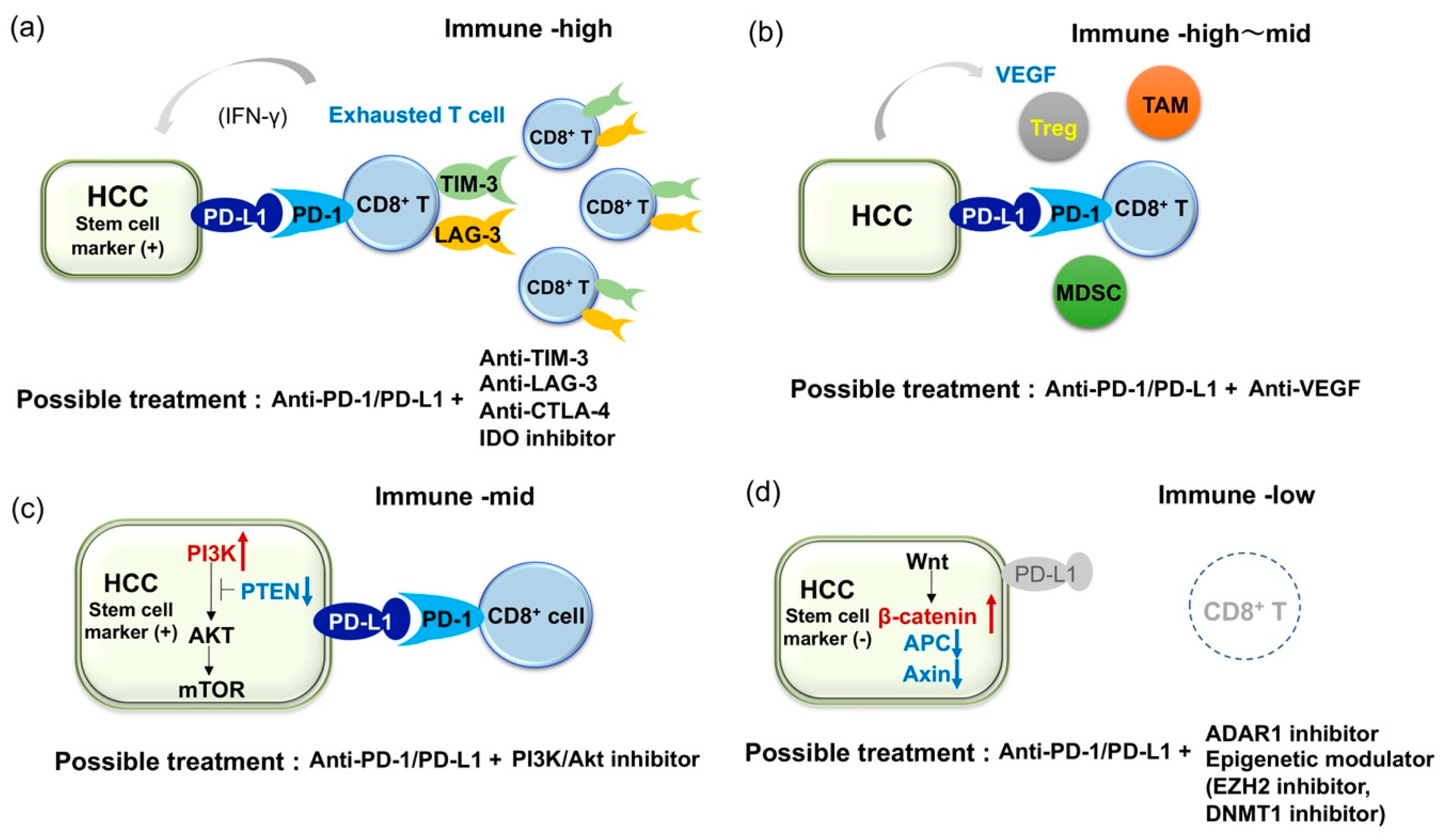
3.2. Combined Immune Checkpoint Blockade Based on Inflammatory Infiltrate Characteristics of HCC
3.3. Combined Blockade of PD-1/PD-L1 and VEGF Axis
3.4. Immune Checkpoint Inhibitors of Cancer Stem Cells
3.5. Current Limitation of Immune Checkpoint Inhibitors and Challenge for HCC with Lack of Immune Infiltrates
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M. Systemic therapy for hepatocellular carcinoma: Latest advances. Cancers 2018, 10, 412. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Kitano, M.; Sakurai, T.; Kudo, M. Molecular mechanism and prediction of sorafenib chemoresistance in human hepatocellular carcinoma. Dig. Dis. 2015, 33, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Arizumi, T.; Hagiwara, S.; Ida, H.; Sakurai, T.; Kudo, M. MicroRNAs for the prediction of early response to sorafenib treatment in human hepatocellular carcinoma. Liver Cancer 2017, 6, 113–125. [Google Scholar] [CrossRef]
- Nishida, N.; Kudo, M. Immune checkpoint blockade for the treatment of human hepatocellular carcinoma. Hepatol. Res. 2018, 48, 622–634. [Google Scholar] [CrossRef]
- Prieto, J.; Melero, I.; Sangro, B. Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 681–700. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.W.; Finn, R.S.; Cheng, A.; Mathurin, P.; Edeline, J.; Kudo, M.; Han, K.; Harding, J.J.; Merle, P.; et al. CheckMate 459: A randomized, multi-center phase 3 study of Nivolumab (NIVO) vs Sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Ann. Oncol. 2019, 30 (Suppl. 5), v851–v934. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in KEYNOTE-240: A randomized, double-blind, phase III trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef]
- Boyault, S.; Rickman, D.S.; de Reynies, A.; Balabaud, C.; Rebouissou, S.; Jeannot, E.; Herault, A.; Saric, J.; Belghiti, J.; Franco, D.; et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology 2007, 45, 42–52. [Google Scholar] [CrossRef]
- Hoshida, Y.; Toffanin, S.; Lachenmayer, A.; Villanueva, A.; Minguez, B.; Llovet, J.M. Molecular classification and novel targets in hepatocellular carcinoma: Recent advancements. Semin. Liver Dis. 2010, 30, 35–51. [Google Scholar] [CrossRef]
- Nishida, N.; Kudo, M. Recent advancements in comprehensive genetic analyses for human hepatocellular carcinoma. Oncology 2013, 84 (Suppl. 1), 93–97. [Google Scholar] [CrossRef]
- Nishida, N.; Kudo, M.; Nishimura, T.; Arizumi, T.; Takita, M.; Kitai, S.; Yada, N.; Hagiwara, S.; Inoue, T.; Minami, Y.; et al. Unique association between global DNA hypomethylation and chromosomal alterations in human hepatocellular carcinoma. PLoS ONE 2013, 8, e72312. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Kudo, M. Oncogenic signal and tumor microenvironment in hepatocellular carcinoma. Oncology 2017, 93 (Suppl. 1), 160–164. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Xing, D.; Luan, L.; Xu, H.; Sharma, R.B.; Popovic, A.; Pawlik, T.M.; Kim, A.K.; Zhu, Q.; Jaffee, E.M.; et al. Characterization of the immune microenvironment in hepatocellular carcinoma. Clin. Cancer Res. 2017, 23, 7333–7339. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Kudo, M. Immunological microenvironment of hepatocellular carcinoma and its clinical implication. Oncology 2017, 92 (Suppl. 1), 40–49. [Google Scholar] [CrossRef] [PubMed]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef]
- Fujimoto, A.; Totoki, Y.; Abe, T.; Boroevich, K.A.; Hosoda, F.; Nguyen, H.H.; Aoki, M.; Hosono, N.; Kubo, M.; Miya, F.; et al. Whole-Genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 2012, 44, 760–764. [Google Scholar] [CrossRef]
- Nishida, N.; Kudo, M.; Nagasaka, T.; Ikai, I.; Goel, A. Characteristic patterns of altered DNA methylation predict emergence of human hepatocellular carcinoma. Hepatology 2012, 56, 994–1003. [Google Scholar] [CrossRef]
- Nishida, N.; Goel, A. Genetic and epigenetic signatures in human hepatocellular carcinoma: A systematic review. Curr. Genom. 2011, 12, 130–137. [Google Scholar] [CrossRef]
- Schulze, K.; Imbeaud, S.; Letouze, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef]
- Nishida, N.; Fukuda, Y.; Kokuryu, H.; Toguchida, J.; Yandell, D.W.; Ikenega, M.; Imura, H.; Ishizaki, K. Role and mutational heterogeneity of the p53 gene in hepatocellular carcinoma. Cancer Res. 1993, 53, 368–372. [Google Scholar] [PubMed]
- Nishida, N.; Nishimura, T.; Nagasaka, T.; Ikai, I.; Goel, A.; Boland, C.R. Extensive methylation is associated with beta-catenin mutations in hepatocellular carcinoma: Evidence for two distinct pathways of human hepatocarcinogenesis. Cancer Res. 2007, 67, 4586–4594. [Google Scholar] [CrossRef]
- Nakayama, J.; Tahara, H.; Tahara, E.; Saito, M.; Ito, K.; Nakamura, H.; Nakanishi, T.; Tahara, E.; Ide, T.; Ishikawa, F. Telomerase activation by hTRT in human normal fibroblasts and hepatocellular carcinomas. Nat. Genet. 1998, 18, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Mallet, M.; Pilati, C.; Calderaro, J.; Bioulac-Sage, P.; Laurent, C.; Laurent, A.; Cherqui, D.; Balabaud, C.; Zucman-Rossi, J. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat. Commun. 2013, 4, 2218. [Google Scholar] [CrossRef] [PubMed]
- Shibata, T.; Aburatani, H. Exploration of liver cancer genomes. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.C.; Llovet, J.M. Genetic landscape and biomarkers of hepatocellular carcinoma. Gastroenterology 2015, 149, 1226–1239. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 2017, 169, 1327–1341. [Google Scholar] [CrossRef]
- Nishida, N.; Nishimura, T.; Kaido, T.; Minaga, K.; Yamao, K.; Kamata, K.; Takenaka, M.; Ida, H.; Hagiwara, S.; Minami, Y.; et al. Molecular scoring of hepatocellular carcinoma for predicting metastatic recurrence and requirements of systemic chemotherapy. Cancers 2018, 10, 367. [Google Scholar] [CrossRef]
- Nishida, N.; Fukuda, Y.; Komeda, T.; Kita, R.; Sando, T.; Furukawa, M.; Amenomori, M.; Shibagaki, I.; Nakao, K.; Ikenaga, M.; et al. Amplification and overexpression of the cyclin D1 gene in aggressive human hepatocellular carcinoma. Cancer Res. 1994, 54, 3107–3110. [Google Scholar]
- Rebouissou, S.; Nault, J.C. Advances in molecular classification and precision oncology in hepatocellular carcinoma. J. Hepatol. 2020, 72, 215–229. [Google Scholar] [CrossRef]
- Nishida, N.; Nagasaka, T.; Nishimura, T.; Ikai, I.; Boland, C.R.; Goel, A. Aberrant methylation of multiple tumor suppressor genes in aging liver, chronic hepatitis, and hepatocellular carcinoma. Hepatology 2008, 47, 908–918. [Google Scholar] [CrossRef]
- Calderaro, J.; Couchy, G.; Imbeaud, S.; Amaddeo, G.; Letouze, E.; Blanc, J.F.; Laurent, C.; Hajji, Y.; Azoulay, D.; Bioulac-Sage, P.; et al. Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. J. Hepatol. 2017, 67, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Desert, R.; Rohart, F.; Canal, F.; Sicard, M.; Desille, M.; Renaud, S.; Turlin, B.; Bellaud, P.; Perret, C.; Clement, B.; et al. Human hepatocellular carcinomas with a periportal phenotype have the lowest potential for early recurrence after curative resection. Hepatology 2017, 66, 1502–1518. [Google Scholar] [CrossRef] [PubMed]
- Kurebayashi, Y.; Ojima, H.; Tsujikawa, H.; Kubota, N.; Maehara, J.; Abe, Y.; Kitago, M.; Shinoda, M.; Kitagawa, Y.; Sakamoto, M. Landscape of immune microenvironment in hepatocellular carcinoma and its additional impact on histological and molecular classification. Hepatology 2018, 68, 1025–1041. [Google Scholar] [CrossRef]
- Gao, Q.; Wang, X.Y.; Qiu, S.J.; Yamato, I.; Sho, M.; Nakajima, Y.; Zhou, J.; Li, B.Z.; Shi, Y.H.; Xiao, Y.S.; et al. Overexpression of PD-L1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin. Cancer Res. 2009, 15, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Umemoto, Y.; Okano, S.; Matsumoto, Y.; Nakagawara, H.; Matono, R.; Yoshiya, S.; Yamashita, Y.; Yoshizumi, T.; Ikegami, T.; Soejima, Y.; et al. Prognostic impact of programmed cell death 1 ligand 1 expression in human leukocyte antigen class I-positive hepatocellular carcinoma after curative hepatectomy. J. Gastroenterol. 2015, 50, 65–75. [Google Scholar] [CrossRef]
- Gabrielson, A.; Wu, Y.; Wang, H.; Jiang, J.; Kallakury, B.; Gatalica, Z.; Reddy, S.; Kleiner, D.; Fishbein, T.; Johnson, L.; et al. Intratumoral CD3 and CD8 T-cell densities associated with relapse-free survival in HCC. Cancer Immunol. Res. 2016, 4, 419–430. [Google Scholar] [CrossRef]
- Calderaro, J.; Rousseau, B.; Amaddeo, G.; Mercey, M.; Charpy, C.; Costentin, C.; Luciani, A.; Zafrani, E.S.; Laurent, A.; Azoulay, D.; et al. Programmed death ligand 1 expression in hepatocellular carcinoma: Relationship with clinical and pathological features. Hepatology 2016, 64, 2038–2046. [Google Scholar] [CrossRef]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; Castro de Moura, M.; Putra, J.; Camprecios, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an immune-specific class of hepatocellular carcinoma, based on molecular features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef]
- Nishida, N.; Sakai, K.; Morita, M.; Aoki, T.; Takita, M.; Hagiwara, S.; Komeda, Y.; Takenaka, M.; Minami, Y.; Ida, H.; et al. Association between genetic and immunological background of hepatocellular carcinoma and expression of programmed cell death-1. Liver Cancer 2020, in press. [Google Scholar] [CrossRef]
- Harding, J.J.; Nandakumar, S.; Armenia, J.; Khalil, D.N.; Albano, M.; Ly, M.; Shia, J.; Hechtman, J.F.; Kundra, R.; El Dika, I.; et al. Prospective genotyping of hepatocellular carcinoma: Clinical implications of next-generation sequencing for matching patients to targeted and immune therapies. Clin. Cancer Res. 2019, 25, 2116–2126. [Google Scholar] [CrossRef] [PubMed]
- Ruiz de Galarreta, M.; Bresnahan, E.; Molina-Sanchez, P.; Lindblad, K.E.; Maier, B.; Sia, D.; Puigvehi, M.; Miguela, V.; Casanova-Acebes, M.; Dhainaut, M.; et al. β-Catenin activation promotes immune escape and resistance to Anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discov. 2019, 9, 1124–1141. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Oh, J.H.; Chun, S.M.; Kim, D.; Ryu, Y.M.; Hwang, H.S.; Kim, S.Y.; An, J.; Cho, E.J.; Lee, H.; et al. Immunogenomic landscape of hepatocellular carcinoma with immune cell stroma and EBV-positive tumor-infiltrating lymphocytes. J. Hepatol. 2019, 71, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Zheng, L.; Yoo, J.K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q.; et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 2017, 169, 1342–1356. [Google Scholar] [CrossRef]
- Zhou, G.; Sprengers, D.; Boor, P.P.C.; Doukas, M.; Schutz, H.; Mancham, S.; Pedroza-Gonzalez, A.; Polak, W.G.; de Jonge, J.; Gaspersz, M.; et al. Antibodies against immune checkpoint molecules restore functions of tumor-infiltrating T cells in hepatocellular carcinomas. Gastroenterology 2017, 153, 1107–1119. [Google Scholar] [CrossRef]
- Yan, W.; Liu, X.; Ma, H.; Zhang, H.; Song, X.; Gao, L.; Liang, X.; Ma, C. Tim-3 fosters HCC development by enhancing TGF-β-mediated alternative activation of macrophages. Gut 2015, 64, 1593–1604. [Google Scholar] [CrossRef]
- Kim, H.D.; Song, G.W.; Park, S.; Jung, M.K.; Kim, M.H.; Kang, H.J.; Yoo, C.; Yi, K.; Kim, K.H.; Eo, S.; et al. Association between expression level of PD1 by tumor-infiltrating CD8(+) T cells and features of hepatocellular carcinoma. Gastroenterology 2018, 155, 1936–1950. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Chang, B.; Shen, L.; Wang, K.; Jin, J.; Huang, T.; Chen, Q.; Li, W.; Wu, P. High number of PD-1 positive intratumoural lymphocytes predicts survival benefit of cytokine-induced killer cells for hepatocellular carcinoma patients. Liver Int. 2018, 38, 1449–1458. [Google Scholar] [CrossRef]
- Roth, G.S.; Decaens, T. Liver immunotolerance and hepatocellular carcinoma: Patho-Physiological mechanisms and therapeutic perspectives. Eur. J. Cancer 2017, 87, 101–112. [Google Scholar] [CrossRef]
- Faivre, S.; Rimassa, L.; Finn, R.S. Molecular therapies for HCC: Looking outside the box. J. Hepatol. 2020, 72, 342–352. [Google Scholar] [CrossRef]
- Arihara, F.; Mizukoshi, E.; Kitahara, M.; Takata, Y.; Arai, K.; Yamashita, T.; Nakamoto, Y.; Kaneko, S. Increase in CD14 + HLA-DR-/low myeloid-derived suppressor cells in hepatocellular carcinoma patients and its impact on prognosis. Cancer Immunol. Immunother. 2013, 62, 1421–1430. [Google Scholar] [CrossRef]
- Chen, K.J.; Lin, S.Z.; Zhou, L.; Xie, H.Y.; Zhou, W.H.; Taki-Eldin, A.; Zheng, S.S. Selective recruitment of regulatory T cell through CCR6-CCL20 in hepatocellular carcinoma fosters tumor progression and predicts poor prognosis. PLoS ONE 2011, 6, e24671. [Google Scholar] [CrossRef] [PubMed]
- Hoechst, B.; Ormandy, L.A.; Ballmaier, M.; Lehner, F.; Kruger, C.; Manns, M.P.; Greten, T.F.; Korangy, F. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology 2008, 135, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Han, Y.; Guo, Q.; Zhang, M.; Cao, X. Cancer-Expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-β1. J. Immunol. 2009, 182, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Quezada, S.A.; Peggs, K.S.; Simpson, T.R.; Allison, J.P. Shifting the equilibrium in cancer immunoediting: From tumor tolerance to eradication. Immunol. Rev. 2011, 241, 104–118. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yang, Y.; Hua, X.; Wang, G.; Liu, W.; Jia, C.; Tai, Y.; Zhang, Q.; Chen, G. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. 2012, 318, 154–161. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, Y.; Reiberger, T.; Duyverman, A.M.; Huang, P.; Samuel, R.; Hiddingh, L.; Roberge, S.; Koppel, C.; Lauwers, G.Y.; et al. Differential effects of sorafenib on liver versus tumor fibrosis mediated by stromal-derived factor 1 α/C-X-C receptor type 4 axis and myeloid differentiation antigen-positive myeloid cell infiltration in mice. Hepatology 2014, 59, 1435–1447. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.R.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Kawaoka, T.; Ando, Y.; Yamauchi, M.; Suehiro, Y.; Yamaoka, K.; Kosaka, Y.; Fuji, Y.; Uchikawa, S.; Morio, K.; Fujino, H.; et al. Incidence of microsatellite instability-high hepatocellular carcinoma among Japanese patients and response to pembrolizumab. Hepatol. Res. 2020. [Google Scholar] [CrossRef]
- Brown, Z.J.; Yu, S.J.; Heinrich, B.; Ma, C.; Fu, Q.; Sandhu, M.; Agdashian, D.; Zhang, Q.; Korangy, F.; Greten, T.F. Indoleamine 2,3-dioxygenase provides adaptive resistance to immune checkpoint inhibitors in hepatocellular carcinoma. Cancer Immunol. Immunother. 2018, 67, 1305–1315. [Google Scholar] [CrossRef]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Mossenta, M.; Busato, D.; Baboci, L.; Cintio, F.D.; Toffoli, G.; Bo, M.D. New insight into therapies targeting angiogenesis in hepatocellular carcinoma. Cancers 2019, 11, 1086. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M. Combination cancer immunotherapy with molecular targeted agents/anti-CTLA-4 antibody for hepatocellular carcinoma. Liver Cancer 2019, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Kang, Y.K.; Yen, C.J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased alpha-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Teng, M.W.; Ngiow, S.F.; Ribas, A.; Smyth, M.J. Classifying cancers based on T-cell infiltration and PD-L1. Cancer Res. 2015, 75, 2139–2145. [Google Scholar] [CrossRef]
- Yi, M.; Jiao, D.; Qin, S.; Chu, Q.; Wu, K.; Li, A. Synergistic effect of immune checkpoint blockade and anti-angiogenesis in cancer treatment. Mol. Cancer 2019, 18, 60. [Google Scholar] [CrossRef]
- Shigeta, K.; Datta, M.; Hato, T.; Kitahara, S.; Chen, I.X.; Matsui, A.; Kikuchi, H.; Mamessier, E.; Aoki, S.; Ramjiawan, R.R.; et al. Dual Programmed death receptor-1 and vascular endothelial growth factor receptor-2 blockade promotes vascular normalization and enhances antitumor immune responses in hepatocellular carcinoma. Hepatology 2019, 1247–1261. [Google Scholar] [CrossRef]
- Nishida, N. Clinical implications of the dual blockade of the PD-1/PD-L1 and vascular endothelial growth factor axes in the treatment of hepatocellular carcinoma. Hepatobiliary Surg. Nutr. 2020, in press. [Google Scholar] [CrossRef]
- Cheng, A.L.; Hsu, C.; Chan, S.L.; Choo, S.P.; Kudo, M. Challenges of combination therapy with immune checkpoint inhibitors for hepatocellular carcinoma. J. Hepatol. 2020, 72, 307–319. [Google Scholar] [CrossRef]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Li, Z.; Cheng, Q.; Wang, Y.; Qian, L.; Gao, J.; Zhu, J.Y. Genetic alterations and expression of PTEN and its relationship with cancer stem cell markers to investigate pathogenesis and to evaluate prognosis in hepatocellular carcinoma. J. Clin. Pathol. 2019, 72, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Yu, C.; Kong, L.; Xu, X.; Yan, J.; Li, Y.; An, T.; Gong, L.; Gong, Y.; Zhu, H.; et al. B591, a novel specific pan-PI3K inhibitor, preferentially targets cancer stem cells. Oncogene 2019, 38, 3371–3386. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, X.; Liu, S.; Guo, L.; Zhang, B.; Zhang, J.; Ye, Q. Programmed cell death-1 (PD-1) checkpoint blockade in combination with a mammalian target of rapamycin inhibitor restrains hepatocellular carcinoma growth induced by hepatoma cell-intrinsic PD-1. Hepatology 2017, 66, 1920–1933. [Google Scholar] [CrossRef] [PubMed]
- Wada, Y.; Nakashima, O.; Kutami, R.; Yamamoto, O.; Kojiro, M. Clinicopathological study on hepatocellular carcinoma with lymphocytic infiltration. Hepatology 1998, 27, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Ishizuka, J.J.; Manguso, R.T.; Cheruiyot, C.K.; Bi, K.; Panda, A.; Iracheta-Vellve, A.; Miller, B.C.; Du, P.P.; Yates, K.B.; Dubrot, J.; et al. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 2019, 565, 43–48. [Google Scholar] [CrossRef]
- Chan, T.H.; Lin, C.H.; Qi, L.; Fei, J.; Li, Y.; Yong, K.J.; Liu, M.; Song, Y.; Chow, R.K.; Ng, V.H.; et al. A disrupted RNA editing balance mediated by ADARs (Adenosine DeAminases that act on RNA) in human hepatocellular carcinoma. Gut 2014, 63, 832–843. [Google Scholar] [CrossRef]
- Hong, Y.K.; Li, Y.; Pandit, H.; Li, S.; Pulliam, Z.; Zheng, Q.; Yu, Y.; Martin, R.C.G. Epigenetic modulation enhances immunotherapy for hepatocellular carcinoma. Cell. Immunol. 2019, 336, 66–74. [Google Scholar] [CrossRef]
- Schonfeld, M.; Zhao, J.; Komatz, A.; Weinman, S.A.; Tikhanovich, I. The polymorphism rs975484 in the protein arginine methyltransferase-1 gene modulates expression of immune checkpoint genes in hepatocellular carcinoma. J. Biol. Chem. 2020, 295, 7126–7137. [Google Scholar] [CrossRef]
- Feun, L.G.; Li, Y.Y.; Wu, C.; Wangpaichitr, M.; Jones, P.D.; Richman, S.P.; Madrazo, B.; Kwon, D.; Garcia-Buitrago, M.; Martin, P.; et al. Phase 2 study of pembrolizumab and circulating biomarkers to predict anticancer response in advanced, unresectable hepatocellular carcinoma. Cancer 2019, 125, 3603–3614. [Google Scholar] [CrossRef]
- Dong, L.Q.; Peng, L.H.; Ma, L.J.; Liu, D.B.; Zhang, S.; Luo, S.Z.; Rao, J.H.; Zhu, H.W.; Yang, S.X.; Xi, S.J.; et al. Heterogeneous immunogenomic features and distinct escape mechanisms in multifocal hepatocellular carcinoma. J. Hepatol. 2020, 72, 896–908. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Kudo, M. Role of immune checkpoint blockade in the treatment for human hepatocellular carcinoma. Dig. Dis. 2017, 35, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Zhang, H.; Sun, B.; Karin, M. The immunobiology of hepatocellular carcinoma in humans and mice: Basic concepts and therapeutic implications. J. Hepatol. 2020, 72, 167–182. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Clinical Trial ID | Trial Name | Agents 1 | Setting 2 | Key Outcome 3 |
|---|---|---|---|---|
| Phase I/II | ||||
| NCT01658878 | CheckMate 040 | Nivolumab | dose-escalation, n = 48, dose-expansion, n = 214 | ORR: 20% 4 DCR: 64%, (37%) 5 OS: 13.2 months (8.6–NE) 6 |
| NCT02702414 | KEYNOTE-224 | Pembrolizumab | second-line n = 104 | ORR: 17% 7 DCR: 62% OS: 12.9 months (9.7–15.5) |
| Phase III | ||||
| NCT03383458 | CheckMate 9DX | Nivolumab versus placebo | adjuvant, randomized, double-blinded (n = 530) | RFS |
| NCT02576509 | CheckMate 459 | Nivolumab versus Sorafenib | first-line, randomized, open label, n = 743 | Median OS: 16.4 months in thenivolumabgroup and 14.7 months in the sorafenib group. 8 Median PFS: 3.7 months for nivolumab and 3.8 months for sorafenib. ORR: 15% in the nivolumab group and 7% in the sorafenib group. |
| NCT03412773 | Rationale-301 | Tislelizumab versus sorafenib | first-line, randomized, open label, (n = 674) | OS |
| NCT02702401 | KEYNOTE-240 | Pembrolizumab versus placebo | second-line, randomized, double-blinded, n = 413 | Median OS: 13.9 months in the pembrolizumab group and 10.6 months in the placebo group; HR 0.781, p = 0.0238. Median PFS: 3.0 months for pembrolizumab and 2.8 months for placebo; HR 0.781, p = 0.0022. 9 ORR: 18.3%, DCR: 62.2% |
| Clinical Trial ID | Trial Name | Agents 1 | Setting 2 | Key Outcome 3 |
|---|---|---|---|---|
| Phase I/II | ||||
| NCT01658878 | CheckMate 040 | Nivolumab + Ipilimumab | n = 50 | ORR: 32% 4 DCR: 54% OS: 22.8 months (9.4–NE) DOR: 17.5 months (4.6–30.5) |
| NCT02519348 | Durvalumab ± Tremelimumab | n = 40 | ORR: 25% 5 DCR: 57.5% | |
| NCT03680508 | TSR-002 + TSR-042 (Dostarlimab) | first-line, (n = 42) | ORR | |
| NCT03250832 | TSR-033 + TSR-042 | dose escalation and dose expansion cohorts (n = 200) | AEs for dose escalation cohort ORR for dose expansion cohort | |
| NCT03695250 | BMS986205 + Nivolumab | first- or second-line, (n = 23) | AEs and ORR | |
| Phase III | ||||
| NCT04039607 | CheckMate9DW | Nivolumab + Ipilimumab versus Sorafenib/Lenvatinib | first-line, randomized, open label, (n = 1084) | OS |
| NCT03298451 | HIMARAYA | Durvalumab ± Tremelimumab versus Sorafenib | first-line, randomized, open label, (n = 1310) | OS |
| Clinical Trial ID | Trial Name | Agents 1 | Setting 2 | Key Outcome 3 |
|---|---|---|---|---|
| Phase I/II | ||||
| NCT03299946 | CaboNivo | Cabozantinib + Nivolumab | neoadjuvant, (n = 15) | AEs and number of patients who complete the treatment. |
| NCT03006926 | Lenvatinib + Pembrolizumab | first-line, (dose-escalation, dose-expansion), n = 30 (n = 97) | ORR: 53.3% (34.3–71.7), DOR: 8.3 months (3.8–11.0) 4 DCR = 90.0%; 73.5–97.9, PFS: 9.7 months 7.7–NE, OS: 14.6 months 9.9–NE. | |
| NCT03289533 | VEGF Liver 100 | Avelumab + Axitinib | AFP ≥400 ng/mL, n = 22 | AE ORR: 13.6% (2.9–34.9) 5 DCR: 68.2 (45.1–86.1) PFS: 5.5 months (1.9–7.4) OS: 12.7 months (0.0–NE) DOR: 5.5 months (3.7–7.3) |
| NCT03418922 | Lenvatinib + Nivolumab | first-line, (n = 30) | DLT, AEs | |
| NCT02715531 | GO30140 | Atezolizumab + Bevacizumab | n = 73 | ORR: 27% 6 PFS: 7.5 months (0.4–23.9+) |
| NCT01658878 | CheckMate 040 | Cabozantinib + Nivolumab ± Ipilimumab | first or second-line, (dose-escalation, dose-expansion), (n = 1097, across all cohorts) | safety, tolerability, ORR |
| NCT03170960 | COSMIC-021 | Cabozantinib + Atezolizumab | first-line, (dose-escalation and dose-expansion), (n = 1732, across all cohorts) | MTD, ORR |
| NCT03347292 | Regorafenib + Pembrolizumab | first-line, (dose-escalation and dose-expansion, n = 57) | TEAE, DLT | |
| NCT03539822 | CAMILLA | Cabozantinib + Durvalumab | second-line, (n = 30) | MTD |
| NCT03475953 | REGOMUNE | Regorafenib + Avelumab | Second-line, (n = 212) | Recommended phase II dose, ORR |
| NCT02572687 | Ramucirumab + Durvalumab | Second-line and AFP ≥1.5x ULN, n = 28 | DLTs ORR: 11% 7 PFS: 4.4 months (1.6–5.7) OS: 10.8 months (5.1–18.4) | |
| NCT3463876 | RESCUE | SHR-121 (Camrelizumab) + Apatinib | n = 18 (n = 40) | ORR: 38.9%8 DCR: 83.3% PFS: 7.2 months (2.6–NE) |
| NCT02423343 | Galunisertib (TGFβ receptor I inhibitor) + Nivolumab | second-line and AFP ≥200 ng/mL, (dose escalation and cohort expansion, n = 75) | MTD | |
| Phase III | ||||
| NCT03847428 | EMERALD-2 | Durvalumab ± Bevacizumab versus placebo | adjuvant, randomized, double-blinded, (n = 888) | RFS |
| NCT03434379 | IMbrave150 | Atezolizumab + Bevacizumab versus sorafenib | first-line, randomized, open label, n = 501 | OS:not reached forAtezolizumab + bevacizumabvs 13.2 months for sorafenib; HR 0.58, p = 0.006 9 PFS: 6.8 monthsforAtezolizumab + bevacizumab versus 4.3 months for sorafenib; HR 0.59, p < 0.0001 ORR: 27% |
| NCT03713593 | LEAP-002 | Lenvatinib + Pembrolizumab versus Lenvatinib | first-line, randomized, double-blinded, (n = 750) | OS, PFS |
| NCT03755791 | COSMIC-312 | Cabozantinib + Atezolizumab versus Sorafenib versus Cabozantinib | first-line, randomized, open label, (n = 740) | OS, PFS |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishida, N.; Kudo, M. Immune Phenotype and Immune Checkpoint Inhibitors for the Treatment of Human Hepatocellular Carcinoma. Cancers 2020, 12, 1274. https://doi.org/10.3390/cancers12051274
Nishida N, Kudo M. Immune Phenotype and Immune Checkpoint Inhibitors for the Treatment of Human Hepatocellular Carcinoma. Cancers. 2020; 12(5):1274. https://doi.org/10.3390/cancers12051274
Chicago/Turabian StyleNishida, Naoshi, and Masatoshi Kudo. 2020. "Immune Phenotype and Immune Checkpoint Inhibitors for the Treatment of Human Hepatocellular Carcinoma" Cancers 12, no. 5: 1274. https://doi.org/10.3390/cancers12051274
APA StyleNishida, N., & Kudo, M. (2020). Immune Phenotype and Immune Checkpoint Inhibitors for the Treatment of Human Hepatocellular Carcinoma. Cancers, 12(5), 1274. https://doi.org/10.3390/cancers12051274