Acquired Resistance to Immune Checkpoint Blockade Therapies



Abstract
1. Introduction
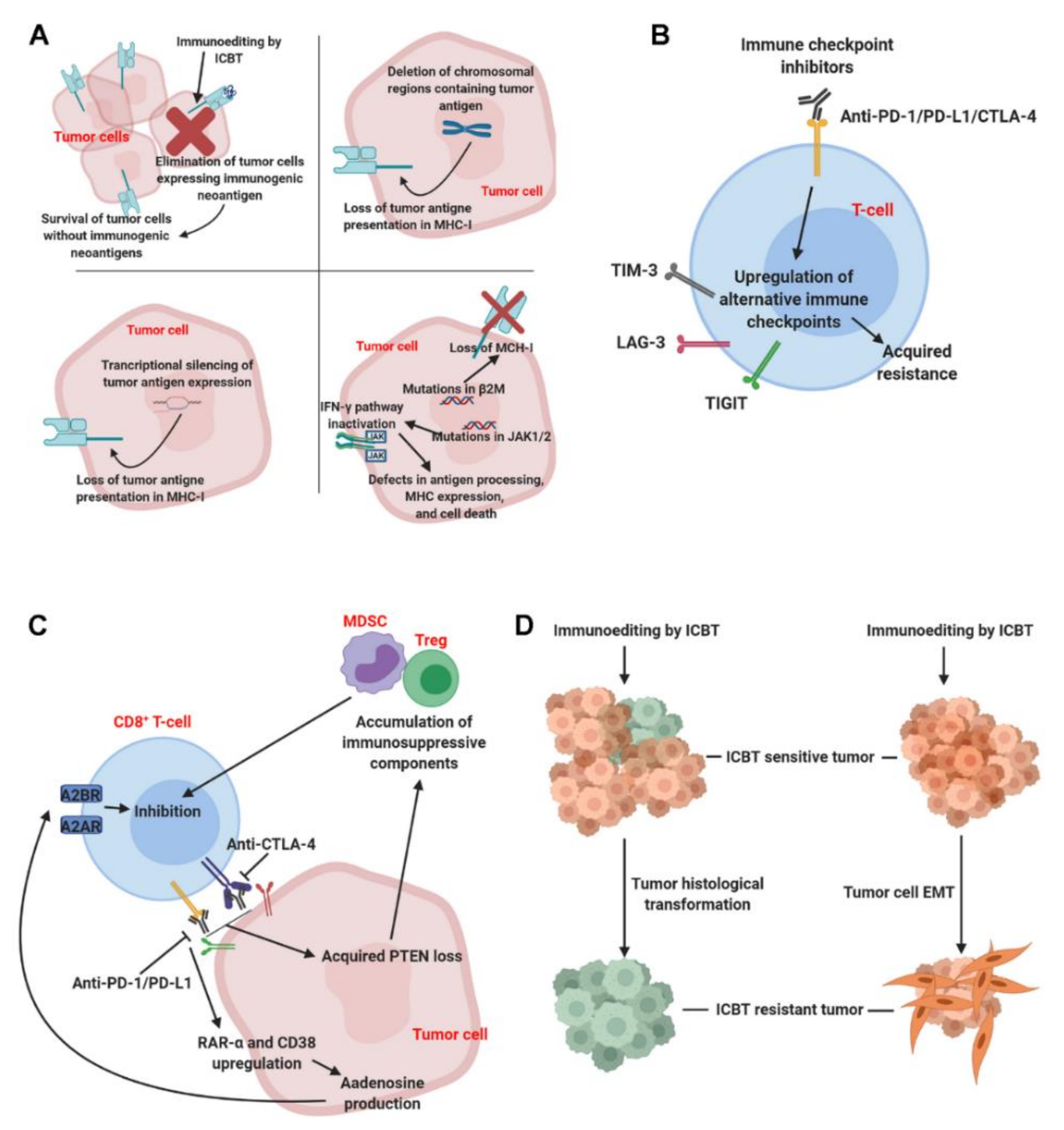
2. Mechanisms of Acquired ICBT Resistance
2.1. Loss of Tumor Cell Visibility to Immune Cells
2.2. Loss of Tumor Cells Sensitivity to Immune Effectors
2.3. Compensation of Alternative Immune Checkpoints on T Cells
2.4. Tumor Cell-Mediated T-Cell Suppression
2.5. Tumor Pathologic Transformation and Epithelial–Mesenchymal Transition
3. Detection of Acquired ICBT Resistance
4. Principles for Overcoming Acquired ICBT Resistance
5. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Busch, W. Aus der Sitzung der medicinischen Section vom 13 November 1867. Berl. Kli. Wochenschr. 1868, 5, 137. (in German). [Google Scholar]
- Coley, W.B. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. 1893. Clin. Orthop. Relat. Res. 1991, 262, 3–11. [Google Scholar]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Palucka, A.K.; Coussens, L.M. The Basis of Oncoimmunology. Cell 2016, 164, 1233–1247. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Zhao, X.; Subramanian, S. Intrinsic Resistance of Solid Tumors to Immune Checkpoint Blockade Therapy. Cancer Res. 2017, 77, 817–822. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Horn, L.; Spigel, D.R.; Vokes, E.E.; Holgado, E.; Ready, N.; Steins, M.; Poddubskaya, E.; Borghaei, H.; Felip, E.; Paz-Ares, L.; et al. Nivolumab Versus Docetaxel in Previously Treated Patients With Advanced Non-Small-Cell Lung Cancer: Two-Year Outcomes From Two Randomized, Open-Label, Phase III Trials (CheckMate 017 and CheckMate 057). J. Clin. Oncol. 2017, 35, 3924–3933. [Google Scholar] [CrossRef]
- Kalbasi, A.; Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020, 20, 25–39. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Syn, N.L.; Teng, M.W.L.; Mok, T.S.K.; Soo, R.A. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017, 18, e731–e741. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: the cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed]
- Mandal, R.; Samstein, R.M.; Lee, K.W.; Havel, J.J.; Wang, H.; Krishna, C.; Sabio, E.Y.; Makarov, V.; Kuo, F.; Blecua, P.; et al. Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science 2019, 364, 485–491. [Google Scholar] [CrossRef]
- Rosenthal, R.; Cadieux, E.L.; Salgado, R.; Bakir, M.A.; Moore, D.A.; Hiley, C.T.; Lund, T.; Tanic, M.; Reading, J.L.; Joshi, K.; et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 2019, 567, 479–485. [Google Scholar] [CrossRef]
- Verdegaal, E.M.; de Miranda, N.F.; Visser, M.; Harryvan, T.; van Buuren, M.M.; Andersen, R.S.; Hadrup, S.R.; van der Minne, C.E.; Schotte, R.; Spits, H.; et al. Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature 2016, 536, 91–95. [Google Scholar] [CrossRef]
- Anagnostou, V.; Smith, K.N.; Forde, P.M.; Niknafs, N.; Bhattacharya, R.; White, J.; Zhang, T.; Adleff, V.; Phallen, J.; Wali, N.; et al. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov. 2017, 7, 264–276. [Google Scholar] [CrossRef]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martin-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949.e916. [Google Scholar] [CrossRef]
- Wylie, B.; Chee, J.; Forbes, C.A.; Booth, M.; Stone, S.R.; Buzzai, A.; Abad, A.; Foley, B.; Cruickshank, M.N.; Waithman, J. Acquired resistance during adoptive cell therapy by transcriptional silencing of immunogenic antigens. Oncoimmunology 2019, 8, 1609874. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F. Molecular mechanisms of IFN-gamma to up-regulate MHC class I antigen processing and presentation. Int. Rev. Immunol. 2009, 28, 239–260. [Google Scholar] [CrossRef] [PubMed]
- Martini, M.; Testi, M.G.; Pasetto, M.; Picchio, M.C.; Innamorati, G.; Mazzocco, M.; Ugel, S.; Cingarlini, S.; Bronte, V.; Zanovello, P.; et al. IFN-gamma-mediated upmodulation of MHC class I expression activates tumor-specific immune response in a mouse model of prostate cancer. Vaccine 2010, 28, 3548–3557. [Google Scholar] [CrossRef] [PubMed]
- Propper, D.J.; Chao, D.; Braybrooke, J.P.; Bahl, P.; Thavasu, P.; Balkwill, F.; Turley, H.; Dobbs, N.; Gatter, K.; Talbot, D.C.; et al. Low-dose IFN-gamma induces tumor MHC expression in metastatic malignant melanoma. Clin. Cancer Res. 2003, 9, 84–92. [Google Scholar]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef]
- Gettinger, S.; Choi, J.; Hastings, K.; Truini, A.; Datar, I.; Sowell, R.; Wurtz, A.; Dong, W.; Cai, G.; Melnick, M.A.; et al. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov. 2017, 7, 1420–1435. [Google Scholar] [CrossRef]
- Hobeika, A.C.; Etienne, W.; Torres, B.A.; Johnson, H.M.; Subramaniam, P.S. IFN-gamma induction of p21(WAF1) is required for cell cycle inhibition and suppression of apoptosis. J. Interferon Cytokine Res. 1999, 19, 1351–1361. [Google Scholar] [CrossRef]
- Harvat, B.L.; Seth, P.; Jetten, A.M. The role of p27Kip1 in gamma interferon-mediated growth arrest of mammary epithelial cells and related defects in mammary carcinoma cells. Oncogene 1997, 14, 2111–2122. [Google Scholar] [CrossRef]
- Mojic, M.; Takeda, K.; Hayakawa, Y. The Dark Side of IFN-gamma: Its Role in Promoting Cancer Immunoevasion. Int. J. Mol. Sci. 2017, 19. [Google Scholar] [CrossRef]
- Abiko, K.; Matsumura, N.; Hamanishi, J.; Horikawa, N.; Murakami, R.; Yamaguchi, K.; Yoshioka, Y.; Baba, T.; Konishi, I.; Mandai, M. IFN-gamma from lymphocytes induces PD-L1 expression and promotes progression of ovarian cancer. Br. J. Cancer 2015, 112, 1501–1509. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, J.M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.H.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 impair tumor antigen-specific CD8(+) T cells in melanoma patients. J. Clin. Invest. 2015, 125, 2046–2058. [Google Scholar] [CrossRef] [PubMed]
- Sasse, S.; Reddemann, K.; Diepstra, A.; Oschlies, I.; Schnitter, A.; Borchmann, S.; Engert, A.; Borchmann, P.; Klapper, W. Programmed cell death protein-1 (PD-1)-expression in the microenvironment of classical Hodgkin lymphoma at relapse during anti-PD-1-treatment. Haematologica 2019, 104, e21–e24. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Bottai, G.; Raschioni, C.; Losurdo, A.; Di Tommaso, L.; Tinterri, C.; Torrisi, R.; Reis-Filho, J.S.; Roncalli, M.; Sotiriou, C.; Santoro, A.; et al. An immune stratification reveals a subset of PD-1/LAG-3 double-positive triple-negative breast cancers. Breast Cancer Res. 2016, 18, 121. [Google Scholar] [CrossRef]
- Chinn, Z.; Stoler, M.H.; Mills, A.M. PD-L1 and IDO expression in cervical and vulvar invasive and intraepithelial squamous neoplasias: implications for combination immunotherapy. Histopathology 2019, 74, 256–268. [Google Scholar] [CrossRef]
- Mills, A.; Zadeh, S.; Sloan, E.; Chinn, Z.; Modesitt, S.C.; Ring, K.L. Indoleamine 2,3-dioxygenase in endometrial cancer: a targetable mechanism of immune resistance in mismatch repair-deficient and intact endometrial carcinomas. Mod. Pathol. 2018, 31, 1282–1290. [Google Scholar] [CrossRef]
- Chen, L.; Diao, L.; Yang, Y.; Yi, X.; Rodriguez, B.L.; Li, Y.; Villalobos, P.A.; Cascone, T.; Liu, X.; Tan, L.; et al. CD38-Mediated Immunosuppression as a Mechanism of Tumor Cell Escape from PD-1/PD-L1 Blockade. Cancer Discov 2018, 8, 1156–1175. [Google Scholar] [CrossRef]
- Miao, Y.; Yang, H.; Levorse, J.; Yuan, S.; Polak, L.; Sribour, M.; Singh, B.; Rosenblum, M.D.; Fuchs, E. Adaptive Immune Resistance Emerges from Tumor-Initiating Stem Cells. Cell 2019, 177, 1172–1186.e1114. [Google Scholar] [CrossRef]
- Sugiura, D.; Maruhashi, T.; Okazaki, I.M.; Shimizu, K.; Maeda, T.K.; Takemoto, T.; Okazaki, T. Restriction of PD-1 function by cis-PD-L1/CD80 interactions is required for optimal T cell responses. Science 2019, 364, 558–566. [Google Scholar] [CrossRef]
- Zhao, X.; Subramanian, S. Oncogenic pathways that affect antitumor immune response and immune checkpoint blockade therapy. Pharmacol. Ther. 2018, 181, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Vidotto, T.; Saggioro, F.P.; Jamaspishvili, T.; Chesca, D.L.; Picanco de Albuquerque, C.G.; Reis, R.B.; Graham, C.H.; Berman, D.M.; Siemens, D.R.; Squire, J.A.; et al. PTEN-deficient prostate cancer is associated with an immunosuppressive tumor microenvironment mediated by increased expression of IDO1 and infiltrating FoxP3+ T regulatory cells. Prostate 2019, 79, 969–979. [Google Scholar] [CrossRef] [PubMed]
- Piro, G.; Carbone, C.; Carbognin, L.; Pilotto, S.; Ciccarese, C.; Iacovelli, R.; Milella, M.; Bria, E.; Tortora, G. Revising PTEN in the Era of Immunotherapy: New Perspectives for an Old Story. Cancers 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, J.A.; Luke, J.J.; Zha, Y.; Segal, J.P.; Ritterhouse, L.L.; Spranger, S.; Matijevich, K.; Gajewski, T.F. Secondary resistance to immunotherapy associated with beta-catenin pathway activation or PTEN loss in metastatic melanoma. J. Immunother. Cancer 2019, 7, 295. [Google Scholar] [CrossRef]
- Oser, M.G.; Niederst, M.J.; Sequist, L.V.; Engelman, J.A. Transformation from non-small-cell lung cancer to small-cell lung cancer: Molecular drivers and cells of origin. Lancet Oncol. 2015, 16, e165–e172. [Google Scholar] [CrossRef]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef]
- Bar, J.; Ofek, E.; Barshack, I.; Gottfried, T.; Zadok, O.; Kamer, I.; Urban, D.; Perelman, M.; Onn, A. Transformation to small cell lung cancer as a mechanism of resistance to immunotherapy in non-small cell lung cancer. Lung Cancer 2019, 138, 109–115. [Google Scholar] [CrossRef]
- Imakita, T.; Fujita, K.; Kanai, O.; Terashima, T.; Mio, T. Small cell lung cancer transformation during immunotherapy with nivolumab: A case report. Respir. Med. Case Rep. 2017, 21, 52–55. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Shields, B.D.; Koss, B.; Taylor, E.M.; Storey, A.J.; West, K.L.; Byrum, S.D.; Mackintosh, S.G.; Edmondson, R.; Mahmoud, F.; Shalin, S.C.; et al. Loss of E-Cadherin Inhibits CD103 Antitumor Activity and Reduces Checkpoint Blockade Responsiveness in Melanoma. Cancer Res. 2019, 79, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Le Floc’h, A.; Jalil, A.; Vergnon, I.; Le Maux Chansac, B.; Lazar, V.; Bismuth, G.; Chouaib, S.; Mami-Chouaib, F. Alpha E beta 7 integrin interaction with E-cadherin promotes antitumor CTL activity by triggering lytic granule polarization and exocytosis. J. Exp. Med. 2007, 204, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Nizard, M.; Roussel, H.; Diniz, M.O.; Karaki, S.; Tran, T.; Voron, T.; Dransart, E.; Sandoval, F.; Riquet, M.; Rance, B.; et al. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat. Commun. 2017, 8, 15221. [Google Scholar] [CrossRef]
- Roberts, E.W.; Broz, M.L.; Binnewies, M.; Headley, M.B.; Nelson, A.E.; Wolf, D.M.; Kaisho, T.; Bogunovic, D.; Bhardwaj, N.; Krummel, M.F. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 2016, 30, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Seymour, L.; Bogaerts, J.; Perrone, A.; Ford, R.; Schwartz, L.H.; Mandrekar, S.; Lin, N.U.; Litiere, S.; Dancey, J.; Chen, A.; et al. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 2017, 18, e143–e152. [Google Scholar] [CrossRef]
- Auslander, N.; Zhang, G.; Lee, J.S.; Frederick, D.T.; Miao, B.; Moll, T.; Tian, T.; Wei, Z.; Madan, S.; Sullivan, R.J.; et al. Robust prediction of response to immune checkpoint blockade therapy in metastatic melanoma. Nat. Med. 2018, 24, 1545–1549. [Google Scholar] [CrossRef]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef]
- Goldberg, S.B.; Narayan, A.; Kole, A.J.; Decker, R.H.; Teysir, J.; Carriero, N.J.; Lee, A.; Nemati, R.; Nath, S.K.; Mane, S.M.; et al. Early Assessment of Lung Cancer Immunotherapy Response via Circulating Tumor DNA. Clin. Cancer Res. 2018, 24, 1872–1880. [Google Scholar] [CrossRef]
- Lee, J.H.; Long, G.V.; Boyd, S.; Lo, S.; Menzies, A.M.; Tembe, V.; Guminski, A.; Jakrot, V.; Scolyer, R.A.; Mann, G.J.; et al. Circulating tumour DNA predicts response to anti-PD1 antibodies in metastatic melanoma. Ann. Oncol. 2017, 28, 1130–1136. [Google Scholar] [CrossRef]
- Cabel, L.; Riva, F.; Servois, V.; Livartowski, A.; Daniel, C.; Rampanou, A.; Lantz, O.; Romano, E.; Milder, M.; Buecher, B.; et al. Circulating tumor DNA changes for early monitoring of anti-PD1 immunotherapy: a proof-of-concept study. Ann. Oncol. 2017, 28, 1996–2001. [Google Scholar] [CrossRef]
- Lipson, E.J.; Velculescu, V.E.; Pritchard, T.S.; Sausen, M.; Pardoll, D.M.; Topalian, S.L.; Diaz, L.A., Jr. Circulating tumor DNA analysis as a real-time method for monitoring tumor burden in melanoma patients undergoing treatment with immune checkpoint blockade. J. Immunother. Cancer 2014, 2, 42. [Google Scholar] [CrossRef] [PubMed]
- Gandara, D.R.; Paul, S.M.; Kowanetz, M.; Schleifman, E.; Zou, W.; Li, Y.; Rittmeyer, A.; Fehrenbacher, L.; Otto, G.; Malboeuf, C.; et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat. Med. 2018, 24, 1441–1448. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.C.; Yarchoan, M.; Durham, J.N.; Yusko, E.C.; Rytlewski, J.A.; Robins, H.S.; Laheru, D.A.; Le, D.T.; Lutz, E.R.; Jaffee, E.M. T cell receptor repertoire features associated with survival in immunotherapy-treated pancreatic ductal adenocarcinoma. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Hogan, S.A.; Courtier, A.; Cheng, P.F.; Jaberg-Bentele, N.F.; Goldinger, S.M.; Manuel, M.; Perez, S.; Plantier, N.; Mouret, J.F.; Nguyen-Kim, T.D.L.; et al. Peripheral Blood TCR Repertoire Profiling May Facilitate Patient Stratification for Immunotherapy against Melanoma. Cancer Immunol. Res. 2019, 7, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Broos, K.; Lecocq, Q.; Raes, G.; Devoogdt, N.; Keyaerts, M.; Breckpot, K. Noninvasive imaging of the PD-1:PD-L1 immune checkpoint: Embracing nuclear medicine for the benefit of personalized immunotherapy. Theranostics 2018, 8, 3559–3570. [Google Scholar] [CrossRef] [PubMed]
- Bensch, F.; van der Veen, E.L.; Lub-de Hooge, M.N.; Jorritsma-Smit, A.; Boellaard, R.; Kok, I.C.; Oosting, S.F.; Schroder, C.P.; Hiltermann, T.J.N.; van der Wekken, A.J.; et al. (89)Zr-atezolizumab imaging as a non-invasive approach to assess clinical response to PD-L1 blockade in cancer. Nat. Med. 2018, 24, 1852–1858. [Google Scholar] [CrossRef]
- Sanmamed, M.; Perez-Gracia, J.; Schalper, K.; Fusco, J.; Gonzalez, A.; Rodriguez-Ruiz, M.; Onate, C.; Perez, G.; Alfaro, C.; Martín-Algarra, S. Changes in serum interleukin-8 (IL-8) levels reflect and predict response to anti-PD-1 treatment in melanoma and non-small-cell lung cancer patients. Ann. Oncol. 2017, 28, 1988–1995. [Google Scholar] [CrossRef]
- Diem, S.; Kasenda, B.; Spain, L.; Martin-Liberal, J.; Marconcini, R.; Gore, M.; Larkin, J. Serum lactate dehydrogenase as an early marker for outcome in patients treated with anti-PD-1 therapy in metastatic melanoma. Br. J. Cancer 2016, 114, 256. [Google Scholar] [CrossRef]
- Tamminga, M.; de Wit, S.; Hiltermann, T.J.N.; Timens, W.; Schuuring, E.; Terstappen, L.W.; Groen, H.J. Circulating tumor cells in advanced non-small cell lung cancer patients are associated with worse tumor response to checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 173. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E. Nivolumab versus docetaxel in advanced nonsquamous non–small-cell lung cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L. Pembrolizumab for the treatment of non–small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.L.; Roh, W.; Reuben, A.; Cooper, Z.A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Bassett, R.L.; Gopalakrishnan, V.; Wani, K. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. 2016, 6, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Schmidt, H.; Nissan, A.; Ridolfi, L.; Aamdal, S.; Hansson, J.; Guida, M.; Hyams, D.M.; Gómez, H.; Bastholt, L. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J. Transl. Med. 2011, 9, 204. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2017, 168, 542. [Google Scholar] [CrossRef]
- Gettinger, S.N.; Wurtz, A.; Goldberg, S.B.; Rimm, D.; Schalper, K.; Kaech, S.; Kavathas, P.; Chiang, A.; Lilenbaum, R.; Zelterman, D.; et al. Clinical Features and Management of Acquired Resistance to PD-1 Axis Inhibitors in 26 Patients With Advanced Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2018, 13, 831–839. [Google Scholar] [CrossRef]
- Aslan, K.; Turco, V.; Blobner, J.; Sonner, J.K.; Liuzzi, A.R.; Nunez, N.G.; De Feo, D.; Kickingereder, P.; Fischer, M.; Green, E.; et al. Heterogeneity of response to immune checkpoint blockade in hypermutated experimental gliomas. Nat. Commun. 2020, 11, 931. [Google Scholar] [CrossRef]
- Williams, J.B.; Li, S.; Higgs, E.F.; Cabanov, A.; Wang, X.; Huang, H.; Gajewski, T.F. Tumor heterogeneity and clonal cooperation influence the immune selection of IFN-gamma-signaling mutant cancer cells. Nat. Commun. 2020, 11, 602. [Google Scholar] [CrossRef]
- Zitvogel, L.; Ma, Y.; Raoult, D.; Kroemer, G.; Gajewski, T.F. The microbiome in cancer immunotherapy: Diagnostic tools and therapeutic strategies. Science 2018, 359, 1366–1370. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Poggio, M.; Hu, T.; Pai, C.C.; Chu, B.; Belair, C.D.; Chang, A.; Montabana, E.; Lang, U.E.; Fu, Q.; Fong, L.; et al. Suppression of Exosomal PD-L1 Induces Systemic Anti-tumor Immunity and Memory. Cell 2019, 177, 414–427. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Approaches | Mechanisms of Approach | Relevance to Anti-Tumor Immune Mechanisms | Reference |
|---|---|---|---|
| Radiographic imaging | Direct measurement of tumor burden | No | [55] |
| Molecular imaging | Measurement of immune protein expression on whole tumor tissues | Yes | [65,66] |
| Serum tumor biomarkers | Estimation of tumor burden by quantifying serum tumor antigens or genetic material | Marker-dependent | [67,68] |
| Circulating tumor cells | Estimation of tumor burden by quantifying and featuring circulating tumor cells | Marker-dependent | [69] |
| PD-L1 expression | Assessment of the proportion of PD-L1-positive tumor cells, immune cells, or both on tumor tissue sections | Yes | [70,71] |
| Tumor-infiltrating lymphocyte | Assessment of T cells at invasive tumor margin or tumor parenchyma | Yes | [72,73] |
| T-cell receptor sequencing | Assessment of T-cell clonality by sequencing of T-cell receptor β-chain | Yes | [74] |
| Mutational and neoantigen burden | Exome sequencing to assess non-synonymous somatic mutations with antigenic prediction | Yes | [15] |
| DNA mismatch repair genes status | Assessment of mismatch repair genes status in tumor parenchyma | Yes | [75] |
| Immune gene signatures | Assessment of immune gene expression signature from the tumor microenvironment | Yes | [76] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.; Wangmo, D.; Robertson, M.; Subramanian, S. Acquired Resistance to Immune Checkpoint Blockade Therapies. Cancers 2020, 12, 1161. https://doi.org/10.3390/cancers12051161
Zhao X, Wangmo D, Robertson M, Subramanian S. Acquired Resistance to Immune Checkpoint Blockade Therapies. Cancers. 2020; 12(5):1161. https://doi.org/10.3390/cancers12051161
Chicago/Turabian StyleZhao, Xianda, Dechen Wangmo, Matthew Robertson, and Subbaya Subramanian. 2020. "Acquired Resistance to Immune Checkpoint Blockade Therapies" Cancers 12, no. 5: 1161. https://doi.org/10.3390/cancers12051161
APA StyleZhao, X., Wangmo, D., Robertson, M., & Subramanian, S. (2020). Acquired Resistance to Immune Checkpoint Blockade Therapies. Cancers, 12(5), 1161. https://doi.org/10.3390/cancers12051161