Combination of Withaferin-A and CAPE Provides Superior Anticancer Potency: Bioinformatics and Experimental Evidence to Their Molecular Targets and Mechanism of Action


 ,
, 
Abstract
1. Introduction
2. Results
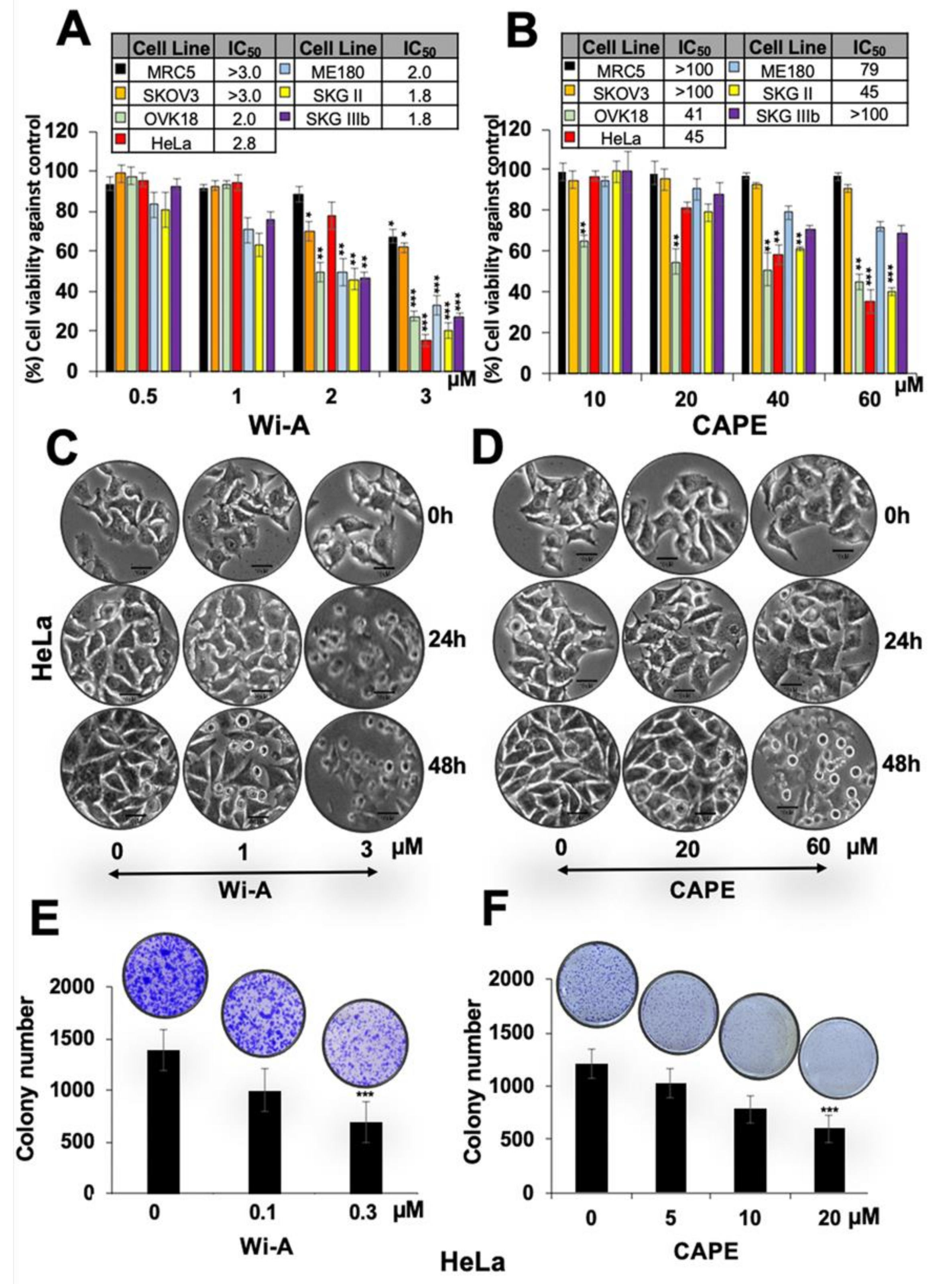
2.1. Wi-A and CAPE Caused Cytotoxicity to Cervical and Ovarian Cancer Cells
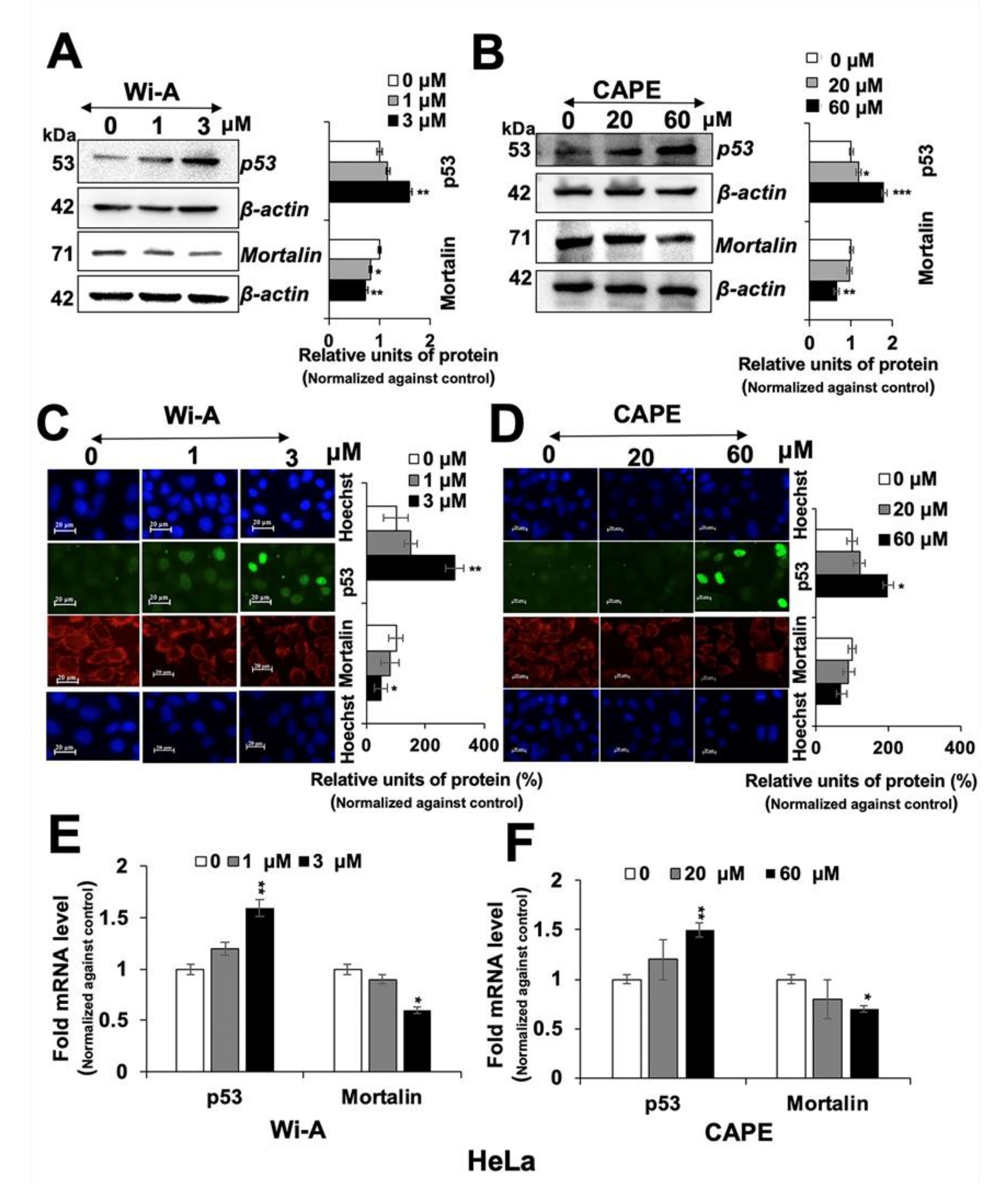
2.2. Wi-A and CAPE Caused Downregulation of Mortalin and Activation of Tumor Suppressor p53 Protein
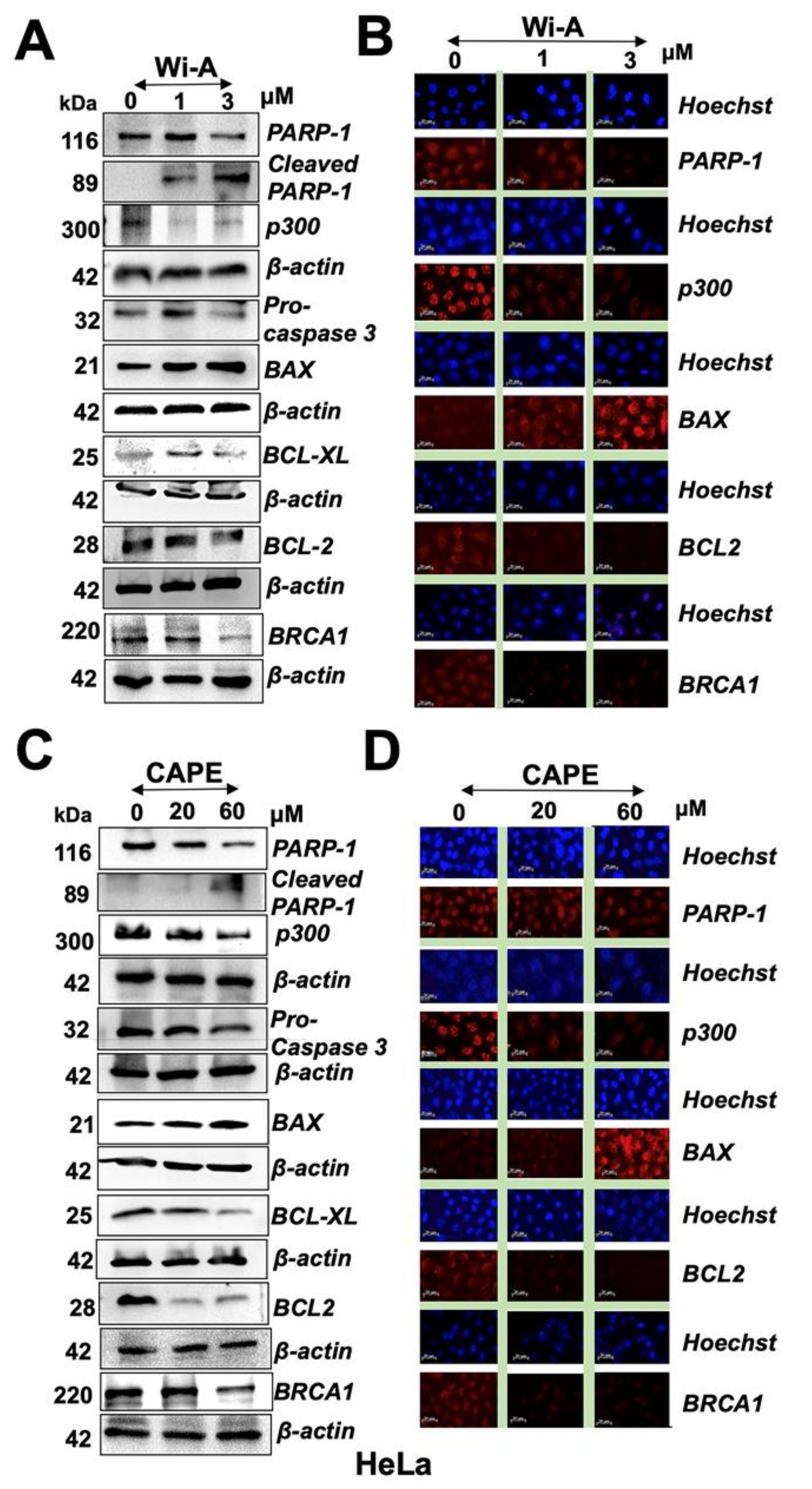
2.3. Wi-A and CAPE Triggered PARP1 Cleavage and Apoptosis Signaling
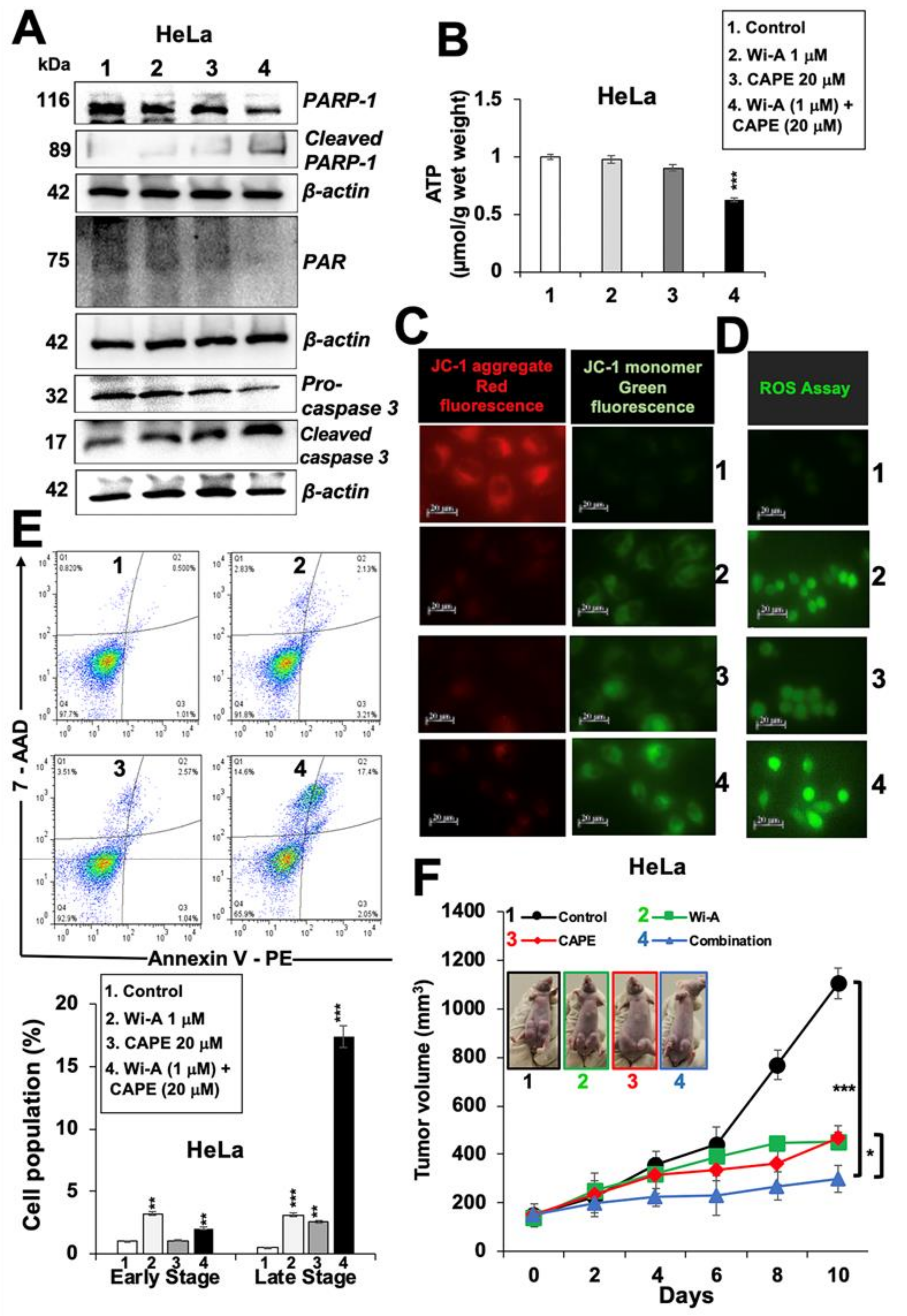
2.4. Combination of Wi-A and CAPE Possesses Stronger Cytotoxicity
2.5. Wi-A and CAPE Directly Interact with PARP1
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. PG-13 Luciferase Reporter Assay
4.3. Cytotoxicity/Growth Inhibition Assay
4.4. Colony Formation Assay
4.5. Western Blot Analysis
4.6. Immunocytochemistry
4.7. Comet Assay
4.8. ROS Assay
4.9. ATP Assay
4.10. Apoptosis Assay
4.11. Trapping Assay
4.12. JC-1 Staining
4.13. Combination Index (CI) Analysis
4.14. RNA Extraction and Quantitative Real-Time Polymerase Chain Reaction
4.15. In Vivo Tumor Suppression
4.16. Bioinformatics Analysis
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Malik, F.; Kumar, A.; Bhushan, S.; Khan, S.; Bhatia, A.; Suri, K.A.; Qazi, G.N.; Singh, J. Reactive oxygen species generation and mitochondrial dysfunction in the apoptotic cell death of human myeloid leukemia HL-60 cells by a dietary compound withaferin A with concomitant protection by N-acetyl cysteine. Apoptosis 2007, 12, 2115–2133. [Google Scholar] [CrossRef] [PubMed]
- Stan, S.D.; Zeng, Y.; Singh, S.V. Ayurvedic medicine constituent withaferin a causes G2 and M phase cell cycle arrest in human breast cancer cells. Nutr. Cancer 2008, 60 (Suppl. 1), 51–60. [Google Scholar] [CrossRef]
- Choi, M.J.; Park, E.J.; Min, K.J.; Park, J.W.; Kwon, T.K. Endoplasmic reticulum stress mediates withaferin A-induced apoptosis in human renal carcinoma cells. Toxicol. Vitr. 2011, 25, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.E.; Shin, J.A.; Jeong, J.H.; Jeon, J.G.; Lee, M.H.; Cho, S.D. Anticancer activity of Ashwagandha against human head and neck cancer cell lines. J. Oral Pathol. Med. 2016, 45, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Suman, S.; Das, T.P.; Sirimulla, S.; Alatassi, H.; Ankem, M.K.; Damodaran, C. Withaferin-A suppress AKT induced tumor growth in colorectal cancer cells. Oncotarget 2016, 7, 13854–13864. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, Y.; Okuzaki, D.; Fukushima, K.; Mukai, S.; Ohno, S.; Ozaki, Y.; Yabuta, N.; Nojima, H. Withaferin A induces cell death selectively in androgen-independent prostate cancer cells but not in normal fibroblast cells. PLoS ONE 2015, 10, e0134137. [Google Scholar] [CrossRef]
- Widodo, N.; Priyandoko, D.; Shah, N.; Wadhwa, R.; Kaul, S.C. Selective killing of cancer cells by Ashwagandha leaf extract and its component withanone involves ROS signaling. PLoS ONE 2010, 5, e13536. [Google Scholar] [CrossRef]
- Yu, Y.; Katiyar, S.P.; Sundar, D.; Kaul, Z.; Miyako, E.; Zhang, Z.; Kaul, S.C.; Reddel, R.R.; Wadhwa, R. Withaferin-A kills cancer cells with and without telomerase: Chemical, computational and experimental evidences. Cell Death Dis. 2017, 8, e2755. [Google Scholar] [CrossRef]
- Sundar, D.; Yu, Y.; Katiyar, S.P.; Putri, J.F.; Dhanjal, J.K.; Wang, J.; Sari, A.N.; Kolettas, E.; Kaul, S.C.; Wadhwa, R. Wild type p53 function in p53(Y220C) mutant harboring cells by treatment with Ashwagandha derived anticancer withanolides: Bioinformatics and experimental evidence. J. Exp. Clin. Cancer Res. 2019, 38, 103. [Google Scholar] [CrossRef]
- Bhargava, P.; Malik, V.; Liu, Y.; Ryu, J.; Kaul, S.C.; Sundar, D.; Wadhwa, R. Molecular insights into Withaferin-A-induced senescence: Bioinformatics and experimental evidence to the role of NFkappaB and CARF. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 183–191. [Google Scholar] [CrossRef]
- Lahat, G.; Zhu, Q.S.; Huang, K.L.; Wang, S.; Bolshakov, S.; Liu, J.; Torres, K.; Langley, R.R.; Lazar, A.J.; Hung, M.C.; et al. Vimentin is a novel anticancer therapeutic target; insights from in vitro and in vivo mice xenograft studies. PLoS ONE 2010, 5, e10105. [Google Scholar] [CrossRef]
- Thaiparambil, J.T.; Bender, L.; Ganesh, T.; Kline, E.; Patel, P.; Liu, Y.; Tighiouart, M.; Vertino, P.M.; Harvey, R.D.; Garcia, A.; et al. Withaferin A inhibits breast cancer invasion and metastasis at sub-cytotoxic doses by inducing vimentin disassembly and serine 56 phosphorylation. Int. J. Cancer 2011, 129, 2744–2755. [Google Scholar] [CrossRef] [PubMed]
- Mohan, R.; Bargagna-Mohan, P. The use of Withaferin A to study intermediate filaments. Methods Enzymol. 2016, 568, 187–218. [Google Scholar] [CrossRef] [PubMed]
- Grin, B.; Mahammad, S.; Wedig, T.; Cleland, M.M.; Tsai, L.; Herrmann, H.; Goldman, R.D. Withaferin A alters intermediate filament organization, cell shape and behavior. PLoS ONE 2012, 7, e39065. [Google Scholar] [CrossRef] [PubMed]
- Bargagna-Mohan, P.; Lei, L.; Thompson, A.; Shaw, C.; Kasahara, K.; Inagaki, M.; Mohan, R. Vimentin phosphorylation underlies myofibroblast sensitivity to withaferin a in vitro and during corneal fibrosis. PLoS ONE 2015, 10, e0133399. [Google Scholar] [CrossRef]
- Munagala, R.; Kausar, H.; Munjal, C.; Gupta, R.C. Withaferin a induces p53-dependent apoptosis by repression of HPV oncogenes and upregulation of tumor suppressor proteins in human cervical cancer cells. Carcinogenesis 2011, 32, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.S.; Park, W.S.; Jung, W.K.; Qian, Z.J.; Lee, D.S.; Choi, J.S.; Lee, D.Y.; Park, S.G.; Seo, S.K.; Kim, H.J.; et al. Caffeic acid phenethyl ester promotes anti-inflammatory effects by inhibiting MAPK and NF-kappaB signaling in activated HMC-1 human mast cells. Pharm. Biol. 2014, 52, 926–932. [Google Scholar] [CrossRef]
- Arasoglu, T.; Derman, S.; Mansuroglu, B. Comparative evaluation of antibacterial activity of caffeic acid phenethyl ester and PLGA nanoparticle formulation by different methods. Nanotechnology 2016, 27, 025103. [Google Scholar] [CrossRef]
- Erdemli, H.K.; Akyol, S.; Armutcu, F.; Akyol, O. Antiviral properties of caffeic acid phenethyl ester and its potential application. J. Intercult. Ethnopharmacol. 2015, 4, 344–347. [Google Scholar] [CrossRef]
- Wadhwa, R.; Nigam, N.; Bhargava, P.; Dhanjal, J.K.; Goyal, S.; Grover, A.; Sundar, D.; Ishida, Y.; Terao, K.; Kaul, S.C. Molecular characterization and enhancement of anticancer activity of caffeic acid phenethyl ester by gamma cyclodextrin. J. Cancer 2016, 7, 1755–1771. [Google Scholar] [CrossRef]
- Frenkel, K.; Wei, H.; Bhimani, R.; Ye, J.; Zadunaisky, J.A.; Huang, M.T.; Ferraro, T.; Conney, A.H.; Grunberger, D. Inhibition of tumor promoter-mediated processes in mouse skin and bovine lens by caffeic acid phenethyl ester. Cancer Res. 1993, 53, 1255–1261. [Google Scholar] [PubMed]
- Su, Z.Z.; Lin, J.; Grunberger, D.; Fisher, P.B. Growth suppression and toxicity induced by caffeic acid phenethyl ester (CAPE) in type 5 adenovirus-transformed rat embryo cells correlate directly with transformation progression. Cancer Res. 1994, 54, 1865–1870. [Google Scholar] [PubMed]
- Jin, U.H.; Chung, T.W.; Kang, S.K.; Suh, S.J.; Kim, J.K.; Chung, K.H.; Gu, Y.H.; Suzuki, I.; Kim, C.H. Caffeic acid phenyl ester in propolis is a strong inhibitor of matrix metalloproteinase-9 and invasion inhibitor: Isolation and identification. Clin. Chim. Acta 2005, 362, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Jin, U.H.; Song, K.H.; Motomura, M.; Suzuki, I.; Gu, Y.H.; Kang, Y.J.; Moon, T.C.; Kim, C.H. Caffeic acid phenethyl ester induces mitochondria-mediated apoptosis in human myeloid leukemia U937 cells. Mol. Cell. Biochem. 2008, 310, 43–48. [Google Scholar] [CrossRef]
- Beauregard, A.P.; Harquail, J.; Lassalle-Claux, G.; Belbraouet, M.; Jean-Francois, J.; Touaibia, M.; Robichaud, G.A. CAPE analogs induce growth arrest and apoptosis in breast cancer cells. Molecules 2015, 20, 12576–12589. [Google Scholar] [CrossRef]
- Firat, F.; Ozgul, M.; Turkoz Uluer, E.; Inan, S. Effects of caffeic acid phenethyl ester (CAPE) on angiogenesis, apoptosis and oxidative stress in various cancer cell lines. Biotech. Histochem. 2019, 94, 491–497. [Google Scholar] [CrossRef]
- Chen, M.J.; Chang, W.H.; Lin, C.C.; Liu, C.Y.; Wang, T.E.; Chu, C.H.; Shih, S.C.; Chen, Y.J. Caffeic acid phenethyl ester induces apoptosis of human pancreatic cancer cells involving caspase and mitochondrial dysfunction. Pancreatology 2008, 8, 566–576. [Google Scholar] [CrossRef]
- Guarini, L.; Su, Z.Z.; Zucker, S.; Lin, J.; Grunberger, D.; Fisher, P.B. Growth inhibition and modulation of antigenic phenotype in human melanoma and glioblastoma multiforme cells by caffeic acid phenethyl ester (CAPE). Cell. Mol. Biol. 1992, 38, 513–527. [Google Scholar]
- Ferreira, R.S.; Dos Santos, N.A.G.; Martins, N.M.; Fernandes, L.S.; Dos Santos, A.C. Caffeic ccid phenethyl ester (CAPE) protects PC12 cells from cisplatin-induced neurotoxicity by activating the NGF-signaling pathway. Neurotox. Res. 2018, 34, 32–46. [Google Scholar] [CrossRef]
- Kim, H.G.; Han, E.H.; Im, J.H.; Lee, E.J.; Jin, S.W.; Jeong, H.G. Caffeic acid phenethyl ester inhibits 3-MC-induced CYP1A1 expression through induction of hypoxia-inducible factor-1alpha. Biochem. Biophys. Res. Commun. 2015, 465, 562–568. [Google Scholar] [CrossRef]
- Lee, K.W.; Chun, K.S.; Lee, J.S.; Kang, K.S.; Surh, Y.J.; Lee, H.J. Inhibition of cyclooxygenase-2 expression and restoration of gap junction intercellular communication in H-ras-transformed rat liver epithelial cells by caffeic acid phenethyl ester. Ann. N. Y. Acad. Sci. 2004, 1030, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, K.; Singh, S.; Burke, T.R., Jr.; Grunberger, D.; Aggarwal, B.B. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-kappa B. Proc. Natl. Acad. Sci. USA 1996, 93, 9090–9095. [Google Scholar] [CrossRef]
- Song, Y.S.; Park, E.H.; Hur, G.M.; Ryu, Y.S.; Lee, Y.S.; Lee, J.Y.; Kim, Y.M.; Jin, C. Caffeic acid phenethyl ester inhibits nitric oxide synthase gene expression and enzyme activity. Cancer Lett. 2002, 175, 53–61. [Google Scholar] [CrossRef]
- Na, H.K.; Wilson, M.R.; Kang, K.S.; Chang, C.C.; Grunberger, D.; Trosko, J.E. Restoration of gap junctional intercellular communication by caffeic acid phenethyl ester (CAPE) in a ras-transformed rat liver epithelial cell line. Cancer Lett. 2000, 157, 31–38. [Google Scholar] [CrossRef]
- Messerli, S.M.; Ahn, M.R.; Kunimasa, K.; Yanagihara, M.; Tatefuji, T.; Hashimoto, K.; Mautner, V.; Uto, Y.; Hori, H.; Kumazawa, S.; et al. Artepillin c (ARC) in brazilian green propolis selectively blocks oncogenic PAK1 signaling and suppresses the growth of NF tumors in mice. Phytother. Res. 2009, 23, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Kuo, H.C.; Chu, C.Y.; Wang, C.J.; Lin, W.C.; Tseng, T.H. Involvement of tumor suppressor protein p53 and p38 MAPK in caffeic acid phenethyl ester-induced apoptosis of C6 glioma cells. Biochem. Pharmacol. 2003, 66, 2281–2289. [Google Scholar] [CrossRef] [PubMed]
- Chuu, C.P.; Lin, H.P.; Ciaccio, M.F.; Kokontis, J.M.; Hause, R.J., Jr.; Hiipakka, R.A.; Liao, S.; Jones, R.B. Caffeic acid phenethyl ester suppresses the proliferation of human prostate cancer cells through inhibition of p70S6K and Akt signaling networks. Cancer Prev. Res. (Phila) 2012, 5, 788–797. [Google Scholar] [CrossRef]
- Demestre, M.; Messerli, S.M.; Celli, N.; Shahhossini, M.; Kluwe, L.; Mautner, V.; Maruta, H. CAPE (caffeic acid phenethyl ester)-based propolis extract (Bio 30) suppresses the growth of human neurofibromatosis (NF) tumor xenografts in mice. Phytother. Res. 2009, 23, 226–230. [Google Scholar] [CrossRef]
- Wu, J.; Omene, C.; Karkoszka, J.; Bosland, M.; Eckard, J.; Klein, C.B.; Frenkel, K. Caffeic acid phenethyl ester (CAPE), derived from a honeybee product propolis, exhibits a diversity of anti-tumor effects in pre-clinical models of human breast cancer. Cancer Lett. 2011, 308, 43–53. [Google Scholar] [CrossRef]
- Liao, H.F.; Chen, Y.Y.; Liu, J.J.; Hsu, M.L.; Shieh, H.J.; Liao, H.J.; Shieh, C.J.; Shiao, M.S.; Chen, Y.J. Inhibitory effect of caffeic acid phenethyl ester on angiogenesis, tumor invasion, and metastasis. J. Agric. Food Chem. 2003, 51, 7907–7912. [Google Scholar] [CrossRef]
- Izuta, H.; Shimazawa, M.; Tsuruma, K.; Araki, Y.; Mishima, S.; Hara, H. Bee products prevent VEGF-induced angiogenesis in human umbilical vein endothelial cells. BMC Complement Altern. Med. 2009, 9, 45. [Google Scholar] [CrossRef]
- Chen, M.J.; Shih, S.C.; Wang, H.Y.; Lin, C.C.; Liu, C.Y.; Wang, T.E.; Chu, C.H.; Chen, Y.J. Caffeic acid phenethyl ester inhibits epithelial-mesenchymal transition of human pancreatic cancer cells. Evid. Based Complement. Altern. Med. 2013, 2013, 270906. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.Y.; Lin, H.P.; Huo, C.; Su, L.C.; Yang, J.; Hsiao, P.H.; Chiang, H.C.; Chung, C.J.; Wang, H.D.; Chang, J.Y.; et al. Caffeic acid phenethyl ester suppresses proliferation and survival of TW2.6 human oral cancer cells via inhibition of Akt signaling. Int. J. Mol. Sci. 2013, 14, 8801–8817. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.Y.; Jim, W.T.; Su, L.C.; Chung, C.J.; Lin, C.Y.; Huo, C.; Tseng, J.C.; Huang, S.H.; Lai, C.J.; Chen, B.C.; et al. Caffeic Acid phenethyl ester is a potential therapeutic agent for oral cancer. Int. J. Mol. Sci. 2015, 16, 10748–10766. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Hsu, J.M.; Kuo, L.K.; Chuu, C.P. Caffeic acid phenethyl ester as an adjuvant therapy for advanced prostate cancer. Med. Hypotheses 2013, 80, 617–619. [Google Scholar] [CrossRef]
- Omene, C.; Kalac, M.; Wu, J.; Marchi, E.; Frenkel, K.; O’Connor, O.A. Propolis and its active component, caffeic acid phenethyl ester (CAPE), modulate breast cancer therapeutic targets via an epigenetically mediated mechanism of action. J. Cancer Sci. Ther. 2013, 5, 334–342. [Google Scholar] [CrossRef]
- Tseng, J.C.; Lin, C.Y.; Su, L.C.; Fu, H.H.; Yang, S.D.; Chuu, C.P. CAPE suppresses migration and invasion of prostate cancer cells via activation of non-canonical Wnt signaling. Oncotarget 2016, 7, 38010–38024. [Google Scholar] [CrossRef]
- Hwang, H.J.; Park, H.J.; Chung, H.J.; Min, H.Y.; Park, E.J.; Hong, J.Y.; Lee, S.K. Inhibitory effects of caffeic acid phenethyl ester on cancer cell metastasis mediated by the down-regulation of matrix metalloproteinase expression in human HT1080 fibrosarcoma cells. J. Nutr. Biochem. 2006, 17, 356–362. [Google Scholar] [CrossRef]
- Lee, K.W.; Kang, N.J.; Kim, J.H.; Lee, K.M.; Lee, D.E.; Hur, H.J.; Lee, H.J. Caffeic acid phenethyl ester inhibits invasion and expression of matrix metalloproteinase in SK-Hep1 human hepatocellular carcinoma cells by targeting nuclear factor kappa B. Genes Nutr. 2008, 2, 319–322. [Google Scholar] [CrossRef]
- Chen, Y.J.; Liao, H.F.; Tsai, T.H.; Wang, S.Y.; Shiao, M.S. Caffeic acid phenethyl ester preferentially sensitizes CT26 colorectal adenocarcinoma to ionizing radiation without affecting bone marrow radioresponse. Int. J. Radiat. Oncol. Biol. Phys. 2005, 63, 1252–1261. [Google Scholar] [CrossRef]
- Lee, Y.Y.; Kao, C.L.; Tsai, P.H.; Tsai, T.H.; Chiou, S.H.; Wu, W.F.; Ku, H.H.; Wong, T.T. Caffeic acid phenethyl ester preferentially enhanced radiosensitizing and increased oxidative stress in medulloblastoma cell line. Childs Nerv. Syst. 2008, 24, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Anjaly, K.; Tiku, A.B. Radio-modulatory potential of caffeic acid phenethyl ester: A therapeutic perspective. Anticancer Agents Med. Chem. 2018, 18, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.J.; Choi, J.H.; Hwang, Y.P.; Chung, Y.C.; Jeong, H.G. Protective effect of caffeic acid phenethyl ester on tert-butyl hydroperoxide-induced oxidative hepatotoxicity and DNA damage. Food Chem. Toxicol. 2008, 46, 2445–2450. [Google Scholar] [CrossRef] [PubMed]
- Albukhari, A.A.; Gashlan, H.M.; El-Beshbishy, H.A.; Nagy, A.A.; Abdel-Naim, A.B. Caffeic acid phenethyl ester protects against tamoxifen-induced hepatotoxicity in rats. Food Chem. Toxicol. 2009, 47, 1689–1695. [Google Scholar] [CrossRef]
- Motawi, T.K.; Abdelazim, S.A.; Darwish, H.A.; Elbaz, E.M.; Shouman, S.A. Could caffeic acid phenethyl ester expand the antitumor effect of tamoxifen in breast carcinoma? Nutr. Cancer 2016, 68, 435–445. [Google Scholar] [CrossRef]
- Motawi, T.K.; Abdelazim, S.A.; Darwish, H.A.; Elbaz, E.M.; Shouman, S.A. Modulation of tamoxifen cytotoxicity by caffeic acid phenethyl ester in MCF-7 breast cancer cells. Oxid. Med. Cell. Longev. 2016, 2016, 3017108. [Google Scholar] [CrossRef]
- Matsunaga, T.; Tsuchimura, S.; Azuma, N.; Endo, S.; Ichihara, K.; Ikari, A. Caffeic acid phenethyl ester potentiates gastric cancer cell sensitivity to doxorubicin and cisplatin by decreasing proteasome function. Anti-Cancer Drugs 2019, 30, 251–259. [Google Scholar] [CrossRef]
- O’Cearbhaill, R.E. Using PARP inhibitors in advanced ovarian cancer. Oncology (Williston Park) 2018, 32, 339–343. [Google Scholar]
- Wu, L.; Zhong, L. Budget impact analysis of niraparib and olaparib for maintenance treatment of platinum-sensitive, recurrent ovarian cancer in the US. J. Med. Econ. 2019, 22, 187–195. [Google Scholar] [CrossRef]
- Putri, J.F.; Bhargava, P.; Dhanjal, J.K.; Yaguchi, T.; Sundar, D.; Kaul, S.C.; Wadhwa, R. Mortaparib, a novel dual inhibitor of mortalin and PARP1, is a potential drug candidate for ovarian and cervical cancers. J. Expt. Clin. Cancer Res. 2019, 38, 499. [Google Scholar] [CrossRef]
- Vaishnavi, K.; Saxena, N.; Shah, N.; Singh, R.; Manjunath, K.; Uthayakumar, M. Differential activities of the two closely related withanolides, withaferin A and withanone: Bioinformatics and experimental evidences. PLoS ONE 2012, 7, e44419. [Google Scholar] [CrossRef] [PubMed]
- Hegan, D.S.; Lu, Y.; Stachelek, G.C.; Crosby, M.E.; Bindra, R.S.; Glazer, P.M. Inhibition of poly(ADP-ribose) polymerase down-regulates BRCA1 and RAD51 in a pathway mediated by E2F4 and p130. Proc. Natl. Acad. Sci. USA 2010, 107, 2201–2206. [Google Scholar] [CrossRef] [PubMed]
- Mweempwa, A.; Wilson, M.K. Mechanisms of resistance to PARP inhibitors - an evolving challenge in oncology. Cancer Drug. Resist. 2019, 2, 608–617, . [Google Scholar] [CrossRef]
- Reilly, S.W.; Puentes, L.N.; Wilson, K.; Hsieh, C.J.; Weng, C.C.; Makvandi, M.; Mach, R.H. Examination of Diazaspiro Cores as Piperazine Bioisosteres in the Olaparib Framework Shows Reduced DNA Damage and Cytotoxicity. J. Med. Chem. 2018, 61, 12, 5367–5379. [Google Scholar] [CrossRef]
- Ridge, K.M.; Shumaker, D.; Robert, A.; Hookway, C.; Gelfand, V.I.; Janmey, P.A.; Lowery, J.; Guo, M.; Weitz, D.A.; Kuczmarski, E.; et al. Methods for determining the cellular functions of vimentin intermediate filaments. Methods Enzymol. 2016, 568, 389–426. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Lim, I.H.; Sung, E.G.; Kim, J.Y.; Song, I.H.; Park, Y.K.; Lee, T.J. Withaferin A inhibits matrix metalloproteinase-9 activity by suppressing the Akt signaling pathway. Oncol. Rep. 2013, 30, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Um, H.J.; Min, K.J.; Kim, D.E.; Kwon, T.K. Withaferin A inhibits JAK/STAT3 signaling and induces apoptosis of human renal carcinoma Caki cells. Biochem. Biophys. Res. Commun. 2012, 427, 24–29. [Google Scholar] [CrossRef]
- Khan, S.; Khamis, I.; Heikkila, J.J. The small heat shock protein, HSP30, is associated with aggresome-like inclusion bodies in proteasomal inhibitor-, arsenite-, and cadmium-treated Xenopus kidney cells. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2015, 189, 130–140. [Google Scholar] [CrossRef]
- Sharda, A.C.; Solomon, F.E.; Devi, P.U.; Udupa, N.; Srinivasan, K.K. Antitumor and radisensitizing effects of withaferin a on mous EHRLICH ascites carcinoma In vivo. Acta Oncol. 1996, 35, 95–100. [Google Scholar] [CrossRef]
- Pires, N.; Gota, V.; Gulia, A.; Hingorani, L.; Agarwal, M.; Puri, A. Safety and pharmacokinetics of withaferin-a in advanced stage high grade osteosarcoma: A phase I trial. J. Ayurveda Integr. Med. 2020, 11, 68–72. [Google Scholar] [CrossRef]
- Gao, R.; Shah, N.; Lee, J.S.; Katiyar, S.P.; Li, L.; Oh, E.; Sundar, D.; Yun, C.O.; Wadhwa, R.; Kaul, S.C. Withanone-rich combination of Ashwagandha withanolides restricts metastasis and angiogenesis through hnRNP-K. Mol. Cancer Ther. 2014, 13, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Kakar, S.S.; Ratajczak, M.Z.; Powell, K.S.; Moghadamfalahi, M.; Miller, D.M.; Batra, S.K.; Singh, S.K. Withaferin a alone and in combination with cisplatin suppresses growth and metastasis of ovarian cancer by targeting putative cancer stem cells. PLoS ONE 2014, 9, e107596. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.; Huifu, H.; Kumari, A.; Sundar, D.; Kaul, S.C.; Wadhwa, R. Induction of senescence in cancer cells by a novel combination of cucurbitacon B and withanone: Molecular mechanism and therapeutic potential. J. Gerontol. A Biol. Sci. Med. Sci. 2019, glz077. [Google Scholar] [CrossRef] [PubMed]
- Jung, B.I.; Kim, M.S.; Kim, H.A.; Kim, D.; Yang, J.; Her, S.; Song, Y.S. Caffeic acid phenethyl ester, a component of beehive propolis, is a novel selective estrogen receptor modulator. Phytother Res. 2010, 24, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Gao, R.; Shah, N.; Bhargava, P.; Furune, T.; Kaul, S.C.; Terao, K.; Wadhwa, R. Anticancer activity in honeybee propolis: Functional insights to the role of caffeic acid phenethyl ester and its complex with gamma-cyclodextrin. Integr. Cancer Ther. 2018, 17, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.H.; Chu, C.C.; Hung, M.W.; Lee, H.J.; Hsu, H.J.; Chang, T.C. Caffeic acid phenethyl ester induces E2F-1-mediated growth inhibition and cell-cycle arrest in human cervical cancer cells. FEBS J. 2013, 280, 2581–2593. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, P.; Kumari, A.; Putri, J.F.; Ishida, Y.; Terao, K.; Kaul, S.C.; Sundar, D.; Wadhwa, R. Caffeic acid phenethyl ester (CAPE) possesses pro-hypoxia and anti-stress activities: Bioinformatics and experimental evidences. Cell Stress Chaperones 2018, 23, 1055–1068. [Google Scholar] [CrossRef]
- Tolba, M.F.; Esmat, A.; Al-Abd, A.M.; Azab, S.S.; Khalifa, A.E.; Mosli, H.A.; Abdel-Rahman, S.Z.; Abdel-Naim, A.B. Caffeic acid phenethyl ester synergistically enhances docetaxel and paclitaxel cytotoxicity in prostate cancer cells. IUBMB Life 2013, 65, 716–729. [Google Scholar] [CrossRef]
- Widodo, N.; Kaur, K.; Shrestha, B.G.; Takagi, Y.; Ishii, T.; Wadhwa, R.; Kaul, S.C. Selective killing of cancer cells by leaf extract of Ashwagandha: Identification of a tumor-inhibitory factor and the first molecular insights to its effect. Clin. Cancer Res. 2007, 13, 2298–2306. [Google Scholar] [CrossRef]
- Lu, W.J.; Lee, N.P.; Kaul, S.C.; Lan, F.; Poon, R.T.; Wadhwa, R.; Luk, J.M. Mortalin-p53 interaction in cancer cells is stress dependent and constitutes a selective target for cancer therapy. Cell Death Differ. 2011, 18, 1046–1056. [Google Scholar] [CrossRef]
- Lu, W.J.; Lee, N.P.; Kaul, S.C.; Lan, F.; Poon, R.T.; Wadhwa, R.; Luk, J.M. Induction of mutant p53-dependent apoptosis in human hepatocellular carcinoma by targeting stress protein mortalin. Int. J. Cancer 2011, 129, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Flachbartova, Z.; Kovacech, B. Mortalin—A multipotent chaperone regulating cellular processes ranging from viral infection to neurodegeneration. Acta Virol. 2013, 57, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Kostecka, A.; Sznarkowska, A.; Meller, K.; Acedo, P.; Shi, Y.; Sakil, H.M.; Kawiak, A.; Lion, M.; Królicka, A.; Wilhelm, M.; et al. JNK–NQO1 axis drives TAp73-mediated tumor suppression upon oxidative and proteasomal stress. Cell Death Dis. 2014, 5, e1484. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, R.; Priyandoko, D.; Gao, R.; Widodo, N.; Nigam, N.; Li, L.; Ahn, H.M.; Yun, C.O.; Ando, N.; Mahe, C.; et al. Stress chaperone mortalin regulates human melanogenesis. Cell Stress Chaperones 2016, 21, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, L.M.; Jiang, Y.; Drobnitzky, N.; Green, M.; Cahill, F.; Patel, A.; Shanneik, Y.; Moore, J.; Ryan, A.J. PARP inhibition combined with thoracic irradiation exacerbates esophageal and skin toxicity in C57BL6 mice. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 767–775. [Google Scholar] [CrossRef]
- Fang, P.; Madden, J.A.; Neums, L.; Moulder, R.K.; Forrest, M.L.; Chien, J. Olaparib-induced adaptive response is disrupted by FOXM1 targeting that enhances sensitivity to PARP inhibition. Mol. Cancer Res. 2018, 16, 961–973. [Google Scholar] [CrossRef]
- Griguolo, G.; Dieci, M.V.; Guarneri, V.; Conte, P. Olaparib for the treatment of breast cancer. Expert Rev. Anticancer Ther. 2018, 18, 519–530. [Google Scholar] [CrossRef]
- Wang, X.; Shi, Y.; Huang, D.; Guan, X. Emerging therapeutic modalities of PARP inhibitors in breast cancer. Cancer Treat. Rev. 2018, 68, 62–68. [Google Scholar] [CrossRef]
- Wu, M.; Liu, J.; Hu, C.; Li, D.; Yang, J.; Wu, Z.; Yang, L.; Chen, Y.; Fu, S.; Wu, J. Olaparib nanoparticles potentiated radiosensitization effects on lung cancer. Int. J. Nanomed. 2018, 13, 8461–8472. [Google Scholar] [CrossRef]
- Vel Szic, K.S.; De Beeck, K.O.; Ratman, D.; Wouters, A.; Beck, I.M.; Declerck, K.; Heyninck, K.; Fransen, E.; Bracke, M.; De Bosscher, K.; et al. Pharmacological levels of withaferin A (Withania somnifera) trigger clinically relevant anticancer effects specific to triple negative breast cancer cells. PLoS ONE 2014, 9, e87850. [Google Scholar] [CrossRef]
- Oben, K.Z.; Alhakeem, S.S.; McKenna, M.K.; Brandon, J.A.; Mani, R.; Noothi, S.K.; Jinpeng, L.; Akunuru, S.; Dhar, S.K.; Singh, I.P.; et al. Oxidative stress-induced JNK/AP-1 signaling is a major pathway involved in selective apoptosis of myelodysplastic syndrome cells by Withaferin-A. Oncotarget 2017, 8, 77436–77452. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Rammeloo, A.W.; Heikkila, J.J. Withaferin-A induces proteasome inhibition, endoplasmic reticulum stress, the heat shock response and acquisition of thermotolerance. PLoS ONE 2012, 7, e50547. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-1: Maestro 019-3 SR, Glide, LigPrep, ProteinPreparation Wizard, Prime, Molecular Dynamics System; Schrödinger: New York, NY, USA, 2020.
- Dawicki-McKenna, J.M.; Langelier, M.F.; DeNizio, J.E.; Riccio, A.A.; Cao, C.D.; Karch, K.R.; McCauley, M.; Steffen, J.D.; Black, B.E.; Pascal, J.M. PARP1 activation requires local unfolding of an autoinhibitory domain. Mol. Cell. 2015, 60, 755–768. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene (Human) | Sequence (5′-3′) |
|---|---|
| p53 forward | GTTCCGAGAGCTGAATGAGG |
| p53 reverse | TCTGAGTCAGGCCCTTCTGT |
| Mortalin forward | AGCTGGAATGGCCTTAGTCAT |
| Mortalin reverse | CAGGAGTTGGTAGTACCCAAATC |
| PARP1 forward | TCAGCCTCCTTGCTACAGAGG |
| PARP1 reverse | GGTCGTTCTGAGCCTTTAGGG |
| p300 forward | AAACCCACCAGATGAGGAC |
| p300 reverse | TATGCACTAGATGGCTCCGCAG |
| 18S forward | CAGGGTTCGATTCCGTAGAG |
| 18S reverse | CCTCCAGTGGATCCTCGTTA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sari, A.N.; Bhargava, P.; Dhanjal, J.K.; Putri, J.F.; Radhakrishnan, N.; Shefrin, S.; Ishida, Y.; Terao, K.; Sundar, D.; Kaul, S.C.; et al. Combination of Withaferin-A and CAPE Provides Superior Anticancer Potency: Bioinformatics and Experimental Evidence to Their Molecular Targets and Mechanism of Action. Cancers 2020, 12, 1160. https://doi.org/10.3390/cancers12051160
Sari AN, Bhargava P, Dhanjal JK, Putri JF, Radhakrishnan N, Shefrin S, Ishida Y, Terao K, Sundar D, Kaul SC, et al. Combination of Withaferin-A and CAPE Provides Superior Anticancer Potency: Bioinformatics and Experimental Evidence to Their Molecular Targets and Mechanism of Action. Cancers. 2020; 12(5):1160. https://doi.org/10.3390/cancers12051160
Chicago/Turabian StyleSari, Anissa Nofita, Priyanshu Bhargava, Jaspreet Kaur Dhanjal, Jayarani F. Putri, Navaneethan Radhakrishnan, Seyad Shefrin, Yoshiyuki Ishida, Keiji Terao, Durai Sundar, Sunil C. Kaul, and et al. 2020. "Combination of Withaferin-A and CAPE Provides Superior Anticancer Potency: Bioinformatics and Experimental Evidence to Their Molecular Targets and Mechanism of Action" Cancers 12, no. 5: 1160. https://doi.org/10.3390/cancers12051160
APA StyleSari, A. N., Bhargava, P., Dhanjal, J. K., Putri, J. F., Radhakrishnan, N., Shefrin, S., Ishida, Y., Terao, K., Sundar, D., Kaul, S. C., & Wadhwa, R. (2020). Combination of Withaferin-A and CAPE Provides Superior Anticancer Potency: Bioinformatics and Experimental Evidence to Their Molecular Targets and Mechanism of Action. Cancers, 12(5), 1160. https://doi.org/10.3390/cancers12051160