Natural Killer Cell Responses in Hepatocellular Carcinoma: Implications for Novel Immunotherapeutic Approaches

 , ,
, , 
Abstract
1. Introduction
2. Alteration of the NKG2D-MICA/B Axis in HCC
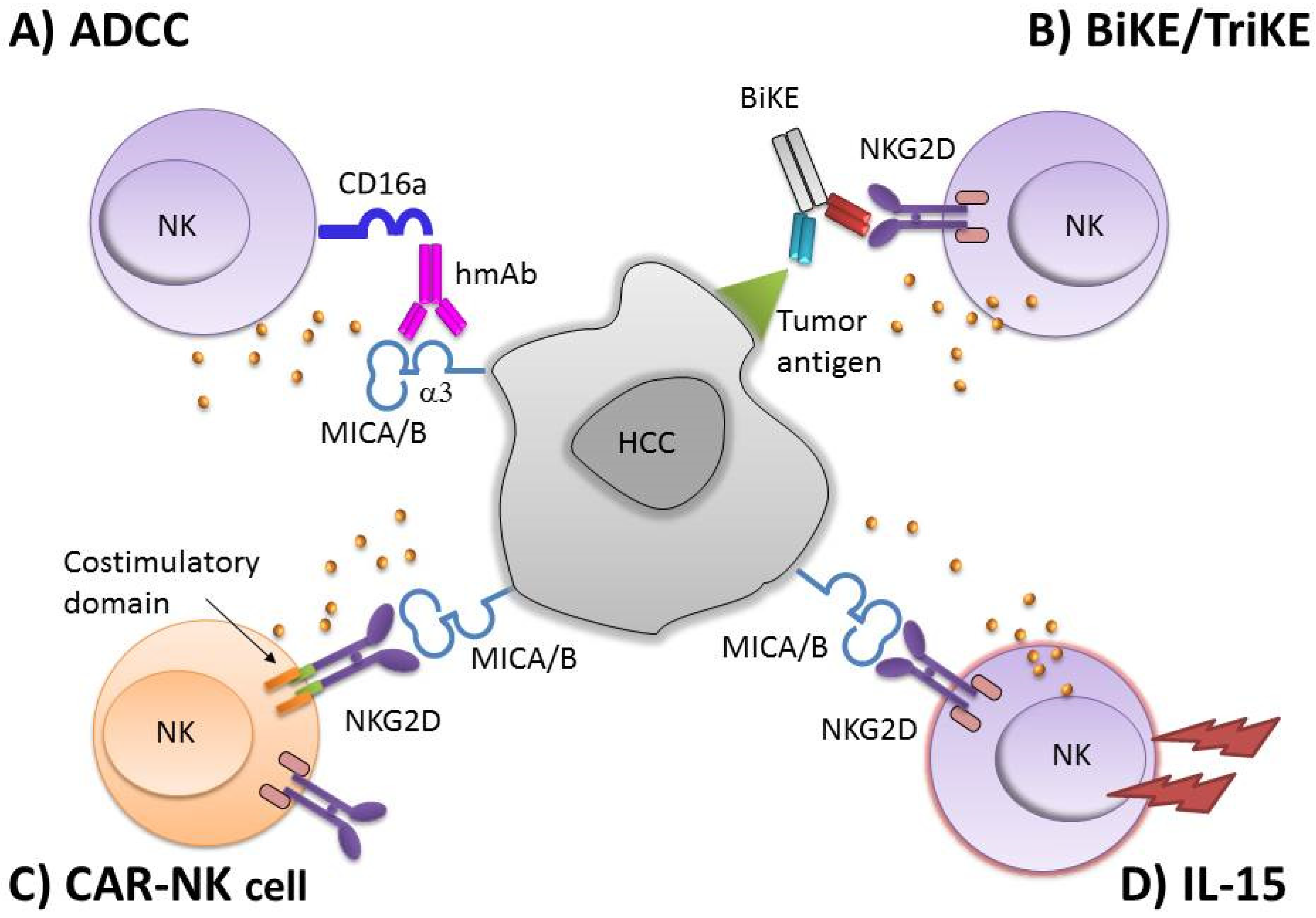
3. NK-Based Immunotherapy
4. NKG2D Based CAR-NK Cells
5. NKG2DL-Specific Antibodies for NK Cell-Mediated ADCC
6. Conclusions
Funding
Conflicts of Interest
References
- Matičič, M.; Lombardi, A.; Mondelli, M.U.; Colombo, M.; ESCMID Study Group for Viral Hepatitis (ESGVH). Elimination of hepatitis C in Europe: Can WHO targets be achieved? Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Coulouarn, C.; Factor, V.M.; Conner, E.A.; Thorgeirsson, S.S. Genomic modeling of tumor onset and progression in a mouse model of aggressive human liver cancer. Carcinogenesis 2011, 32, 1434–1440. [Google Scholar] [CrossRef] [PubMed]
- Chew, V.; Chen, J.; Lee, D.; Loh, E.; Lee, J.; Lim, K.H.; Weber, A.; Slankamenac, K.; Poon, R.T.; Yang, H.; et al. Chemokine-driven lymphocyte infiltration: An early intratumoural event determining long-term survival in resectable hepatocellular carcinoma. Gut 2012, 61, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Fang, L.; Liu, R.; Wang, Y.; Xing, J.; Chen, Y.; Zhuang, R.; Zhang, Y.; Zhang, C.; Yang, A.; et al. UPR decreases CD226 ligand CD155 expression and sensitivity to NK cell-mediated cytotoxicity in hepatoma cells. Eur. J. Immunol. 2014, 44, 3758–3767. [Google Scholar] [CrossRef]
- Mantovani, S.; Oliviero, B.; Lombardi, A.; Varchetta, S.; Mele, D.; Sangiovanni, A.; Rossi, G.; Donadon, M.; Torzilli, G.; Soldani, C.; et al. Deficient Natural Killer Cell NKp30-Mediated Function and Altered NCR3 Splice Variants in Hepatocellular Carcinoma. Hepatology 2019, 69, 1165–1179. [Google Scholar] [CrossRef] [PubMed]
- Vitale, M.; Cantoni, C.; Pietra, G.; Mingari, M.C.; Moretta, L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur. J. Immunol. 2014, 44, 1582–1592. [Google Scholar] [CrossRef] [PubMed]
- Vacca, P.; Munari, E.; Tumino, N.; Moretta, F.; Pietra, G.; Vitale, M.; Del Zotto, G.; Mariotti, F.R.; Mingari, M.C.; Moretta, L. Human natural killer cells and other innate lymphoid cells in cancer: Friends or foes? Immunol. Lett. 2018, 201, 14–19. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, Z.; Zhou, X.; Zhang, H.; Yang, N.; Wu, Y.; Chen, Y.; Yang, G.; Ren, T. Loss of expression of MHC class I-related chain A (MICA) is a frequent event and predicts poor survival in patients with hepatocellular carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 3123–3131. [Google Scholar]
- Zhang, J.; Zheng, H.; Diao, Y. Natural Killer Cells and Current Applications of Chimeric Antigen Receptor-Modified NK-92 Cells in Tumor Immunotherapy. Int. J. Mol. Sci. 2019, 20, 317. [Google Scholar] [CrossRef]
- Xiao, L.; Cen, D.; Gan, H.; Sun, Y.; Huang, N.; Xiong, H.; Jin, Q.; Su, L.; Liu, X.; Wang, K.; et al. Adoptive Transfer of NKG2D CAR mRNA-Engineered Natural Killer Cells in Colorectal Cancer Patients. Mol. Ther. J. Am. Soc. Gene Ther. 2019, 27, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Garrity, D.; Call, M.E.; Feng, J.; Wucherpfennig, K.W. The activating NKG2D receptor assembles in the membrane with two signaling dimers into a hexameric structure. Proc. Natl. Acad. Sci. USA 2005, 102, 7641–7646. [Google Scholar] [CrossRef] [PubMed]
- Molfetta, R.; Quatrini, L.; Zitti, B.; Capuano, C.; Galandrini, R.; Santoni, A.; Paolini, R. Regulation of NKG2D Expression and Signaling by Endocytosis. Trends Immunol. 2016, 37, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, C.L.; Chalupny, N.J.; Schooley, K.; VandenBos, T.; Kubin, M.; Cosman, D. UL16-binding proteins, novel MHC class I-related proteins, bind to NKG2D and activate multiple signaling pathways in primary NK cells. J. Immunol. 2002, 168, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, E.; Koch, J.; Cerwenka, A.; Steinle, A. New prospects on the NKG2D/NKG2DL system for oncology. Oncoimmunolog 2013, 2, e26097. [Google Scholar] [CrossRef]
- Lanier, L.L. NKG2D Receptor and Its Ligands in Host Defense. Cancer Immunol. Res. 2015, 3, 575–582. [Google Scholar] [CrossRef]
- Rook, A.H.; Kehrl, J.H.; Wakefield, L.M.; Roberts, A.B.; Sporn, M.B.; Burlington, D.B.; Lane, H.C.; Fauci, A.S. Effects of transforming growth factor beta on the functions of natural killer cells: Depressed cytolytic activity and blunting of interferon responsiveness. J. Immunol. 1986, 136, 3916–3920. [Google Scholar]
- Bellone, G.; Aste-Amezaga, M.; Trinchieri, G.; Rodeck, U. Regulation of NK cell functions by TGF-beta 1. J. Immunol. 1995, 155, 1066–1073. [Google Scholar]
- Lazarova, M.; Steinle, A. Impairment of NKG2D-Mediated Tumor Immunity by TGF-β. Front. Immunol. 2019, 10, 2689. [Google Scholar] [CrossRef]
- Espinoza, J.L.; Takami, A.; Yoshioka, K.; Nakata, K.; Sato, T.; Kasahara, Y.; Nakao, S. Human microRNA-1245 down-regulates the NKG2D receptor in natural killer cells and impairs NKG2D-mediated functions. Haematologica 2012, 97, 1295–1303. [Google Scholar] [CrossRef]
- Park, Y.P.; Choi, S.C.; Kiesler, P.; Gil-Krzewska, A.; Borrego, F.; Weck, J.; Krzewski, K.; Coligan, J.E. Complex regulation of human NKG2D-DAP10 cell surface expression: Opposing roles of the γc cytokines and TGF-β1. Blood 2011, 118, 3019–3027. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Ménard, C.; Terme, M.; Flament, C.; Taieb, J.; Chaput, N.; Puig, P.E.; Novault, S.; Escudier, B.; Vivier, E.; et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J. Exp. Med. 2005, 202, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.; Mitchell, J.P.; Court, J.; Linnane, S.; Mason, M.D.; Tabi, Z. Human tumor-derived exosomes down-modulate NKG2D expression. J. Immunol. 2008, 180, 7249–7258. [Google Scholar] [CrossRef]
- Carambia, A.; Freund, B.; Schwinge, D.; Heine, M.; Laschtowitz, A.; Huber, S.; Wraith, D.C.; Korn, T.; Schramm, C.; Lohse, A.W.; et al. TGF-β-dependent induction of CD4+CD25+Foxp3+ Tregs by liver sinusoidal endothelial cells. J. Hepatol. 2014, 61, 594–599. [Google Scholar] [CrossRef]
- Faivre, S.; Rimassa, L.; Finn, R.S. Molecular therapies for HCC: Looking outside the box. J. Hepatol. 2020, 72, 342–352. [Google Scholar] [CrossRef]
- Ye, L.; Zhang, Q.; Cheng, Y.; Chen, X.; Wang, G.; Shi, M.; Zhang, T.; Cao, Y.; Pan, H.; Zhang, L.; et al. Tumor-derived exosomal HMGB1 fosters hepatocellular carcinoma immune evasion by promoting TIM-1+ regulatory B cell expansion. J. Immunother. Cancer 2018, 6, 145. [Google Scholar] [CrossRef]
- Sène, D.; Levasseur, F.; Abel, M.; Lambert, M.; Camous, X.; Hernandez, C.; Pène, V.; Rosenberg, A.R.; Jouvin-Marche, E.; Marche, P.N.; et al. Hepatitis C virus (HCV) evades NKG2D-dependent NK cell responses through NS5A-mediated imbalance of inflammatory cytokines. PLoS Pathog. 2010, 6, e1001184. [Google Scholar] [CrossRef]
- Piñeiro, F.J.; Luddy, K.A.; Harmon, C.; O’Farrelly, C. Hepatic Tumor Microenvironments and Effects on NK Cell Phenotype and Function. Int. J. Mol. Sci. 2019, 20, 4131. [Google Scholar] [CrossRef]
- Hoechst, B.; Voigtlaender, T.; Ormandy, L.; Gamrekelashvili, J.; Zhao, F.; Wedemeyer, H.; Lehner, F.; Manns, M.P.; Greten, T.F.; Korangy, F. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology 2009, 50, 799–807. [Google Scholar] [CrossRef]
- Cariani, E.; Missale, G. Immune landscape of hepatocellular carcinoma microenvironment: Implications for prognosis and therapeutic applications. Liver Int. 2019, 39, 1608–1621. [Google Scholar] [CrossRef]
- Easom, N.; Stegmann, K.A.; Swadling, L.; Pallett, L.J.; Burton, A.R.; Odera, D.; Schmidt, N.; Huang, W.C.; Fusai, G.; Davidson, B.; et al. IL-15 Overcomes Hepatocellular Carcinoma-Induced NK Cell Dysfunction. Front. Immunol. 2018, 9, 1009. [Google Scholar] [CrossRef]
- Quatrini, L.; Molfetta, R.; Zitti, B.; Peruzzi, G.; Fionda, C.; Capuano, C.; Galandrini, R.; Cippitelli, M.; Santoni, A.; Paolini, R. Ubiquitin-dependent endocytosis of NKG2D-DAP10 receptor complexes activates signaling and functions in human NK cells. Sci. Signal. 2015, 8, ra108. [Google Scholar] [CrossRef]
- Raulet, D.H.; Gasser, S.; Gowen, B.G.; Deng, W.; Jung, H. Regulation of ligands for the NKG2D activating receptor. Annu. Rev. Immunol. 2013, 31, 413–441. [Google Scholar] [CrossRef]
- Champsaur, M.; Lanier, L.L. Effect of NKG2D ligand expression on host immune responses. Immunol. Rev. 2010, 235, 267–285. [Google Scholar] [CrossRef]
- Ashiru, O.; López-Cobo, S.; Fernández-Messina, L.; Pontes-Quero, S.; Pandolfi, R.; Reyburn, H.T.; Valés-Gómez, M. A GPI anchor explains the unique biological features of the common NKG2D-ligand allele MICA*008. Biochem. J. 2013, 454, 295–302. [Google Scholar] [CrossRef]
- Diefenbach, A.; Raulet, D.H. Strategies for target cell recognition by natural killer cells. Immunol. Rev. 2001, 181, 170–184. [Google Scholar] [CrossRef]
- Guan, Y.; Li, W.; Hou, Z.; Han, Q.; Lan, P.; Zhang, J.; Tian, Z.; Zhang, C. HBV suppresses expression of MICA/B on hepatoma cells through up-regulation of transcription factors GATA2 and GATA3 to escape from NK cell surveillance. Oncotarget 2016, 7, 56107–56119. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.; He, X.; Ma, H.; Hou, N.; Wei, C.; Song, T.; Zhang, Y.; Sun, L.; Ma, Q.; Zhong, H. Hepatitis C virus infection downregulates the ligands of the activating receptor NKG2D. Cell. Mol. Immunol. 2008, 5, 475–478. [Google Scholar] [CrossRef]
- Dassa, L.; Seidel, E.; Oiknine-Djian, E.; Yamin, R.; Wolf, D.G.; Le-Trilling, V.; Mandelboim, O. The Human Cytomegalovirus Protein UL148A Downregulates the NK Cell-Activating Ligand MICA To Avoid NK Cell Attack. J. Virol. 2018, 92, e00162-18. [Google Scholar] [CrossRef]
- Kamimura, H.; Yamagiwa, S.; Tsuchiya, A.; Takamura, M.; Matsuda, Y.; Ohkoshi, S.; Inoue, M.; Wakai, T.; Shirai, Y.; Nomoto, M.; et al. Reduced NKG2D ligand expression in hepatocellular carcinoma correlates with early recurrence. J. Hepatol. 2012, 56, 381–388. [Google Scholar] [CrossRef]
- Chen, Y.; Cheng, M.; Tian, Z. Hepatitis B virus down-regulates expressions of MHC class I molecules on hepatoplastoma cell line. Cell. Mol. Immunol. 2006, 3, 373–378. [Google Scholar]
- Wu, J.; Zhang, X.J.; Shi, K.Q.; Chen, Y.P.; Ren, Y.F.; Song, Y.J.; Li, G.; Xue, Y.F.; Fang, Y.X.; Deng, Z.J.; et al. Hepatitis B surface antigen inhibits MICA and MICB expression via induction of cellular miRNAs in hepatocellular carcinoma cells. Carcinogenesis 2014, 35, 155–163. [Google Scholar] [CrossRef]
- Kishikawa, T.; Otsuka, M.; Yoshikawa, T.; Ohno, M.; Takata, A.; Shibata, C.; Kondo, Y.; Akanuma, M.; Yoshida, H.; Koike, K. Regulation of the expression of the liver cancer susceptibility gene MICA by microRNAs. Sci. Rep. 2013, 3, 2739. [Google Scholar] [CrossRef]
- Zunke, F.; Rose-John, S. The shedding protease ADAM17: Physiology and pathophysiology. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2059–2070. [Google Scholar] [CrossRef]
- Reiss, K.; Bhakdi, S. The plasma membrane: Penultimate regulator of ADAM sheddase function. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2082–2087. [Google Scholar] [CrossRef]
- Zingoni, A.; Cecere, F.; Vulpis, E.; Fionda, C.; Molfetta, R.; Soriani, A.; Petrucci, M.T.; Ricciardi, M.R.; Fuerst, D.; Amendola, M.G.; et al. Genotoxic Stress Induces Senescence-Associated ADAM10-Dependent Release of NKG2D MIC Ligands in Multiple Myeloma Cells. J. Immunol. 2015, 195, 736–748. [Google Scholar] [CrossRef]
- Boutet, P.; Agüera-González, S.; Atkinson, S.; Pennington, C.J.; Edwards, D.R.; Murphy, G.; Reyburn, H.T.; Valés-Gómez, M. Cutting edge: The metalloproteinase ADAM17/TNF-alpha-converting enzyme regulates proteolytic shedding of the MHC class I-related chain B protein. J. Immunol. 2009, 182, 49–53. [Google Scholar] [CrossRef]
- Waldhauer, I.; Steinle, A. Proteolytic release of soluble UL16-binding protein 2 from tumor cells. Cancer Res. 2006, 66, 2520–2526. [Google Scholar] [CrossRef]
- Kohga, K.; Takehara, T.; Tatsumi, T.; Ishida, H.; Miyagi, T.; Hosui, A.; Hayashi, N. Sorafenib inhibits the shedding of major histocompatibility complex class I-related chain A on hepatocellular carcinoma cells by down-regulating a disintegrin and metalloproteinase 9. Hepatology 2010, 51, 1264–1273. [Google Scholar] [CrossRef]
- Arai, J.; Goto, K.; Tanoue, Y.; Ito, S.; Muroyama, R.; Matsubara, Y.; Nakagawa, R.; Kaise, Y.; Lim, L.A.; Yoshida, H.; et al. Enzymatic inhibition of MICA sheddase ADAM17 by lomofungin in hepatocellular carcinoma cells. Int. J. Cancer 2018, 143, 2575–2583. [Google Scholar] [CrossRef]
- Oliviero, B.; Mantovani, S.; Varchetta, S.; Mele, D.; Grossi, G.; Ludovisi, S.; Nuti, E.; Rossello, A.; Mondelli, M.U. Hepatitis C virus-induced NK cell activation causes metzincin-mediated CD16 cleavage and impaired antibody-dependent cytotoxicity. J. Hepatol. 2017, 66, 1130–1137. [Google Scholar] [CrossRef]
- Arai, J.; Goto, K.; Stephanou, A.; Tanoue, Y.; Ito, S.; Muroyama, R.; Matsubara, Y.; Nakagawa, R.; Morimoto, S.; Kaise, Y.; et al. Predominance of regorafenib over sorafenib: Restoration of membrane-bound MICA in hepatocellular carcinoma cells. J. Gastroenterol. Hepatol. 2018, 33, 1075–1081. [Google Scholar] [CrossRef]
- Goto, K.; Arai, J.; Stephanou, A.; Kato, N. Novel therapeutic features of disulfiram against hepatocellular carcinoma cells with inhibitory effects on a disintegrin and metalloproteinase 10. Oncotarget 2018, 9, 18821–18831. [Google Scholar] [CrossRef]
- Luo, Q.; Luo, W.; Zhu, Q.; Huang, H.; Peng, H.; Liu, R.; Xie, M.; Li, S.; Li, M.; Hu, X.; et al. Tumor-Derived Soluble MICA Obstructs the NKG2D Pathway to Restrain NK Cytotoxicity. Aging Dis. 2020, 11, 118–128. [Google Scholar] [CrossRef]
- Kumar, V.; Yi Lo, P.H.; Sawai, H.; Kato, N.; Takahashi, A.; Deng, Z.; Urabe, Y.; Mbarek, H.; Tokunaga, K.; Tanaka, Y.; et al. Soluble MICA and a MICA variation as possible prognostic biomarkers for HBV-induced hepatocellular carcinoma. PLoS ONE 2012, 7, e44743. [Google Scholar] [CrossRef]
- Duffy, M.J.; Mullooly, M.; O’Donovan, N.; Sukor, S.; Crown, J.; Pierce, A.; McGowan, P.M. The ADAMs family of proteases: New biomarkers and therapeutic targets for cancer? Clin Proteom. 2011, 8, 9. [Google Scholar] [CrossRef]
- Xiao, W.; Dong, W.; Zhang, C.; Saren, G.; Geng, P.; Zhao, H.; Li, Q.; Zhu, J.; Li, G.; Zhang, S.; et al. Effects of the epigenetic drug MS-275 on the release and function of exosome-related immune molecules in hepatocellular carcinoma cells. Eur. J. Med. Res. 2013, 18, 61. [Google Scholar] [CrossRef]
- Ashiru, O.; Boutet, P.; Fernández-Messina, L.; Agüera-González, S.; Skepper, J.N.; Valés-Gómez, M.; Reyburn, H.T. Natural killer cell cytotoxicity is suppressed by exposure to the human NKG2D ligand MICA*008 that is shed by tumor cells in exosomes. Cancer Res. 2010, 70, 481–489. [Google Scholar] [CrossRef]
- Fang, L.; Gong, J.; Wang, Y.; Liu, R.; Li, Z.; Wang, Z.; Zhang, Y.; Zhang, C.; Song, C.; Yang, A.; et al. MICA/B expression is inhibited by unfolded protein response and associated with poor prognosis in human hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2014, 33, 76. [Google Scholar] [CrossRef]
- Ma, Y.; Hendershot, L.M. The role of the unfolded protein response in tumour development: Friend or foe? Nat. Rev. Cancer 2004, 4, 966–977. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, J.H.; Lim, Y.S.; Yeon, J.E.; Song, T.J.; Yu, S.J.; Gwak, G.Y.; Kim, K.M.; Kim, Y.J.; Lee, J.W.; et al. Adjuvant immunotherapy with autologous cytokine-induced killer cells for hepatocellular carcinoma. Gastroenterology 2015, 148, 1383–1391.e6. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.A. The shared and contrasting roles of IL2 and IL15 in the life and death of normal and neoplastic lymphocytes: Implications for cancer therapy. Cancer Immunol. Res. 2015, 3, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Rautela, J.; Huntington, N.D. IL-15 signaling in NK cell cancer immunotherapy. Curr. Opin. Immunol. 2017, 44, 1–6. [Google Scholar] [CrossRef]
- Mortier, E.; Quéméner, A.; Vusio, P.; Lorenzen, I.; Boublik, Y.; Grötzinger, J.; Plet, A.; Jacques, Y. Soluble interleukin-15 receptor alpha (IL-15R alpha)-sushi as a selective and potent agonist of IL-15 action through IL-15R beta/gamma. Hyperagonist IL-15 x IL-15R alpha fusion proteins. J. Biol. Chem. 2006, 281, 1612–1619. [Google Scholar] [CrossRef]
- Bessard, A.; Solé, V.; Bouchaud, G.; Quéméner, A.; Jacques, Y. High antitumor activity of RLI, an interleukin-15 (IL-15)-IL-15 receptor alpha fusion protein, in metastatic melanoma and colorectal cancer. Mol. Cancer Ther. 2009, 8, 2736–2745. [Google Scholar] [CrossRef]
- Rosario, M.; Liu, B.; Kong, L.; Collins, L.I.; Schneider, S.E.; Chen, X.; Han, K.; Jeng, E.K.; Rhode, P.R.; Leong, J.W.; et al. The IL-15-Based ALT-803 Complex Enhances FcγRIIIa-Triggered NK Cell Responses and In Vivo Clearance of B Cell Lymphomas. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 596–608. [Google Scholar] [CrossRef] [PubMed]
- Mathios, D.; Park, C.K.; Marcus, W.D.; Alter, S.; Rhode, P.R.; Jeng, E.K.; Wong, H.C.; Pardoll, D.M.; Lim, M. Therapeutic administration of IL-15 superagonist complex ALT-803 leads to long-term survival and durable antitumor immune response in a murine glioblastoma model. Int. J. Cancer 2016, 138, 187–194. [Google Scholar] [CrossRef]
- Jiang, W.; Zhang, C.; Tian, Z.; Zhang, J. hIL-15 gene-modified human natural killer cells (NKL-IL15) augments the anti-human hepatocellular carcinoma effect in vivo. Immunobiology 2014, 219, 547–553. [Google Scholar] [CrossRef]
- Sadelain, M.; Rivière, I.; Riddell, S. Therapeutic T cell engineering. Nature 2017, 545, 423–431. [Google Scholar] [CrossRef]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef]
- Morgan, M.A.; Schambach, A. Chimeric Antigen Receptor T Cells: Extending Translation from Liquid to Solid Tumors. Hum. Gene Ther. 2018, 29, 1083–1097. [Google Scholar] [CrossRef]
- Knochelmann, H.M.; Smith, A.S.; Dwyer, C.J.; Wyatt, M.M.; Mehrotra, S.; Paulos, C.M. CAR T Cells in Solid Tumors: Blueprints for Building Effective Therapies. Front. Immunol. 2018, 9, 1740. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Rose, S. First-Ever CAR T-cell Therapy Approved in U.S. Cancer Discov. 2017, 7, OF1. [Google Scholar] [CrossRef]
- FDA Approves Second CAR T-cell Therapy. Cancer Discov. 2018, 8, 5–6. [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Santomasso, B.D.; Park, J.H.; Salloum, D.; Riviere, I.; Flynn, J.; Mead, E.; Halton, E.; Wang, X.; Senechal, B.; Purdon, T.; et al. Clinical and Biological Correlates of Neurotoxicity Associated with CAR T-cell Therapy in Patients with B-cell Acute Lymphoblastic Leukemia. Cancer Discov. 2018, 8, 958–971. [Google Scholar] [CrossRef]
- Mehta, R.S.; Rezvani, K. Chimeric Antigen Receptor Expressing Natural Killer Cells for the Immunotherapy of Cancer. Front. Immunol. 2018, 9, 283. [Google Scholar] [CrossRef]
- Zhang, C.; Oberoi, P.; Oelsner, S.; Waldmann, A.; Lindner, A.; Tonn, T.; Wels, W.S. Chimeric Antigen Receptor-Engineered NK-92 Cells: An Off-the-Shelf Cellular Therapeutic for Targeted Elimination of Cancer Cells and Induction of Protective Antitumor Immunity. Front. Immunol. 2017, 8, 533. [Google Scholar] [CrossRef]
- Raulet, D.H.; Marcus, A.; Coscoy, L. Dysregulated cellular functions and cell stress pathways provide critical cues for activating and targeting natural killer cells to transformed and infected cells. Immunol. Rev. 2017, 280, 93–101. [Google Scholar] [CrossRef]
- Barrow, A.D.; Martin, C.J.; Colonna, M. The Natural Cytotoxicity Receptors in Health and Disease. Front. Immunol. 2019, 10, 909. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Erbe, A.K.; Hank, J.A.; Morris, Z.S.; Sondel, P.M. NK Cell-Mediated Antibody-Dependent Cellular Cytotoxicity in Cancer Immunotherapy. Front. Immunol. 2015, 6, 368. [Google Scholar] [CrossRef]
- Chang, Y.H.; Connolly, J.; Mimura, K.; Kono, K.; Campana, D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. 2013, 73, 1777–1786. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Wang, X.; Zhang, H.; Zhi, L.; Lv, T.; Li, M.; Lu, C.; Zhu, W. Structure-based rational design of a novel chimeric PD1-NKG2D receptor for natural killer cells. Mol. Immunol. 2019, 114, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Barber, A.; Rynda, A.; Sentman, C.L. Chimeric NKG2D expressing T cells eliminate immunosuppression and activate immunity within the ovarian tumor microenvironment. J. Immunol. 2009, 183, 6939–6947. [Google Scholar] [CrossRef]
- Babic, M.; Romagnani, C. The Role of Natural Killer Group 2, Member D in Chronic Inflammation and Autoimmunity. Front. Immunol. 2018, 9, 1219. [Google Scholar] [CrossRef]
- Meehan, K.R.; Talebian, L.; Tosteson, T.D.; Hill, J.M.; Szczepiorkowski, Z.; Sentman, C.L.; Ernstoff, M.S. Adoptive cellular therapy using cells enriched for NKG2D+CD3+CD8+T cells after autologous transplantation for myeloma. Biol. Blood Marrow Transplant. J. Am. Soc. Blood Marrow Transplant. 2013, 19, 129–137. [Google Scholar] [CrossRef]
- Abe, Y.; Muto, M.; Nieda, M.; Nakagawa, Y.; Nicol, A.; Kaneko, T.; Goto, S.; Yokokawa, K.; Suzuki, K. Clinical and immunological evaluation of zoledronate-activated Vgamma9gammadelta T-cell-based immunotherapy for patients with multiple myeloma. Exp. Hematol. 2009, 37, 956–968. [Google Scholar] [CrossRef]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; Defor, T.E.; Burns, L.J.; et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef]
- Nakajima, J.; Murakawa, T.; Fukami, T.; Goto, S.; Kaneko, T.; Yoshida, Y.; Takamoto, S.; Kakimi, K. A phase I study of adoptive immunotherapy for recurrent non-small-cell lung cancer patients with autologous gammadelta T cells. Eur. J. Cardio-Thorac. Surg. Off. J. Eur. Assoc. Cardio-Thorac. Surg. 2010, 37, 1191–1197. [Google Scholar] [CrossRef]
- Vivier, E.; Nunès, J.A.; Vély, F. Natural killer cell signaling pathways. Science 2004, 306, 1517–1519. [Google Scholar] [CrossRef] [PubMed]
- Hatjiharissi, E.; Xu, L.; Santos, D.D.; Hunter, Z.R.; Ciccarelli, B.T.; Verselis, S.; Modica, M.; Cao, Y.; Manning, R.J.; Leleu, X.; et al. Increased natural killer cell expression of CD16, augmented binding and ADCC activity to rituximab among individuals expressing the Fc{gamma}RIIIa-158 V/V and V/F polymorphism. Blood 2007, 110, 2561–2564. [Google Scholar] [CrossRef] [PubMed]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef]
- Shields, R.L.; Lai, J.; Keck, R.; O’Connell, L.Y.; Hong, K.; Meng, Y.G.; Weikert, S.H.; Presta, L.G. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J. Biol. Chem. 2002, 277, 26733–26740. [Google Scholar] [CrossRef] [PubMed]
- Shinkawa, T.; Nakamura, K.; Yamane, N.; Shoji-Hosaka, E.; Kanda, Y.; Sakurada, M.; Uchida, K.; Anazawa, H.; Satoh, M.; Yamasaki, M.; et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J. Biol. Chem. 2003, 278, 3466–3473. [Google Scholar] [CrossRef] [PubMed]
- Cartron, G.; Watier, H. Obinutuzumab: What is there to learn from clinical trials? Blood 2017, 130, 581–589. [Google Scholar] [CrossRef]
- Navid, F.; Santana, V.M.; Barfield, R.C. Anti-GD2 antibody therapy for GD2-expressing tumors. Curr. Cancer Drug Targets 2010, 10, 200–209. [Google Scholar] [CrossRef]
- Arienti, C.; Pignatta, S.; Tesei, A. Epidermal Growth Factor Receptor Family and its Role in Gastric Cancer. Front. Oncol. 2019, 9, 1308. [Google Scholar] [CrossRef]
- Rimawi, M.F.; Schiff, R.; Osborne, C.K. Targeting HER2 for the treatment of breast cancer. Annu. Rev. Med. 2015, 66, 111–128. [Google Scholar] [CrossRef]
- Messersmith, W.A.; Ahnen, D.J. Targeting EGFR in colorectal cancer. N. Engl. J. Med. 2008, 359, 1834–1836. [Google Scholar] [CrossRef]
- Nakano, K.; Orita, T.; Nezu, J.; Yoshino, T.; Ohizumi, I.; Sugimoto, M.; Furugaki, K.; Kinoshita, Y.; Ishiguro, T.; Hamakubo, T.; et al. Anti-glypican 3 antibodies cause ADCC against human hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2009, 378, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Wang, H.; Tan, Z.; Hu, S.; Wang, H.; Shi, B.; Yang, L.; Li, P.; Gu, J.; Wang, H.; et al. Growth suppression of human hepatocellular carcinoma xenografts by a monoclonal antibody CH12 directed to epidermal growth factor receptor variant III. J. Biol. Chem. 2011, 286, 5913–5920. [Google Scholar] [CrossRef] [PubMed]
- Duffy, A.G.; Ma, C.; Ulahannan, S.V.; Rahma, O.E.; Makarova-Rusher, O.; Cao, L.; Yu, Y.; Kleiner, D.E.; Trepel, J.; Lee, M.J.; et al. Phase I and Preliminary Phase II Study of TRC105 in Combination with Sorafenib in Hepatocellular Carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 4633–4641. [Google Scholar] [CrossRef] [PubMed]
- Ferrari de Andrade, L.; Tay, R.E.; Pan, D.; Luoma, A.M.; Ito, Y.; Badrinath, S.; Tsoucas, D.; Franz, B.; May, K.F., Jr.; Harvey, C.J.; et al. Antibody-mediated inhibition of MICA and MICB shedding promotes NK cell-driven tumor immunity. Science 2018, 359, 1537–1542. [Google Scholar] [CrossRef]
- Morisaki, T.; Hirano, T.; Koya, N.; Kiyota, A.; Tanaka, H.; Umebayashi, M.; Onishi, H.; Katano, M. NKG2D-directed cytokine-activated killer lymphocyte therapy combined with gemcitabine for patients with chemoresistant metastatic solid tumors. Anticancer Res. 2014, 34, 4529–4538. [Google Scholar]
- Vallera, D.A.; Felices, M.; McElmurry, R.; McCullar, V.; Zhou, X.; Schmohl, J.U.; Zhang, B.; Lenvik, A.J.; Panoskaltsis-Mortari, A.; Verneris, M.R.; et al. IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing Persistence, In Vivo Expansion, and Enhanced Function. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 3440–3450. [Google Scholar] [CrossRef]
- Davis, Z.B.; Vallera, D.A.; Miller, J.S.; Felices, M. Natural killer cells unleashed: Checkpoint receptor blockade and BiKE/TriKE utilization in NK-mediated anti-tumor immunotherapy. Semin. Immunol. 2017, 31, 64–75. [Google Scholar] [CrossRef]
- Chan, W.K.; Kang, S.; Youssef, Y.; Glankler, E.N.; Barrett, E.R.; Carter, A.M.; Ahmed, E.H.; Prasad, A.; Chen, L.; Zhang, J.; et al. A CS1-NKG2D Bispecific Antibody Collectively Activates Cytolytic Immune Cells against Multiple Myeloma. Cancer Immunol. Res. 2018, 6, 776–787. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| ID | Study Phase | Clinical Trial | Interventions | Conditions | Status |
|---|---|---|---|---|---|
| NCT02854839 | 2 | A Study of MG4101 (Allogeneic Natural Killer Cell) for Intermediate-stage of Hepatocellular Carcinoma | Biological: MG4101 | HCC | C |
| NCT04162158 | 1 2 | Safety and Efficacy of Allogeneic NK Cells Therapy in Patients With Advanced Hepatocellular Carcinoma | Biological: allogeneic NK cells therapy | HCC | R |
| NCT01147380 | 1 | Safety Study of Liver Natural Killer Cell Therapy for Hepatoma Liver Transplantation (MIAMINK) | Biological: Liver NK cell inoculation | Liver Cirrhosis HCCE vidence of Liver Transplantation | C |
| NCT02008929 | 2 | To Evaluate the Efficacy and Safety of MG4101(Ex Vivo Expanded Allogeneic NK Cell) (MG4101) | Biological: MG4101 | HCC | C |
| NCT00769106 | 3 | Study of Cytokine-induced Killer Cell (CIK) Treatment in Patients After Resection of Liver Cancer (HCC-CIK) | Biological: CIK treatment | HCC | C |
| NCT03515252 | 1 | Study of Autologous Immune Killer Cells in Patients With Late Stage Hepatocellular Carcinoma or Lung Cancer | Biological: IKC | NSCLC Stage IIIB NSCLC Stage IV HCC by BCLC Stage Lung Cancer Liver Cancer | C |
| NCT03592706 | 2 3 | Autologous Immune Killer Cells to Treat Liver Cancer Patients as an Adjunct Therapy | Biological: IKC Procedure: TACE | HCC Liver Cancer | R |
| NCT01749865 | 3 | CIK Treatment for HCC Patient Underwent Radical Resection | Biological: CIK | HCC | C |
| NCT04106167 | Obser | Long-term, Non-interventional, Observational Study Following Treatment With Fate Therapeutics FT500 Cellular Immunotherapy | Genetic: Allogeneic NK cell No study drug is administered in this study. Subjects who received an allogeneic, iPSC-derived NK cell in a previous trial will be evaluated in this trial for long-term safety and efficacy | Advanced Solid Tumor Lymphoma Gastric Cancer Colorectal Cancer Head and Neck Cancer Squamous Cell Carcinoma EGFR Positive Solid Tumor HER2-positive Breast Cancer HCC SCLC Renal Cell Carcinoma Pancreas Cancer Melanoma NSCLC Urothelial Carcinoma Cervical Cancer Microsatellite Instability Merkel Cell Carcinoma | R |
| NCT03008343 | 1 2 | Combination of Irreversible Electroporation and NK Immunotherapy for Recurrent Liver Cancer | Device: Irreversible Electroporation Biological: NK | Recurrent Liver Carcinoma | C |
| NCT03841110 | 1 | FT500 as Monotherapy and in Combination With Immune Checkpoint Inhibitors in Subjects With Advanced Solid Tumors | Drug: FT500 Drug: Nivolumab Drug: Pembrolizumab Drug: Atezolizumab Drug: Cyclophosphamide Drug: Fludarabine | Advanced Solid Tumor Lymphoma Gastric Cancer Colorectal Cancer Head and Neck Cancer Squamous Cell Carcinoma EGFR Positive Solid Tumor HER2-positive Breast Cancer HCC SCLC Renal Cell Carcinoma Pancreas Cancer Melanoma NSCLC Urothelial Carcinoma Cervical Cancer Microsatellite Instability Merkel Cell Carcinoma | R |
| NCT03319459 | 1 | FATE-NK100 as Monotherapy and in Combination With Monoclonal Antibody in Subjects With Advanced Solid Tumors | FATE-NK100 as Monotherapy and in Combination With Monoclonal Antibody in Subjects With Advanced Solid Tumors | HER2 Positive Gastric Cancer Colorectal Cancer Head and Neck Squamous Cell Carcinoma EGFR Positive Solid Tumor Advanced Solid Tumors HER2-positive Breast Cancer HCC NSCLC Renal Cell Carcinoma Pancreatic Cancer Melanoma | A |
| NCT00909558 | 1 | Safety and Effectiveness Study of Autologous Natural Killer and Natural Killer T Cells on Cancer | Biological: Autologous NK/NKT Cell Immunotherapy | Breast Cancer Glioma HCC Squamous Cell Lung Cancer Pancreatic Cancer Colon Cancer Prostate Cancer | S |
| NCT02725996 | 2 | By Using Adoptive Transfer of Autologous NK Cells to Prevent Recurrence of Hepatocellular Carcinoma After Curative Therapy | Biological: NK cells Other: Curative therapy | HCC | U |
| NCT02399735 | 1 | Safety Study of NK Cells From Sibship to Treat the Recurrence of HCC After Liver Transplantation | Biological: Low Dose NK cells ×4 times Biological: Normal Dose NK cells ×4 times Biological: Normal Dose NK cells ×8 times | HCC | U |
| NCT02839954 | 1 2 | CAR-pNK Cell Immunotherapy in MUC1 Positive Relapsed or Refractory Solid Tumor | Biological: anti-MUC1 CAR-pNK cells | HCC NSCLC Pancreatic Carcinoma Triple-Negative Invasive Breast Carcinoma Malignant Glioma of Brain Colorectal Carcinoma Gastric Carcinoma | U |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mantovani, S.; Oliviero, B.; Varchetta, S.; Mele, D.; Mondelli, M.U. Natural Killer Cell Responses in Hepatocellular Carcinoma: Implications for Novel Immunotherapeutic Approaches. Cancers 2020, 12, 926. https://doi.org/10.3390/cancers12040926
Mantovani S, Oliviero B, Varchetta S, Mele D, Mondelli MU. Natural Killer Cell Responses in Hepatocellular Carcinoma: Implications for Novel Immunotherapeutic Approaches. Cancers. 2020; 12(4):926. https://doi.org/10.3390/cancers12040926
Chicago/Turabian StyleMantovani, Stefania, Barbara Oliviero, Stefania Varchetta, Dalila Mele, and Mario U. Mondelli. 2020. "Natural Killer Cell Responses in Hepatocellular Carcinoma: Implications for Novel Immunotherapeutic Approaches" Cancers 12, no. 4: 926. https://doi.org/10.3390/cancers12040926
APA StyleMantovani, S., Oliviero, B., Varchetta, S., Mele, D., & Mondelli, M. U. (2020). Natural Killer Cell Responses in Hepatocellular Carcinoma: Implications for Novel Immunotherapeutic Approaches. Cancers, 12(4), 926. https://doi.org/10.3390/cancers12040926