Thioholgamide A, a New Anti-Proliferative Anti-Tumor Agent, Modulates Macrophage Polarization and Metabolism

 , , ,
, , ,  , , and
, , and
Abstract
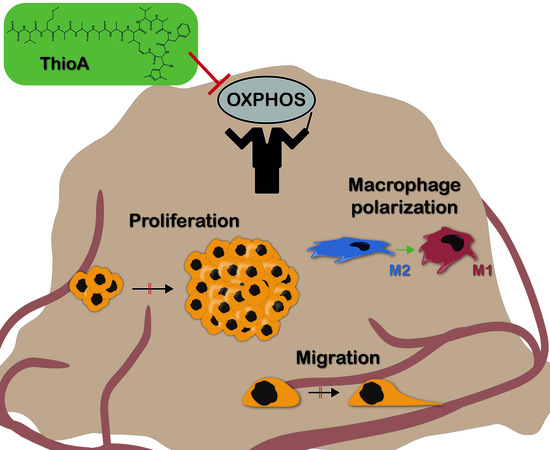
1. Introduction
2. Results
2.1. Thioholgamide A Impairs Tumor Cell Viability and Proliferation
2.2. Thioholgamide A Inhibits Oxidative Phosphorylation and Affects Mitochondrial Mass and Morphology
2.3. Thioholgamide A Inhibits Tumor Cell Proliferation In Vitro, in Tumor Spheroids, and In Vivo
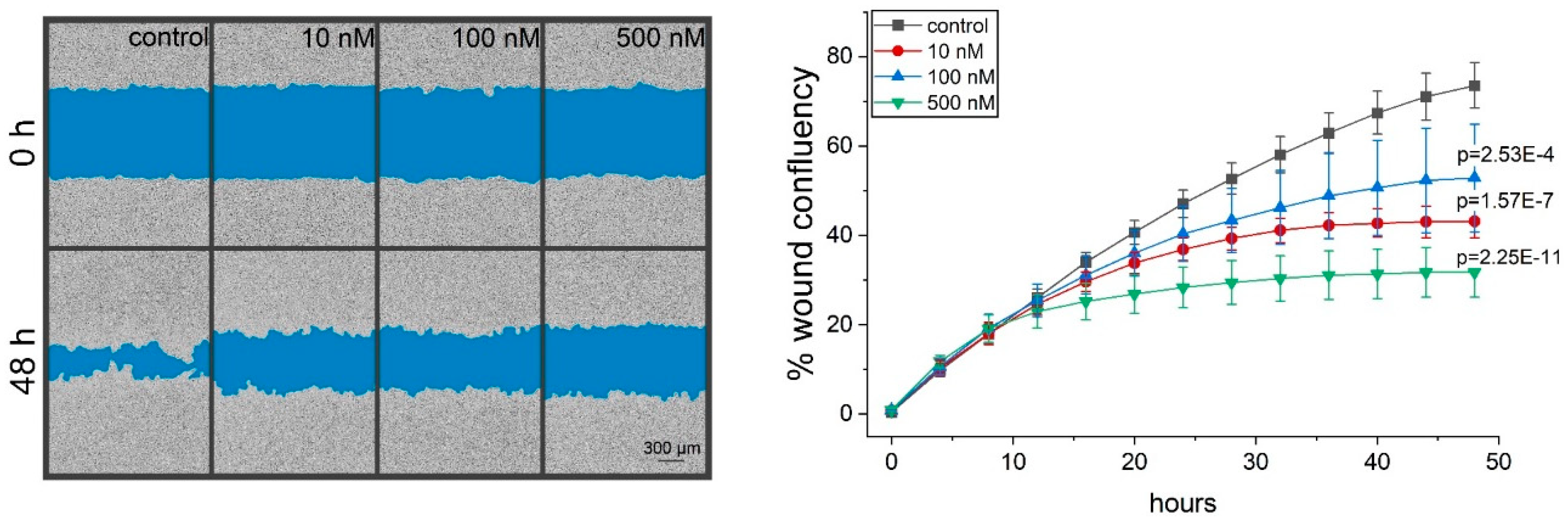
2.4. Thioholgamide A Inhibits Tumor Cell Migration
2.5. Thioholgamide A Inhibits OXPHOS-Dependent Atp Production in Macrophages
2.6. Thioholgamide A Alters the Macrophage Phenotype
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cultivation of Human Monocyte-Derived Macrophages
4.3. Thioholgamide A Isolation and Purification
4.4. Endotoxin Assay
4.5. Viability Measurements
4.5.1. MTT Assay
4.5.2. APH Assay
4.5.3. Annexin V/PI Staining
4.5.4. Time-Dependent Cell Death Measurement
4.6. Western Blot
4.7. Mitochondrial Mass
4.8. Seahorse Measurement
4.9. Immunofluorescence
4.10. Rna Isolation, Reverse Transcription, and Quantitative Pcr
4.11. In Vitro Proliferation Measurement
4.12. Zebrafish Xenograft Model And In Vivo Proliferation Measurement
4.13. Migration Measurement
4.14. Macrophage Surface Marker Expression
4.15. Macrophage Morphology Analysis
4.16. Macrophage Phagocytosis Assay
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, M.; Bruner, S.D.; Ding, Y. Heterologous production of microbial ribosomally synthesized and post-translationally modified peptides. Front. Microbiol. 2018, 9, 1801. [Google Scholar] [CrossRef] [PubMed]
- Kjaerulff, L.; Sikandar, A.; Zaburannyi, N.; Adam, S.; Herrmann, J.; Koehnke, J.; Müller, R. Thioholgamides: Thioamide-containing cytotoxic RiPP natural products. ACS Chem. Biol. 2017, 12, 2837–2841. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting tumor microenvironment for cancer therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Ostuni, R.; Kratochvill, F.; Murray, P.J.; Natoli, G. Macrophages and cancer: From mechanisms to therapeutic implications. Trends Immunol. 2015, 36, 229–239. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting tumor-associated macrophages in cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef] [PubMed]
- Jänicke, R.U.; Sprengart, M.L.; Wati, M.R.; Porter, A.G. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J. Biol. Chem. 1998, 273, 9357–9360. [Google Scholar] [CrossRef] [PubMed]
- Takase, S.; Kurokawa, R.; Kondoh, Y.; Honda, K.; Suzuki, T.; Kawahara, T.; Ikeda, H.; Dohmae, N.; Osada, H.; Shin-ya, K.; et al. Mechanism of action of prethioviridamide, an anticancer ribosomally synthesized and post-translationally modified peptide with a polythioamide structure. ACS Chem. Biol. 2019, 14, 1819–1828. [Google Scholar] [CrossRef] [PubMed]
- Plitzko, B.; Kaweesa, E.N.; Loesgen, X.S. The natural product mensacarcin induces mitochondrial toxicity and apoptosis in melanoma cells. J. Biol. Chem. 2017, 292, 21102–21116. [Google Scholar] [CrossRef]
- Kirchberger, S.; Sturtzel, C.; Pascoal, S.; Distel, M. Quo natas, Danio?—Recent progress in modeling cancer in zebrafish. Front. Oncol. 2017, 7. [Google Scholar] [CrossRef]
- El-Shamy, A.; Eng, F.J.; Doyle, E.H.; Klepper, A.L.; Sun, X.; Sangiovanni, A.; Iavarone, M.; Colombo, M.; Schwartz, R.E.; Hoshida, Y.; et al. A cell culture system for distinguishing hepatitis C viruses with and without liver cancer-related mutations in the viral core gene. J. Hepatol. 2015, 63, 1323–1333. [Google Scholar] [CrossRef]
- Divakaruni, A.S.; Paradyse, A.; Ferrick, D.A.; Murphy, A.N.; Jastroch, M. Analysis and Interpretation of Microplate-Based Oxygen Consumption and pH Data, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2014; Volume 547, ISBN 9780128014158. [Google Scholar]
- Hayakawa, Y.; Sasaki, K.; Adachi, H.; Furihata, K.; Nagai, K.; Shin-ya, K. Thioviridamide, a novel apoptosis inducer in transformed cells from streptomyces olivoviridis. J. Antibiot. (Tokyo). 2006, 59, 1–5. [Google Scholar] [CrossRef]
- Frattaruolo, L.; Fiorillo, M.; Brindisi, M.; Curcio, R.; Dolce, V.; Lacret, R.; Truman, A.; Sotgia, F.; Lisanti, M.P.; Cappello, A.R. Thioalbamide, a thioamidated peptide from amycolatopsis alba, affects tumor growth and stemness by inducing metabolic dysfunction and oxidative stress. Cells 2019, 8, 1408. [Google Scholar] [CrossRef]
- Yan, C.; Brunson, D.C.; Tang, Q.; Do, D.; Iftimia, N.A.; Moore, J.C.; Hayes, M.N.; Welker, A.M.; Garcia, E.G.; Dubash, T.D.; et al. Visualizing engrafted human cancer and therapy responses in immunodeficient zebrafish. Cell 2019, 177, 1903–1914.e14. [Google Scholar] [CrossRef]
- Veinotte, C.J.; Dellaire, G.; Berman, J.N. Hooking the big one: The potential of zebrafish xenotransplantation to reform cancer drug screening in the genomic era. DMM Dis. Model. Mech. 2014, 7, 745–754. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.L.; Li, F.; Yeo, J.Z.; Yong, K.J.; Bassal, M.A.; Ng, G.H.; Lee, M.Y.; Leong, C.Y.; Tan, H.K.; Wu, C.; et al. New high-throughput screening identifies compounds that reduce viability specifically in liver cancer cells that express high levels of SALL4 by inhibiting oxidative phosphorylation. Gastroenterology 2019, 157, 1615–1629.e17. [Google Scholar] [CrossRef] [PubMed]
- Matassa, D.S.; Amoroso, M.R.; Lu, H.; Avolio, R.; Arzeni, D.; Procaccini, C.; Faicchia, D.; Maddalena, F.; Simeon, V.; Agliarulo, I.; et al. Oxidative metabolism drives inflammation-induced platinum resistance in human ovarian cancer. Cell Death Differ. 2016, 23, 1542–1554. [Google Scholar] [CrossRef]
- Ashton, T.M.; Gillies McKenna, W.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative phosphorylation as an emerging target in cancer therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef]
- Pakos-zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Carter, N.J.; Keam, S.J. Trabectedin: A review of its use in the management of soft tissue sarcoma and ovarian cancer. Drugs 2007, 67, 2257–2276. [Google Scholar] [CrossRef]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef]
- Müller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017, 18, 234. [Google Scholar] [CrossRef]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell 2018, 174, 1293–1308.e36. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Manic, G.; Coussens, L.M.; Kroemer, G.; Galluzzi, L. Macrophages and metabolism in the tumor microenvironment. Cell Metab. 2019, 30, 36–50. [Google Scholar] [CrossRef]
- Van den Bossche, J.; O’Neill, L.A.; Menon, D. Macrophage immunometabolism: Where are we (Going)? Trends Immunol. 2017, 38, 395–406. [Google Scholar] [CrossRef]
- Zeni, E.; Mazzetti, L.; Miotto, D.; Lo Cascio, N.; Maestrelli, P.; Querzoli, P.; Pedriali, M.; De Rosa, E.; Fabbri, L.M.; Mapp, C.E.; et al. Macrophage expression of interleukin-10 is a prognostic factor in nonsmall cell lung cancer. Eur. Respir. J. 2007, 30, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Wu, D.; Wu, P.; Wang, Z.; Huang, J.; Gao, J.X. Serum IL-10 predicts worse outcome in cancer patients: A meta-analysis. PLoS ONE 2015, 10, e0139598. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Huang, H.; Wang, Z.; Zhang, G. The inflammatory CXC chemokines, GROαhigh, IP-10low, and MIGlow, in tumor microenvironment can be used as new indicators for non-small cell lung cancer progression. Immunol. Invest. 2017, 46, 361–374. [Google Scholar] [CrossRef]
- Yang, C.; Wei, C.; Wang, S.; Shi, D.; Zhang, C.; Lin, X.; Dou, R.; Xiong, B. Elevated CD163+/CD68+ ratio at tumor invasive front is closely associated with aggressive phenotype and poor prognosis in colorectal cancer. Int. J. Biol. Sci. 2019, 15, 984–998. [Google Scholar] [CrossRef]
- Ye, L.; Zhang, T.; Kang, Z.; Guo, G.; Sun, Y.; Lin, K.; Huang, Q.; Shi, X.; Ni, Z.; Ding, N.; et al. Tumor-infiltrating immune cells act as a marker for prognosis in colorectal cancer. Front. Immunol. 2019, 10, 2368. [Google Scholar] [CrossRef]
- Chen, W.; Sandoval, H.; Kubiak, J.Z.; Li, X.C.; Ghobrial, R.M.; Kloc, M. The phenotype of peritoneal mouse macrophages depends on the mitochondria and ATP/ADP homeostasis. Cell. Immunol. 2018, 324, 1–7. [Google Scholar] [CrossRef]
- Weigert, A.; Mora, J.; Sekar, D.; Syed, S.; Brüne, B. Killing is not enough: How apoptosis hijacks tumor-associated macrophages to promote cancer progression. In Apoptosis in Cancer Pathogenesis and Anti-Cancer Therapy; Gregory, C.D., Ed.; Springer International Publishing: Cham, Switzerland, 2016; Volume 930, pp. 205–239, ISBN 978-3-319-39404-6, 978-3-319-39406-0. [Google Scholar]
- Dehne, N.; Mora, J.; Namgaladze, D.; Weigert, A.; Brüne, B. Cancer cell and macrophage cross-talk in the tumor microenvironment. Curr. Opin. Pharmacol. 2017, 35, 12–19. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Schaper, F.; de Leeuw, K.; Horst, G.; Bootsma, H.; Limburg, P.C.; Heeringa, P.; Bijl, M.; Westra, J. High mobility group box 1 skews macrophage polarization and negatively influences phagocytosis of apoptotic cells. Rheumatology 2016, 55, 2260–2270. [Google Scholar] [CrossRef] [PubMed]
- Rey-Giraud, F.; Hafner, M.; Ries, C.H. In vitro generation of monocyte-derived macrophages under serum-free conditions improves their tumor promoting functions. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Roos, A.; Schlagwein, N.; Woltman, A.M.; Daha, M.R.; Van Kooten, C. IL-10–producing macrophages preferentially clear early apoptotic cells. Blood 2006, 107, 4930–4937. [Google Scholar] [CrossRef]
- Krysko, O.; Holtappels, G.; Zhang, N.; Kubica, M.; Deswarte, K.; Derycke, L.; Claeys, S.; Hammad, H.; Brusselle, G.G.; Vandenabeele, P.; et al. Alternatively activated macrophages and impaired phagocytosis of S. aureus in chronic rhinosinusitis. Allergy Eur. J. Allergy Clin. Immunol. 2011, 66, 396–403. [Google Scholar] [CrossRef]
- Varin, A.; Mukhopadhyay, S.; Herbein, G.; Gordon, S. Alternative activation of macrophages by IL-4 impairs phagocytosis of pathogens but potentiates microbial-induced signalling and cytokine secretion. Blood 2010, 115, 353–362. [Google Scholar] [CrossRef]
- Li, Y.; Jia, A.; Wang, Y.; Dong, L.; Wang, Y.; He, Y.; Wang, S.; Cao, Y.; Yang, H.; Bi, Y.; et al. Immune effects of glycolysis or oxidative phosphorylation metabolic pathway in protecting against bacterial infection. J. Cell. Physiol. 2019, 234, 20298–20309. [Google Scholar] [CrossRef]
- Astanina, K.; Koch, M.; Jüngst, C.; Zumbusch, A.; Kiemer, A.K. Lipid droplets as a novel cargo of tunnelling nanotubes in endothelial cells. Sci. Rep. 2015, 5, 11453. [Google Scholar] [CrossRef]
- Ahmed, Y.; Rebets, Y.; Estévez, M.R.; Zapp, J.; Myronovskyi, M.; Luzhetskyy, A. Engineering of Streptomyces lividans for heterologous expression of secondary metabolite gene clusters. Microb. Cell Fact. 2020, 19, 5. [Google Scholar] [CrossRef]
- Gürtler, A.; Kunz, N.; Gomolka, M.; Hornhardt, S.; Friedl, A.A.; McDonald, K.; Kohn, J.E.; Posch, A. Stain-Free technology as a normalization tool in Western blot analysis. Anal. Biochem. 2013, 433, 105–111. [Google Scholar] [CrossRef]
- Chopra, A.; Willmore, W.G.; Biggar, K.K. Protein quantification and visualization via ultraviolet-dependent labeling with 2,2,2-trichloroethanol. Sci. Rep. 2019, 9, 13923. [Google Scholar] [CrossRef]
- Westerfield, M. The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Danio Rerio), 4th ed.; University of Oregon Press: Eugene, OR, USA, 2000. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | IC50 |
|---|---|
| HCT116 | 176 nM |
| Huh7 | 141 nM |
| MCF7 | 480 nM |
| A549 | 1.16 μM |
| RIL175 | 157 nM |
| Cell Line | Caspase 3/7 Activity | Membrane Permeability | Proliferation |
|---|---|---|---|
| HCT116 | 412.7 ± 27.5 | 840.8 ± 163.6 | 90.8 ± 5.5 |
| Huh7 | 197.2 ± 44.8 | 578.8 ± 55.8 | 52.1 ± 5.7 |
| MCF7 | n.d. | n.d. | 489.9 ± 9.3 |
| Gene | Accession Number | Primer Forward Sequence | Primer Forward Sequence |
|---|---|---|---|
| IL10 | NM_000572 | CAACAGAAGCTTCCATTCCA | AGCAGTTAGGAAGCCCCAAG |
| MMP9 | NM_004994.3 | CTTTGAGTCCGGTGGACGAT | TCGCCAGTACTTCCCATCCT |
| TNF | NM_000594.4 | CTCCACCCATGTGCTCCTCA | CTCTGGCAGGGGCTCTTGAT |
| IP10 | NM_001565.4 | GAGCCTACAGCAGAGGAACC | AAGGCAGCAAATCAGAATCG |
| RNA18S | NR_003286.4 | AGGTCTGTGATGCCCTTAGA | GAATGGGGTTCAACGGGTTA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dahlem, C.; Siow, W.X.; Lopatniuk, M.; Tse, W.K.F.; Kessler, S.M.; Kirsch, S.H.; Hoppstädter, J.; Vollmar, A.M.; Müller, R.; Luzhetskyy, A.; et al. Thioholgamide A, a New Anti-Proliferative Anti-Tumor Agent, Modulates Macrophage Polarization and Metabolism. Cancers 2020, 12, 1288. https://doi.org/10.3390/cancers12051288
Dahlem C, Siow WX, Lopatniuk M, Tse WKF, Kessler SM, Kirsch SH, Hoppstädter J, Vollmar AM, Müller R, Luzhetskyy A, et al. Thioholgamide A, a New Anti-Proliferative Anti-Tumor Agent, Modulates Macrophage Polarization and Metabolism. Cancers. 2020; 12(5):1288. https://doi.org/10.3390/cancers12051288
Chicago/Turabian StyleDahlem, Charlotte, Wei Xiong Siow, Maria Lopatniuk, William K. F. Tse, Sonja M. Kessler, Susanne H. Kirsch, Jessica Hoppstädter, Angelika M. Vollmar, Rolf Müller, Andriy Luzhetskyy, and et al. 2020. "Thioholgamide A, a New Anti-Proliferative Anti-Tumor Agent, Modulates Macrophage Polarization and Metabolism" Cancers 12, no. 5: 1288. https://doi.org/10.3390/cancers12051288
APA StyleDahlem, C., Siow, W. X., Lopatniuk, M., Tse, W. K. F., Kessler, S. M., Kirsch, S. H., Hoppstädter, J., Vollmar, A. M., Müller, R., Luzhetskyy, A., Bartel, K., & Kiemer, A. K. (2020). Thioholgamide A, a New Anti-Proliferative Anti-Tumor Agent, Modulates Macrophage Polarization and Metabolism. Cancers, 12(5), 1288. https://doi.org/10.3390/cancers12051288