Cell Intrinsic and Systemic Metabolism in Tumor Immunity and Immunotherapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Microenvironmental Metabolic Complexity
3. Tumor Cells
4. Fibroblasts
5. Macrophages
6. Dendritic Cells
7. Lymphocytes
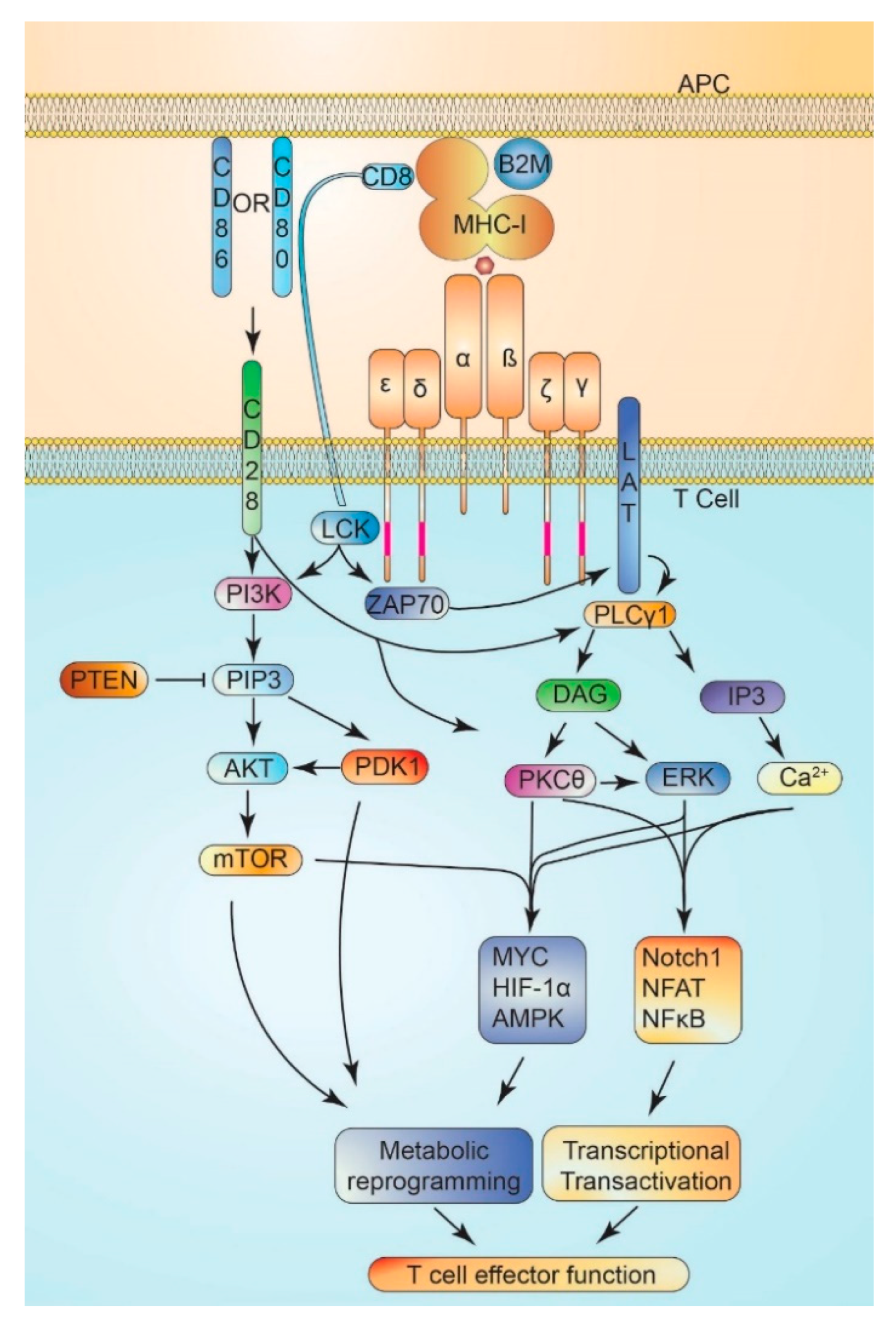
8. T Cell Activation and Metabolism
8.1. T Cell Activation
8.2. Costimulatory Signaling
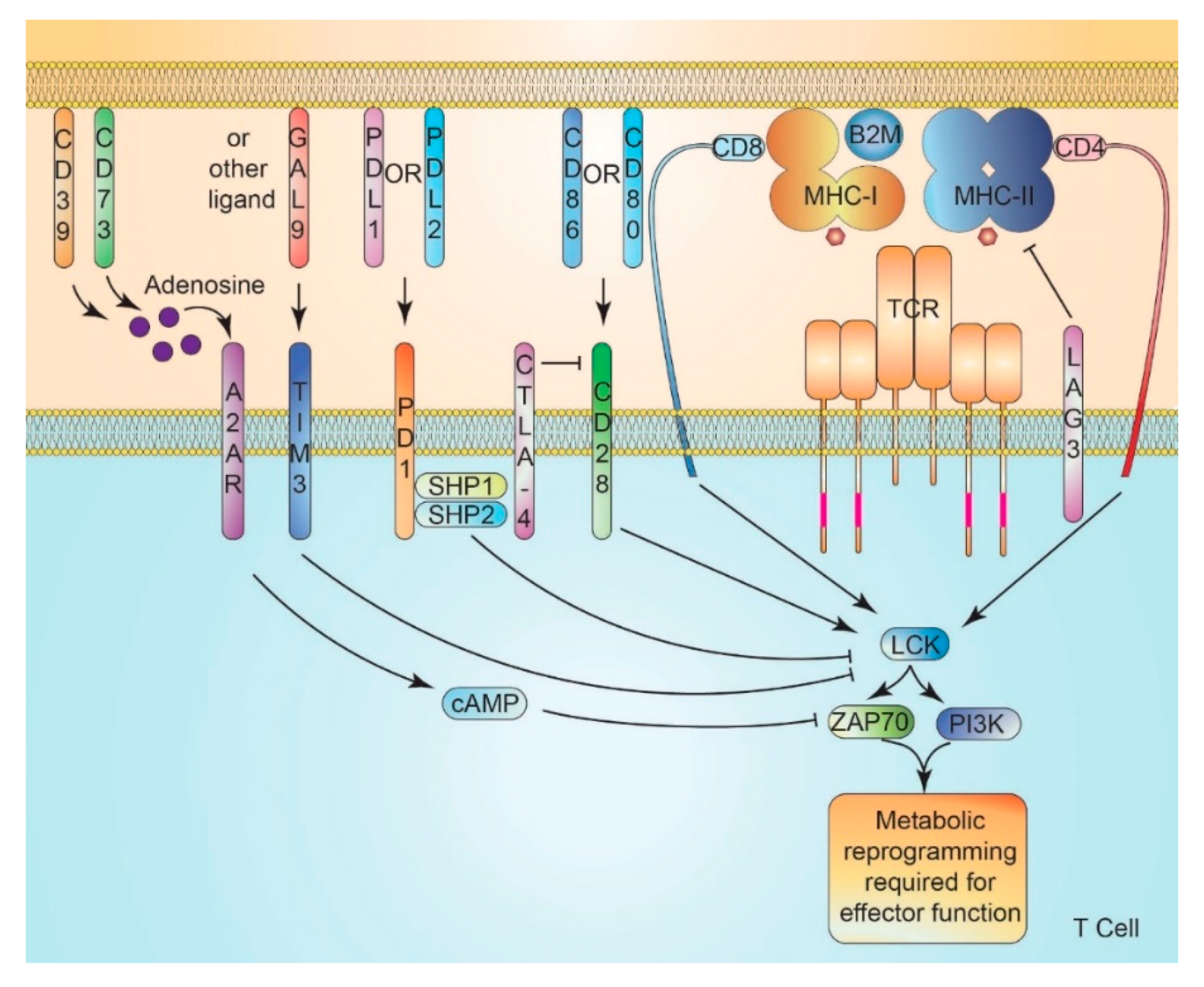
9. Immune Checkpoint Signaling
10. Metabolically-Mediated TME Immunomodulation
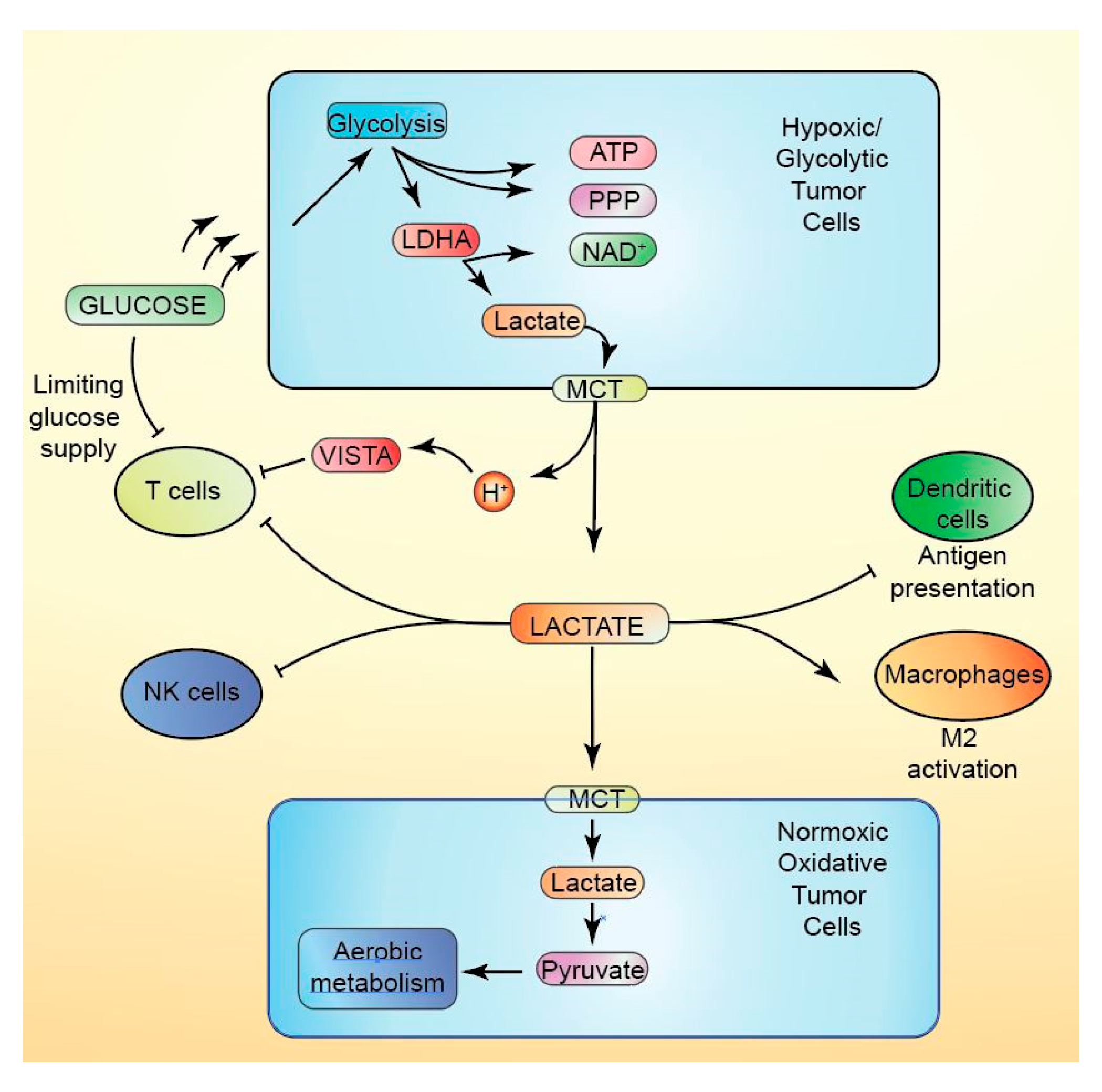
10.1. Lactate
10.2. Hypoxia
10.3. Arachidonate Metabolism
10.4. Short-Chain Fatty Acid Metabolism
11. Tumor Cell-Intrinsic Immunotherapy Resistance
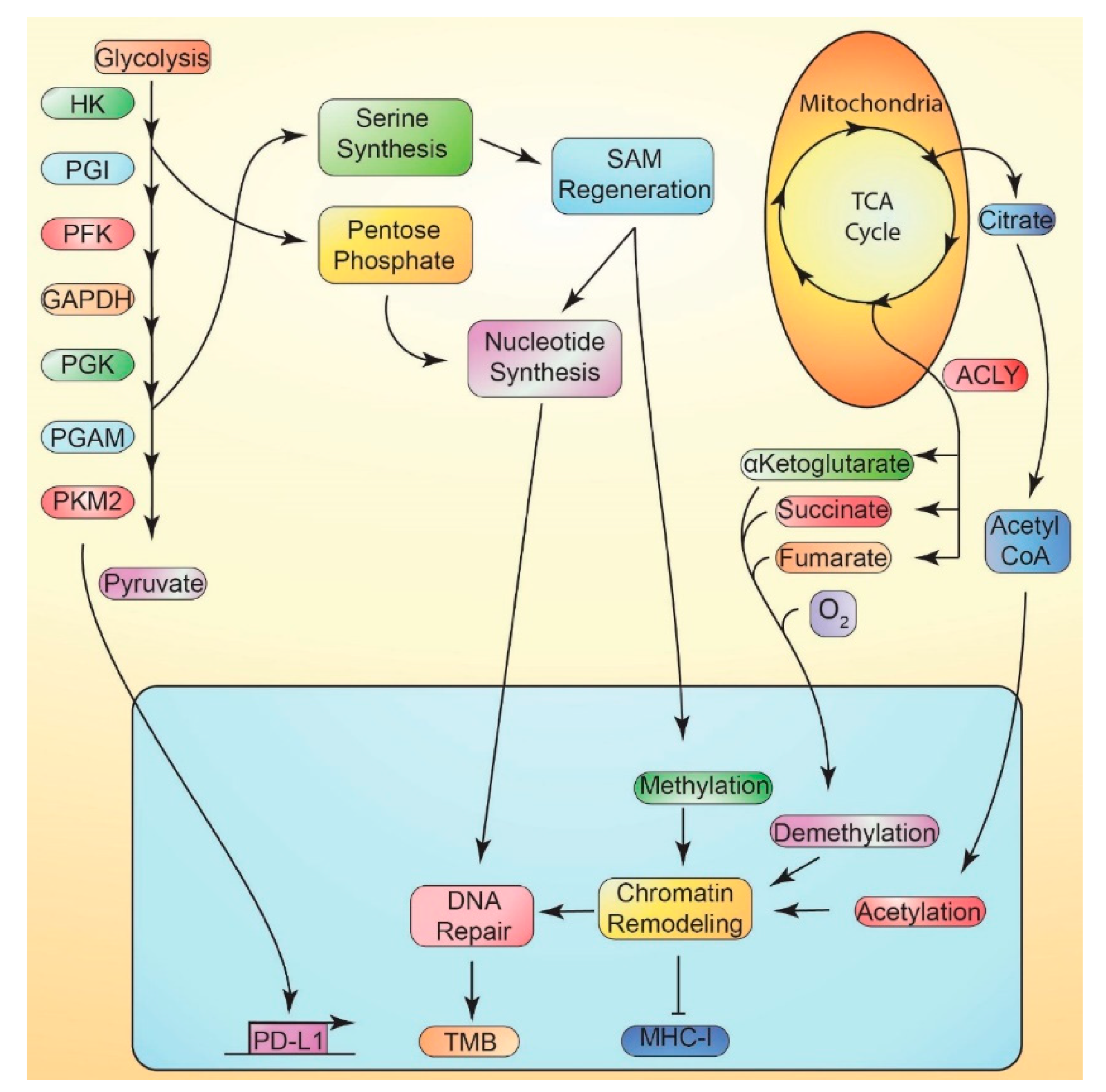
11.1. Metabolically-Mediated Immune Evasion
11.2. Suppression of MHC-I vs. Metabolism
11.3. Tumor Mutational Burden
12. Interactions between Systemic Metabolic Perturbations and Intrinsic Immunometabolism
12.1. Obesity
12.2. Caloric Deficit
13. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pianko, M.J.; Goldberg, A.D.; Lesokhin, A.M. Clinical Development of PD-1 Blockade in Hematologic Malignancies. Cancer J. (Sudbury, Mass.) 2018, 24, 31–35. [Google Scholar] [CrossRef]
- Thallinger, C.; Füreder, T.; Preusser, M.; Heller, G.; Müllauer, L.; Höller, C.; Prosch, H.; Frank, N.; Swierzewski, R.; Berger, W.; et al. Review of cancer treatment with immune checkpoint inhibitors. Wien. Klin. Wochenschr. 2018, 130, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Roy, A.; Dwarakanath, B.S. Metabolic Cooperation and Competition in the Tumor Microenvironment: Implications for Therapy. Front. Oncol. 2017, 7, e68. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.R.; Danai, L.V.; Lewis, C.A.; Chan, S.H.; Gui, D.Y.; Kunchok, T.; Dennstedt, E.A.; Vander Heiden, M.G.; Muir, A. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife 2019, 8, e44235. [Google Scholar] [CrossRef]
- Chang, C.H.; Qiu, J.; O'Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef]
- Brahimi-Horn, M.C.; Chiche, J.; Pouysségur, J. Hypoxia and cancer. J. Mol. Med. 2007, 85, 1301–1307. [Google Scholar] [CrossRef]
- Renner, K.; Singer, K.; Koehl, G.E.; Geissler, E.K.; Peter, K.; Siska, P.J.; Kreutz, M. Metabolic Hallmarks of Tumor and Immune Cells in the Tumor Microenvironment. Front. Immunol. 2017, 8, e248. [Google Scholar] [CrossRef]
- Buck, M.D.; O'Sullivan, D.; Pearce, E.L. T cell metabolism drives immunity. J. Exp. Med. 2015, 212, 1345–1360. [Google Scholar] [CrossRef]
- Le Bourgeois, T.; Strauss, L.; Aksoylar, H.-I.; Daneshmandi, S.; Seth, P.; Patsoukis, N.; Boussiotis, V.A. Targeting T Cell Metabolism for Improvement of Cancer Immunotherapy. Front. Oncol. 2018, 8, e237. [Google Scholar] [CrossRef]
- Whiteside, T. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904. [Google Scholar] [CrossRef]
- Li, H.; Fan, X.; Houghton, J. Tumor microenvironment: the role of the tumor stroma in cancer. J. Cell. Biochem. 2007, 101, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Lyssiotis, C.A.; Kimmelman, A.C. Metabolic interactions in the tumor microenvironment. Trends Cell Biol. 2017, 27, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.S.; Ostrowski, M.; Balderson, B.; Christian, N.; Crowe, S.M. Glucose metabolism regulates T cell activation, differentiation, and functions. Front. Immunol. 2015, 6, e1. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.L. Metabolism in T cell activation and differentiation. Curr. Opin. Immunol. 2010, 22, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.-C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.-C.; Cui, G.; Micevic, G.; Perales, J.C. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [PubMed]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kurupati, R.; Liu, L.; Zhou, X.Y.; Zhang, G.; Hudaihed, A.; Filisio, F.; Giles-Davis, W.; Xu, X.; Karakousis, G.C.; et al. Enhancing CD8(+) T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell 2017, 32, 377–391. [Google Scholar] [CrossRef]
- Chowdhury, P.S.; Chamoto, K.; Kumar, A.; Honjo, T. PPAR-Induced Fatty Acid Oxidation in T Cells Increases the Number of Tumor-Reactive CD8+ T Cells and Facilitates Anti–PD-1 Therapy. Cancer Immunol. Res. 2018, 6, e1375. [Google Scholar]
- Wan, H.; Xu, B.; Zhu, N.; Ren, B. PGC-1α activator–induced fatty acid oxidation in tumor-infiltrating CTLs enhances effects of PD-1 blockade therapy in lung cancer. Tumori J. 2019, 106, 55–63. [Google Scholar] [CrossRef]
- Chamoto, K.; Chowdhury, P.S.; Kumar, A.; Sonomura, K.; Matsuda, F.; Fagarasan, S.; Honjo, T. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc. Natl. Acad. Sci. USA 2017, 114, e761. [Google Scholar] [CrossRef]
- Biswas, S.K. Metabolic reprogramming of immune cells in cancer progression. Immunity 2015, 43, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, C.M.; Holowka, T.; Sun, J.; Blagih, J.; Amiel, E.; DeBerardinis, R.J.; Cross, J.R.; Jung, E.; Thompson, C.B.; Jones, R.G. Toll-like receptor–induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 2010, 115, 4742–4749. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.J.; Everts, B. Dendritic cell metabolism. Nat. Rev. Immunol. 2015, 15, e18. [Google Scholar] [CrossRef] [PubMed]
- Malinarich, F.; Duan, K.; Hamid, R.A.; Bijin, A.; Lin, W.X.; Poidinger, M.; Fairhurst, A.-M.; Connolly, J.E. High mitochondrial respiration and glycolytic capacity represent a metabolic phenotype of human tolerogenic dendritic cells. J. Immunol. 2015, 194, 5174–5186. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: the next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef]
- Mori, K.; Uchida, T.; Yoshie, T.; Mizote, Y.; Ishikawa, F.; Katsuyama, M.; Shibanuma, M. A mitochondrial ROS pathway controls matrix metalloproteinase 9 levels and invasive properties in RAS-activated cancer cells. FEBS J. 2019, 286, 459–478. [Google Scholar] [CrossRef]
- Ishikawa, K.; Koshikawa, N.; Takenaga, K.; Nakada, K.; Hayashi, J. Reversible regulation of metastasis by ROS-generating mtDNA mutations. Mitochondrion 2008, 8, 339–344. [Google Scholar] [CrossRef]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef]
- Pelicano, H.; Lu, W.; Zhou, Y.; Zhang, W.; Chen, Z.; Hu, Y.; Huang, P. Mitochondrial dysfunction and reactive oxygen species imbalance promote breast cancer cell motility through a CXCL14-mediated mechanism. Cancer Res. 2009, 69, 2375–2383. [Google Scholar] [CrossRef] [PubMed]
- Pelicano, H.; Zhang, W.; Liu, J.; Hammoudi, N.; Dai, J.; Xu, R.H.; Pusztai, L.; Huang, P. Mitochondrial dysfunction in some triple-negative breast cancer cell lines: role of mTOR pathway and therapeutic potential. Breast Cancer Res. 2014, 16, e434. [Google Scholar] [CrossRef] [PubMed]
- Diehl, F.F.; Lewis, C.A.; Fiske, B.P.; Vander Heiden, M.G. Cellular redox state constrains serine synthesis and nucleotide production to impact cell proliferation. Nat. Metab. 2019, 1, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Santidrian, A.F.; Matsuno-Yagi, A.; Ritland, M.; Seo, B.B.; LeBoeuf, S.E.; Gay, L.J.; Yagi, T.; Felding-Habermann, B. Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J. Clin Invest. 2013, 123, 1068–1081. [Google Scholar] [CrossRef]
- Cardaci, S.; Zheng, L.; MacKay, G.; van den Broek, N.J.; MacKenzie, E.D.; Nixon, C.; Stevenson, D.; Tumanov, S.; Bulusu, V.; Kamphorst, J.J.; et al. Pyruvate carboxylation enables growth of SDH-deficient cells by supporting aspartate biosynthesis. Nat. Cell Biol. 2015, 17, 1317–1326. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef]
- Arner, E.N.; Du, W.; Brekken, R.A. Behind the Wheel of Epithelial Plasticity in KRAS-Driven Cancers. Front. Oncol. 2019, 9, e1049. [Google Scholar] [CrossRef]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Gaglio, D.; Metallo, C.M.; Gameiro, P.A.; Hiller, K.; Danna, L.S.; Balestrieri, C.; Alberghina, L.; Stephanopoulos, G.; Chiaradonna, F. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol. Syst. Biol. 2011, 7, e523. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.B.; Aiba, I.; Long, Y.; Lin, H.K.; Feun, L.; Savaraj, N.; Kuo, M.T. Activation of Ras/PI3K/ERK pathway induces c-Myc stabilization to upregulate argininosuccinate synthetase, leading to arginine deiminase resistance in melanoma cells. Cancer Res. 2012, 72, 2622–2633. [Google Scholar] [CrossRef] [PubMed]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism, and Cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Ben-Sahra, I.; Lockwood, S.E.; Timson, R.C.; Byles, V.; Henning, G.T.; Gao, P.; Selfors, L.M.; Asara, J.M.; Manning, B.D. Direct stimulation of NADP(+) synthesis through Akt-mediated phosphorylation of NAD kinase. Science 2019, 363, 1088–1092. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nature Reviews Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Jones, R.G.; Thompson, C.B. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009, 23, 537–548. [Google Scholar] [CrossRef]
- Vousden, K.H.; Ryan, K.M. p53 and metabolism. Nat. Rev. Cancer 2009, 9, 691–700. [Google Scholar] [CrossRef]
- Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Karnieli, E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004, 64, 2627–2633. [Google Scholar] [CrossRef]
- Geng, J.; Yuan, X.; Wei, M.; Wu, J.; Qin, Z.H. The diverse role of TIGAR in cellular homeostasis and cancer. Free Radic Res. 2018, 52, 1240–1249. [Google Scholar] [CrossRef]
- Tajan, M.; Hock, A.K.; Blagih, J.; Robertson, N.A.; Labuschagne, C.F.; Kruiswijk, F.; Humpton, T.J.; Adams, P.D.; Vousden, K.H. A Role for p53 in the Adaptation to Glutamine Starvation through the Expression of SLC1A3. Cell Metab. 2018, 28, 721–736 e6. [Google Scholar] [CrossRef]
- Puzio-Kuter, A.M. The Role of p53 in Metabolic Regulation. Genes Cancer 2011, 2, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Molina, A.; Serrano, M. PTEN in cancer, metabolism, and aging. Trends Endocrinol. Metab. 2013, 24, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Mathur, D.; Stratikopoulos, E.; Ozturk, S.; Steinbach, N.; Pegno, S.; Schoenfeld, S.; Yong, R.; Murty, V.V.; Asara, J.M.; Cantley, L.C.; et al. PTEN Regulates Glutamine Flux to Pyrimidine Synthesis and Sensitivity to Dihydroorotate Dehydrogenase Inhibition. Cancer Discov. 2017, 7, 380–390. [Google Scholar] [CrossRef]
- Garcia-Cao, I.; Song, M.S.; Hobbs, R.M.; Laurent, G.; Giorgi, C.; de Boer, V.C.; Anastasiou, D.; Ito, K.; Sasaki, A.T.; Rameh, L.; et al. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell 2012, 149, 49–62. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef]
- Marin, T.L.; Gongol, B.; Zhang, F.; Martin, M.; Johnson, D.A.; Xiao, H.; Wang, Y.; Subramaniam, S.; Chien, S.; Shyy, J.Y. AMPK promotes mitochondrial biogenesis and function by phosphorylating the epigenetic factors DNMT1, RBBP7, and HAT1. Sci. Signal. 2017, 10, eaaf7478. [Google Scholar] [CrossRef]
- LeBoeuf, S.E.; Wu, W.L.; Karakousi, T.R.; Karadal, B.; Jackson, S.R.; Davidson, S.M.; Wong, K.-K.; Koralov, S.B.; Sayin, V.I.; Papagiannakopoulos, T. Activation of Oxidative Stress Response in Cancer Generates a Druggable Dependency on Exogenous Non-essential Amino Acids. Cell Metab. 2020, 31, 339–350. [Google Scholar] [CrossRef]
- Sanford-Crane, H.; Abrego, J.; Sherman, M.H. Fibroblasts as Modulators of Local and Systemic Cancer Metabolism. Cancers (Basel) 2019, 11, e619. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Lisanti, M.P.; Sotgia, F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin. Cancer Biol. 2014, 25, 47–60. [Google Scholar] [CrossRef]
- Khalili, J.S.; Liu, S.; Rodriguez-Cruz, T.G.; Whittington, M.; Wardell, S.; Liu, C.; Zhang, M.; Cooper, Z.A.; Frederick, D.T.; Li, Y.; et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin. Cancer Res. 2012, 18, 5329–5340. [Google Scholar] [CrossRef]
- Nazareth, M.R.; Broderick, L.; Simpson-Abelson, M.R.; Kelleher, R.J., Jr.; Yokota, S.J.; Bankert, R.B. Characterization of human lung tumor-associated fibroblasts and their ability to modulate the activation of tumor-associated T cells. J. Immunol. 2007, 178, 5552–5562. [Google Scholar] [CrossRef]
- Pinchuk, I.V.; Saada, J.I.; Beswick, E.J.; Boya, G.; Qiu, S.M.; Mifflin, R.C.; Raju, G.S.; Reyes, V.E.; Powell, D.W. PD-1 ligand expression by human colonic myofibroblasts/fibroblasts regulates CD4+ T-cell activity. Gastroenterology 2008, 135, 1228–1237. [Google Scholar] [CrossRef]
- Franklin, R.A.; Liao, W.; Sarkar, A.; Kim, M.V.; Bivona, M.R.; Liu, K.; Pamer, E.G.; Li, M.O. The cellular and molecular origin of tumor-associated macrophages. Science 2014, 344, 921–925. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef]
- Arlauckas, S.P.; Garris, C.S.; Kohler, R.H.; Kitaoka, M.; Cuccarese, M.F.; Yang, K.S.; Miller, M.A.; Carlson, J.C.; Freeman, G.J.; Anthony, R.M.; et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci. Transl. Med. 2017, 9, eaal3604. [Google Scholar] [CrossRef]
- Wenes, M.; Shang, M.; Di Matteo, M.; Goveia, J.; Martin-Perez, R.; Serneels, J.; Prenen, H.; Ghesquiere, B.; Carmeliet, P.; Mazzone, M. Macrophage Metabolism Controls Tumor Blood Vessel Morphogenesis and Metastasis. Cell Metab. 2016, 24, 701–715. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Taheri, F.; Ochoa, J.B.; Faghiri, Z.; Culotta, K.; Park, H.J.; Lan, M.S.; Zea, A.H.; Ochoa, A.C. L-Arginine regulates the expression of the T-cell receptor zeta chain (CD3zeta) in Jurkat cells. Clin. Cancer Res. 2001, 7, 958–965. [Google Scholar]
- Rodriguez, P.C.; Zea, A.H.; DeSalvo, J.; Culotta, K.S.; Zabaleta, J.; Quiceno, D.G.; Ochoa, J.B.; Ochoa, A.C. L-arginine consumption by macrophages modulates the expression of CD3 zeta chain in T lymphocytes. J. Immunol. 2003, 171, 1232–1239. [Google Scholar] [CrossRef]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842 e13. [Google Scholar] [CrossRef]
- Platten, M.; von Knebel Doeberitz, N.; Oezen, I.; Wick, W.; Ochs, K. Cancer Immunotherapy by Targeting IDO1/TDO and Their Downstream Effectors. Front. Immunol. 2014, 5, e673. [Google Scholar] [CrossRef]
- Zhao, Q.; Kuang, D.M.; Wu, Y.; Xiao, X.; Li, X.F.; Li, T.J.; Zheng, L. Activated CD69+ T cells foster immune privilege by regulating IDO expression in tumor-associated macrophages. J. Immunol. 2012, 188, 1117–1124. [Google Scholar] [CrossRef]
- Mondanelli, G.; Bianchi, R.; Pallotta, M.T.; Orabona, C.; Albini, E.; Iacono, A.; Belladonna, M.L.; Vacca, C.; Fallarino, F.; Macchiarulo, A.; et al. A Relay Pathway between Arginine and Tryptophan Metabolism Confers Immunosuppressive Properties on Dendritic Cells. Immunity 2017, 46, 233–244. [Google Scholar] [CrossRef]
- Du, X.; Chapman, N.M.; Chi, H. Emerging Roles of Cellular Metabolism in Regulating Dendritic Cell Subsets and Function. Front. Cell Dev. Biol. 2018, 6, e152. [Google Scholar] [CrossRef]
- Tibúrcio, R.; Nunes, S.; Nunes, I.; Rosa Ampuero, M.; Silva, I.B.; Lima, R.; Machado Tavares, N.; Brodskyn, C. Molecular Aspects of Dendritic Cell Activation in Leishmaniasis: An Immunobiological View. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Everts, B.; Amiel, E.; Huang, S.C.-C.; Smith, A.M.; Chang, C.-H.; Lam, W.Y.; Redmann, V.; Freitas, T.C.; Blagih, J.; van der Windt, G.J.W.; et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKɛ supports the anabolic demands of dendritic cell activation. Nat. Immunol. 2014, 15, 323–332. [Google Scholar] [CrossRef]
- Nasi, A.; Fekete, T.; Krishnamurthy, A.; Snowden, S.; Rajnavölgyi, E.; Catrina, A.I.; Wheelock, C.E.; Vivar, N.; Rethi, B. Dendritic Cell Reprogramming by Endogenously Produced Lactic Acid. J. Immunol. 2013, 191, e3090. [Google Scholar] [CrossRef]
- O'Sullivan, D.; van der Windt, G.J.; Huang, S.C.; Curtis, J.D.; Chang, C.H.; Buck, M.D.; Qiu, J.; Smith, A.M.; Lam, W.Y.; DiPlato, L.M.; et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity 2014, 41, 75–88. [Google Scholar] [CrossRef]
- van der Windt, G.J.; Everts, B.; Chang, C.H.; Curtis, J.D.; Freitas, T.C.; Amiel, E.; Pearce, E.J.; Pearce, E.L. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 2012, 36, 68–78. [Google Scholar] [CrossRef]
- Wang, K.; Shen, T.; Siegal, G.P.; Wei, S. The CD4/CD8 ratio of tumor-infiltrating lymphocytes at the tumor-host interface has prognostic value in triple-negative breast cancer. Hum. Pathol. 2017, 69, 110–117. [Google Scholar] [CrossRef]
- Han, S.; Zhang, C.; Li, Q.; Dong, J.; Liu, Y.; Huang, Y.; Jiang, T.; Wu, A. Tumour-infiltrating CD4+ and CD8+ lymphocytes as predictors of clinical outcome in glioma. Br. J. Cancer 2014, 110, 2560–2568. [Google Scholar] [CrossRef]
- Badoual, C.; Hans, S.; Rodriguez, J.; Peyrard, S.; Klein, C.; Agueznay, N.E.H.; Mosseri, V.; Laccourreye, O.; Bruneval, P.; Fridman, W.H.; et al. Prognostic Value of Tumor-Infiltrating CD4+ T-Cell Subpopulations in Head and Neck Cancers. Clin. Cancer Res. 2006, 12, e465. [Google Scholar]
- Oja, A.E.; Piet, B.; van der Zwan, D.; Blaauwgeers, H.; Mensink, M.; de Kivit, S.; Borst, J.; Nolte, M.A.; van Lier, R.A.W.; Stark, R.; et al. Functional Heterogeneity of CD4+ Tumor-Infiltrating Lymphocytes With a Resident Memory Phenotype in NSCLC. Front. Immunol. 2018, 9, e2654. [Google Scholar] [CrossRef]
- Zhao, Y.-J.; Zhang, J.; Shi, F.; Hu, Z.-P.; Wu, J.-P.; Wu, G.-J.; Wang, R.-B.; Zhou, Q.; Chang, H.; Li, Y.-N.; et al. Expression of PD-1 on CD4+ Tumor-Infiltrating Lymphocytes in Tumor Microenvironment Associated with Pathological Characteristics of Breast Cancer. J. Immunol. Res. 2018, 2018, e8. [Google Scholar] [CrossRef]
- Droeser, R.; Zlobec, I.; Kilic, E.; Güth, U.; Heberer, M.; Spagnoli, G.; Oertli, D.; Tapia, C. Differential pattern and prognostic significance of CD4+, FOXP3+ and IL-17+tumor infiltrating lymphocytes in ductal and lobular breast cancers. BMC Cancer 2012, 12, e134. [Google Scholar] [CrossRef]
- Pan, Y.; Tian, T.; Park, C.O.; Lofftus, S.Y.; Mei, S.; Liu, X.; Luo, C.; O'Malley, J.T.; Gehad, A.; Teague, J.E.; et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 2017, 543, 252–256. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Sena, L.A.; Chandel, N.S. Mitochondria in the Regulation of Innate and Adaptive Immunity. Immunity 2015, 42, 406–417. [Google Scholar] [CrossRef]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.-R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef]
- Kamiński, M.M.; Sauer, S.W.; Kamiński, M.; Opp, S.; Ruppert, T.; Grigaravičius, P.; Grudnik, P.; Gröne, H.-J.; Krammer, P.H.; Gülow, K. T cell Activation Is Driven by an ADP-Dependent Glucokinase Linking Enhanced Glycolysis with Mitochondrial Reactive Oxygen Species Generation. Cell Rep. 2012, 2, 1300–1315. [Google Scholar] [CrossRef]
- Desdín-Micó, G.; Soto-Heredero, G.; Mittelbrunn, M. Mitochondrial activity in T cells. Mitochondrion 2018, 41, 51–57. [Google Scholar] [CrossRef]
- McNamee, E.N.; Korns Johnson, D.; Homann, D.; Clambey, E.T. Hypoxia and hypoxia-inducible factors as regulators of T cell development, differentiation, and function. Immunol. Res. 2013, 55, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Siska, P.J.; Beckermann, K.E.; Mason, F.M.; Andrejeva, G.; Greenplate, A.R.; Sendor, A.B.; Chiang, Y.-C.J.; Corona, A.L.; Gemta, L.F.; Vincent, B.G.; et al. Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight 2017, 2, e93411. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-s.; Liu, W.; Ly, D.; Xu, H.; Qu, L.; Zhang, L. Tumor-infiltrating B cells: Their role and application in anti-tumor immunity in lung cancer. Cell. Mol. Immunol. 2019, 16, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Nelson, B.H. CD20+ B cells: the other tumor-infiltrating lymphocytes. J. Immunol. 2010, 185, 4977–4982. [Google Scholar] [CrossRef] [PubMed]
- Franchina, D.G.; Grusdat, M.; Brenner, D. B-Cell Metabolic Remodeling and Cancer. Trends Cancer 2018, 4, 138–150. [Google Scholar] [CrossRef]
- Waters, L.R.; Ahsan, F.M.; Wolf, D.M.; Shirihai, O.; Teitell, M.A. Initial B Cell Activation Induces Metabolic Reprogramming and Mitochondrial Remodeling. iScience 2018, 5, 99–109. [Google Scholar] [CrossRef]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Skaarup Larsen, M.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef]
- Petitprez, F.; de Reyniès, A.; Keung, E.Z.; Chen, T.W.-W.; Sun, C.-M.; Calderaro, J.; Jeng, Y.-M.; Hsiao, L.-P.; Lacroix, L.; Bougoüin, A.; et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 2020, 577, 556–560. [Google Scholar] [CrossRef]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T cell activation. Annu. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef]
- Rossjohn, J.; Gras, S.; Miles, J.J.; Turner, S.J.; Godfrey, D.I.; McCluskey, J. T Cell Antigen Receptor Recognition of Antigen-Presenting Molecules. Annu. Rev. Immunol. 2015, 33, 169–200. [Google Scholar] [CrossRef] [PubMed]
- Rossy, J.; Williamson, D.; Gaus, K. How does the kinase Lck phosphorylate the T cell receptor? Spatial organization as a regulatory mechanism. Front. Immunol. 2012, 3, e167. [Google Scholar] [CrossRef] [PubMed]
- Padhan, K.; Varma, R. Immunological synapse: A multi-protein signalling cellular apparatus for controlling gene expression. Immunology 2010, 129, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.; Zhong, X. Role of diacylglycerol kinases in T cell development and function. Crit. Rev. Immunol. 2013, 33, 97–118. [Google Scholar] [CrossRef]
- Trebak, M.; Kinet, J.-P. Calcium signalling in T cells. Nat. Rev. Immunol. 2019, 19, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Macian, F. NFAT proteins: key regulators of T-cell development and function. Nat. Rev. Immunol. 2005, 5, 472–484. [Google Scholar] [CrossRef]
- Paul, S.; Schaefer, B.C. A new look at T cell receptor signaling to nuclear factor-κB. Trends Immunol. 2013, 34, 269–281. [Google Scholar] [CrossRef]
- Kelliher, M.A.; Roderick, J.E. NOTCH Signaling in T-Cell-Mediated Anti-Tumor Immunity and T-Cell-Based Immunotherapies. Front. Immunol. 2018, 9, e1718. [Google Scholar] [CrossRef]
- Oestreich, K.J.; Read, K.A.; Gilbertson, S.E.; Hough, K.P.; McDonald, P.W.; Krishnamoorthy, V.; Weinmann, A.S. Bcl-6 directly represses the gene program of the glycolysis pathway. Nat. Immunol. 2014, 15, 957–964. [Google Scholar] [CrossRef]
- Man, K.; Miasari, M.; Shi, W.; Xin, A.; Henstridge, D.C.; Preston, S.; Pellegrini, M.; Belz, G.T.; Smyth, G.K.; Febbraio, M.A.; et al. The transcription factor IRF4 is essential for TCR affinity–mediated metabolic programming and clonal expansion of T cells. Nat. Immunol. 2013, 14, 1155–1165. [Google Scholar] [CrossRef]
- Gerriets, V.A.; Kishton, R.J.; Johnson, M.O.; Cohen, S.; Siska, P.J.; Nichols, A.G.; Warmoes, M.O.; de Cubas, A.A.; MacIver, N.J.; Locasale, J.W.; et al. Foxp3 and Toll-like receptor signaling balance Treg cell anabolic metabolism for suppression. Nat. Immunol. 2016, 17, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.R.; Herman, C.E.; Maciver, N.J.; Wofford, J.A.; Wieman, H.L.; Hammen, J.J.; Rathmell, J.C. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J. Immunol. (Baltimore, Md.: 1950) 2008, 180, 4476–4486. [Google Scholar] [CrossRef] [PubMed]
- Klein Geltink, R.I.; O’Sullivan, D.; Corrado, M.; Bremser, A.; Buck, M.D.; Buescher, J.M.; Firat, E.; Zhu, X.; Niedermann, G.; Caputa, G.; et al. Mitochondrial Priming by CD28. Cell 2017, 171, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Pollizzi, K.N.; Powell, J.D. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat. Rev. Immunol. 2014, 14, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Tarasenko, T.N.; Pacheco, S.E.; Koenig, M.K.; Gomez-Rodriguez, J.; Kapnick, S.M.; Diaz, F.; Zerfas, P.M.; Barca, E.; Sudderth, J.; DeBerardinis, R.J.; et al. Cytochrome c Oxidase Activity Is a Metabolic Checkpoint that Regulates Cell Fate Decisions During T Cell Activation and Differentiation. Cell Metab. 2017, 25, 1254–1268. [Google Scholar] [CrossRef] [PubMed]
- Dollins, C.M.; Nair, S.; Boczkowski, D.; Lee, J.; Layzer, J.M.; Gilboa, E.; Sullenger, B.A. Assembling OX40 aptamers on a molecular scaffold to create a receptor-activating aptamer. Chem. Biol. 2008, 15, 675–682. [Google Scholar] [CrossRef]
- Willoughby, J.; Griffiths, J.; Tews, I.; Cragg, M.S. OX40: Structure and function–What questions remain? Mol. Immunol. 2017, 83, 13–22. [Google Scholar] [CrossRef]
- Linch, S.N.; McNamara, M.J.; Redmond, W.L. OX40 Agonists and Combination Immunotherapy: Putting the Pedal to the Metal. Front. Oncol. 2015, 5, e34. [Google Scholar] [CrossRef]
- Vinay, D.S.; Kwon, B.S. 4-1BB (CD137), an inducible costimulatory receptor, as a specific target for cancer therapy. BMB Rep. 2014, 47, 122–129. [Google Scholar] [CrossRef]
- Sanchez-Paulete, A.R.; Labiano, S.; Rodriguez-Ruiz, M.E.; Azpilikueta, A.; Etxeberria, I.; Bolaños, E.; Lang, V.; Rodriguez, M.; Aznar, M.A.; Jure-Kunkel, M.; et al. Deciphering CD137 (4-1BB) signaling in T-cell costimulation for translation into successful cancer immunotherapy. Eur. J. of Immunol. 2016, 46, 513–522. [Google Scholar] [CrossRef]
- Choi, B.K.; Lee, D.Y.; Lee, D.G.; Kim, Y.H.; Kim, S.-H.; Oh, H.S.; Han, C.; Kwon, B.S. 4-1BB signaling activates glucose and fatty acid metabolism to enhance CD8(+) T cell proliferation. Cell. Mol. Immunol. 2017, 14, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Menk, A.V.; Scharping, N.E.; Rivadeneira, D.B.; Calderon, M.J.; Watson, M.J.; Dunstane, D.; Watkins, S.C.; Delgoffe, G.M. 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J. Exp. Med. 2018, 215, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Melero, I.; Shuford, W.W.; Newby, S.A.; Aruffo, A.; Ledbetter, J.A.; Hellström, K.E.; Mittler, R.S.; Chen, L. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat. Med. 1997, 3, 682–685. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.-T.; Bac, N.D.; Nguyen, K.-H.; Tien, N.L.B.; Thanh, V.V.; Nga, V.T.; Ngoc, V.T.N.; Anh Dao, D.T.; Hoan, L.N.; Hung, N.P.; et al. An Update on Anti-CD137 Antibodies in Immunotherapies for Cancer. Int. J. Mol. Sci. 2019, 20, e1822. [Google Scholar] [CrossRef] [PubMed]
- Moran, A.E.; Kovacsovics-Bankowski, M.; Weinberg, A.D. The TNFRs OX40, 4-1BB, and CD40 as targets for cancer immunotherapy. Curr. Opin. Immunol. 2013, 25, 230–237. [Google Scholar] [CrossRef]
- Tsurutani, N.; Mittal, P.; St Rose, M.-C.; Ngoi, S.M.; Svedova, J.; Menoret, A.; Treadway, F.B.; Laubenbacher, R.; Suárez-Ramírez, J.E.; Cauley, L.S.; et al. Costimulation Endows Immunotherapeutic CD8 T Cells with IL-36 Responsiveness during Aerobic Glycolysis. J. Immunol. (Baltimore, Md.: 1950) 2016, 196, 124–134. [Google Scholar] [CrossRef]
- Elgueta, R.; Benson, M.J.; de Vries, V.C.; Wasiuk, A.; Guo, Y.; Noelle, R.J. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev. 2009, 229, 152–172. [Google Scholar] [CrossRef]
- Vonderheide, R.H. CD40 Agonist Antibodies in Cancer Immunotherapy. Annu. Rev. Med. 2020, 71, 47–58. [Google Scholar] [CrossRef]
- Sim, G.C.; Radvanyi, L. The IL-2 cytokine family in cancer immunotherapy. Cytokine Growth Factor Rev. 2014, 25, 377–390. [Google Scholar] [CrossRef]
- Amatore, F.; Gorvel, L.; Olive, D. Inducible Co-Stimulator (ICOS) as a potential therapeutic target for anti-cancer therapy. Expert Opin. Ther. Targets 2018, 22, 343–351. [Google Scholar] [CrossRef]
- Gigoux, M.; Lovato, A.; Leconte, J.; Leung, J.; Sonenberg, N.; Suh, W.-K. Inducible costimulator facilitates T-dependent B cell activation by augmenting IL-4 translation. Mol. Immunol. 2014, 59, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Cohen, S.; Guy, C.; Shrestha, S.; Neale, G.; Brown, S.A.; Cloer, C.; Kishton, R.J.; Gao, X.; Youngblood, B.; et al. mTORC1 and mTORC2 Kinase Signaling and Glucose Metabolism Drive Follicular Helper T Cell Differentiation. Immunity 2016, 45, 540–554. [Google Scholar] [CrossRef] [PubMed]
- Strauss, L.; Bergmann, C.; Szczepanski, M.J.; Lang, S.; Kirkwood, J.M.; Whiteside, T.L. Expression of ICOS on Human Melanoma-Infiltrating CD4+CD25highFoxp3+ T Regulatory Cells: Implications and Impact on Tumor-Mediated Immune Suppression. J. Immunol. 2008, 180, e2967. [Google Scholar]
- Martin-Orozco, N.; Li, Y.; Wang, Y.; Liu, S.; Hwu, P.; Liu, Y.-J.; Dong, C.; Radvanyi, L. Melanoma Cells Express ICOS Ligand to Promote the Activation and Expansion of T-Regulatory Cells. Cancer Res. 2010, 70, 9581. [Google Scholar] [CrossRef]
- Ronchetti, S.; Ricci, E.; Petrillo, M.G.; Cari, L.; Migliorati, G.; Nocentini, G.; Riccardi, C. Glucocorticoid-induced tumour necrosis factor receptor-related protein: A key marker of functional regulatory T cells. J. Immunol. Res. 2015, 2015, e171520. [Google Scholar] [CrossRef]
- Zappasodi, R.; Sirard, C.; Li, Y.; Budhu, S.; Abu-Akeel, M.; Liu, C.; Yang, X.; Zhong, H.; Newman, W.; Qi, J.; et al. Rational design of anti-GITR-based combination immunotherapy. Nat. Med. 2019, 25, 759–766. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252. [Google Scholar] [CrossRef]
- Thompson, C.B.; Allison, J.P. The emerging role of CTLA-4 as an immune attenuator. Immunity 1997, 7, 445–450. [Google Scholar] [CrossRef]
- Tsai, H.-F.; Hsu, P.-N. Cancer immunotherapy by targeting immune checkpoints: mechanism of T cell dysfunction in cancer immunity and new therapeutic targets. J. Biomed. Sci. 2017, 24, e35. [Google Scholar] [CrossRef]
- Teft, W.A.; Kirchhof, M.G.; Madrenas, J. A molecular perspective of CTLA-4 function. Annu. Rev. Immunol. 2006, 24, 65–97. [Google Scholar] [CrossRef]
- Dyck, L.; Mills, K.H. Immune checkpoints and their inhibition in cancer and infectious diseases. Eur. J. Immunol. 2017, 47, 765–779. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, F.; Jiang, F.; Lv, X.; Zhang, R.; Lu, A.; Zhang, G. A mini-review for cancer immunotherapy: molecular understanding of PD-1/PD-L1 pathway & translational blockade of immune checkpoints. Int. J. Mol. Sci. 2016, 17, e1151. [Google Scholar] [PubMed]
- Arasanz, H.; Gato-Cañas, M.; Zuazo, M.; Ibañez-Vea, M.; Breckpot, K.; Kochan, G.; Escors, D. PD1 signal transduction pathways in T cells. Oncotarget 2017, 8, 51936–51945. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, e6692. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yue, C.; Herrmann, A.; Song, J.; Egelston, C.; Wang, T.; Zhang, Z.; Li, W.; Lee, H.; Aftabizadeh, M.; et al. STAT3 Activation-Induced Fatty Acid Oxidation in CD8+ T Effector Cells Is Critical for Obesity-Promoted Breast Tumor Growth. Cell Metab. 2020, 31, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Bengsch, B.; Johnson, A.L.; Kurachi, M.; Odorizzi, P.M.; Pauken, K.E.; Attanasio, J.; Stelekati, E.; McLane, L.M.; Paley, M.A.; Delgoffe, G.M.; et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8+ T Cell Exhaustion. Immunity 2016, 45, 358–373. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Liu, H.; Madeira da Silva, L.; Arora, R.; Liu, Z.; Phillips, J.B.; Schmitt, D.C.; Vu, T.; McClellan, S.; Lin, Y.; et al. Immunoregulatory Protein B7-H3 Reprograms Glucose Metabolism in Cancer Cells by ROS-Mediated Stabilization of HIF1α. Cancer Res. 2016, 76, 2231–2242. [Google Scholar] [CrossRef]
- Escors, D.; Gato-Cañas, M.; Zuazo, M.; Arasanz, H.; García-Granda, M.J.; Vera, R.; Kochan, G. The intracellular signalosome of PD-L1 in cancer cells. Signal. Transduct. Target. Ther. 2018, 3, e26. [Google Scholar] [CrossRef]
- Daassi, D.; Mahoney, K.M.; Freeman, G.J. The importance of exosomal PDL1 in tumour immune evasion. Nat. Rev. Immunol. 2020, 20, 209–215. [Google Scholar] [CrossRef]
- Li, X.; Wenes, M.; Romero, P.; Huang, S.C.-C.; Fendt, S.-M.; Ho, P.-C. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 425–441. [Google Scholar] [CrossRef]
- Strauss, L.; Mahmoud, M.A.A.; Weaver, J.D.; Tijaro-Ovalle, N.M.; Christofides, A.; Wang, Q.; Pal, R.; Yuan, M.; Asara, J.; Patsoukis, N.; et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Science Immunology 2020, 5, eaay1863. [Google Scholar] [CrossRef] [PubMed]
- Prasad, D.V.; Nguyen, T.; Li, Z.; Yang, Y.; Duong, J.; Wang, Y.; Dong, C. Murine B7-H3 is a negative regulator of T cells. J. Immunol. 2004, 173, 2500–2506. [Google Scholar] [CrossRef] [PubMed]
- Hofmeyer, K.A.; Ray, A.; Zang, X. The contrasting role of B7-H3. Proc. Natl. Acad. Sci. USA 2008, 105, 10277–10278. [Google Scholar] [CrossRef] [PubMed]
- Andrews, L.P.; Yano, H.; Vignali, D.A.A. Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: breakthroughs or backups. Nat. Immunol. 2019, 20, 1425–1434. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Li, J.; Liu, D.; Hong, S.; Qiao, Q.; Sun, Q.; Li, P.; Lyu, N.; Sun, T.; Xie, S.; et al. Tumor-expressed B7-H3 mediates the inhibition of antitumor T-cell functions in ovarian cancer insensitive to PD-1 blockade therapy. Cell. Mol. Immunol. 2019, 17, 227–236. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef]
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A.A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 2017, 276, 80–96. [Google Scholar] [CrossRef]
- Hemon, P.; Jean-Louis, F.; Ramgolam, K.; Brignone, C.; Viguier, M.; Bachelez, H.; Triebel, F.; Charron, D.; Aoudjit, F.; Al-Daccak, R.; et al. MHC Class II Engagement by Its Ligand LAG-3 (CD223) Contributes to Melanoma Resistance to Apoptosis. J. Immunol. 2011, 186, e5173. [Google Scholar] [CrossRef]
- Long, L.; Zhang, X.; Chen, F.; Pan, Q.; Phiphatwatchara, P.; Zeng, Y.; Chen, H. The promising immune checkpoint LAG-3: from tumor microenvironment to cancer immunotherapy. Genes cancer 2018, 9, 176–189. [Google Scholar]
- Wolf, Y.; Anderson, A.C.; Kuchroo, V.K. TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunology 2019, 20, 173–185. [Google Scholar] [CrossRef]
- Ferris, R.L.; Lu, B.; Kane, L.P. Too much of a good thing? Tim-3 and TCR signaling in T cell exhaustion. J. Immunol. 2014, 193, 1525–1530. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Lo, Y.-C.; Powell, J.D. A2aR antagonists: Next generation checkpoint blockade for cancer immunotherapy. Comput. Struct. Biotechnol. J. 2015, 13, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol. Rev. 2017, 276, 121–144. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Ngiow, S.F.; Gao, Y.; Patch, A.-M.; Barkauskas, D.S.; Messaoudene, M.; Lin, G.; Coudert, J.D.; Stannard, K.A.; Zitvogel, L. A2AR adenosine signaling suppresses natural killer cell maturation in the tumor microenvironment. Cancer Res. 2018, 78, 1003–1016. [Google Scholar] [CrossRef]
- Nowak, M.; Lynch, L.; Yue, S.; Ohta, A.; Sitkovsky, M.; Balk, S.P.; Exley, M.A. The A2aR adenosine receptor controls cytokine production in iNKT cells. Eur. J. Immunol. 2010, 40, 682–687. [Google Scholar] [CrossRef]
- Leone, R.D.; Emens, L.A. Targeting adenosine for cancer immunotherapy. J. Immunother. Cancer 2018, 6, e57. [Google Scholar] [CrossRef]
- Labadie, B.W.; Bao, R.; Luke, J.J. Reimagining IDO Pathway Inhibition in Cancer Immunotherapy via Downstream Focus on the Tryptophan–Kynurenine–Aryl Hydrocarbon Axis. Clin. Cancer Res. 2019, 25, e1462. [Google Scholar] [CrossRef]
- Grohmann, U.; Fallarino, F.; Puccetti, P. Tolerance, DCs and tryptophan: much ado about IDO. Trends Immunol. 2003, 24, 242–248. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Malachowski, W.P.; DuHadaway, J.B.; Muller, A.J. Discovery of IDO1 inhibitors: from bench to bedside. Cancer Res. 2017, 77, 6795–6811. [Google Scholar] [CrossRef]
- Romero-Garcia, S.; Moreno-Altamirano, M.M.B.; Prado-Garcia, H.; Sánchez-García, F.J. Lactate contribution to the tumor microenvironment: mechanisms, effects on immune cells and therapeutic relevance. Front. Immunol. 2016, 7, e52. [Google Scholar] [CrossRef]
- Holm, E.; Hagmuller, E.; Staedt, U.; Schlickeiser, G.; Gunther, H.J.; Leweling, H.; Tokus, M.; Kollmar, H.B. Substrate balances across colonic carcinomas in humans. Cancer Res. 1995, 55, 1373–1378. [Google Scholar]
- Choi, S.Y.C.; Collins, C.C.; Gout, P.W.; Wang, Y. Cancer-generated lactic acid: A regulatory, immunosuppressive metabolite? J. Pathol. 2013, 230, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Hirschhaeuser, F.; Sattler, U.G.; Mueller-Klieser, W. Lactate: A metabolic key player in cancer. Cancer Res. 2011, 71, 6921–6925. [Google Scholar] [CrossRef]
- Cascone, T.; McKenzie, J.A.; Mbofung, R.M.; Punt, S.; Wang, Z.; Xu, C.; Williams, L.J.; Wang, Z.; Bristow, C.A.; Carugo, A.; et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab. 2018, 27, 977–987. [Google Scholar] [CrossRef]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Walenta, S.; Wetterling, M.; Lehrke, M.; Schwickert, G.; Sundfør, K.; Rofstad, E.K.; Mueller-Klieser, W. High Lactate Levels Predict Likelihood of Metastases, Tumor Recurrence, and Restricted Patient Survival in Human Cervical Cancers. Cancer Res. 2000, 60, e916. [Google Scholar]
- Kato, Y.; Ozawa, S.; Miyamoto, C.; Maehata, Y.; Suzuki, A.; Maeda, T.; Baba, Y. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 2013, 13, e89. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Filipazzi, P.; Grioni, M.; Iero, M.; De Milito, A.; Ricupito, A.; Cova, A.; Canese, R.; Jachetti, E.; Rossetti, M. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 2012, 72, 2746–2756. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.J.; Su, L.J.; Pinckney, J.; Critton, D.; Boyer, E.; Krishnakumar, A.; Corbett, M.; Rankin, A.L.; Dibella, R.; Campbell, L.; et al. VISTA is an acidic pH-selective ligand for PSGL-1. Nature 2019, 574, 565–570. [Google Scholar] [CrossRef]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S. Inhibitory effect of tumor cell–derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Gottfried, E.; Kunz-Schughart, L.A.; Ebner, S.; Mueller-Klieser, W.; Hoves, S.; Andreesen, R.; Mackensen, A.; Kreutz, M. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 2006, 107, 2013–2021. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, T.; Aoki, M.; Tomita, H.; Akazawa, T.; Sato, K.; Kuze, B.; Mizuta, K.; Hara, A.; Nagaoka, H.; Inoue, N. M2-like macrophage polarization in high lactic acid-producing head and neck cancer. Cancer Sci. 2017, 108, 1128–1134. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.R.; Cleveland, J.L. Targeting lactate metabolism for cancer therapeutics. J. Clin. Investig. 2013, 123, 3685–3692. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, T.; Akazawa, T.; Aoki, M.; Kuze, B.; Mizuta, K.; Ito, Y.; Inoue, N. Dichloroacetate improves immune dysfunction caused by tumor-secreted lactic acid and increases antitumor immunoreactivity. Int. J. Cancer 2013, 133, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Pilon-Thomas, S.; Kodumudi, K.N.; El-Kenawi, A.E.; Russell, S.; Weber, A.M.; Luddy, K.; Damaghi, M.; Wojtkowiak, J.W.; Mulé, J.J.; Ibrahim-Hashim, A. Neutralization of tumor acidity improves antitumor responses to immunotherapy. Cancer Res. 2016, 76, 1381–1390. [Google Scholar] [CrossRef]
- Ruoslahti, E. Specialization of tumour vasculature. Nat. Rev. Cancer 2002, 2, e83. [Google Scholar] [CrossRef]
- Thomlinson, R.; Gray, L. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br. J. Cancer 1955, 9, e539. [Google Scholar] [CrossRef]
- Westendorf, A.M.; Skibbe, K.; Adamczyk, A.; Buer, J.; Geffers, R.; Hansen, W.; Pastille, E.; Jendrossek, V. Hypoxia enhances immunosuppression by inhibiting CD4+ effector T cell function and promoting Treg activity. Cell. Physiol. Biochem. 2017, 41, 1271–1284. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, e83. [Google Scholar] [CrossRef]
- Ai, M.; Budhani, P.; Sheng, J.; Balasubramanyam, S.; Bartkowiak, T.; Jaiswal, A.R.; Ager, C.R.; Haria, D.D.; Curran, M.A. Tumor hypoxia drives immune suppression and immunotherapy resistance. J. Immunother. Cancer 2015, 3, P392. [Google Scholar] [CrossRef]
- Triner, D.; Shah, Y.M. Hypoxia-inducible factors: A central link between inflammation and cancer. J. Clin. Investig. 2016, 126, 3689–3698. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.J.; Gribar, S.C.; Li, J.; Kohler, J.W.; Branca, M.F.; Dubowski, T.; Sodhi, C.P.; Hackam, D.J. Hypoxia causes an increase in phagocytosis by macrophages in a HIF-1α-dependent manner. J. Leukoc. Biol. 2007, 82, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Peyssonnaux, C.; Datta, V.; Cramer, T.; Doedens, A.; Theodorakis, E.A.; Gallo, R.L.; Hurtado-Ziola, N.; Nizet, V.; Johnson, R.S. HIF-1α expression regulates the bactericidal capacity of phagocytes. J. Clin. Investig. 2005, 115, 1806–1815. [Google Scholar] [CrossRef]
- Zinkernagel, A.S.; Johnson, R.S.; Nizet, V. Hypoxia inducible factor (HIF) function in innate immunity and infection. J. Mol. Med. 2007, 85, 1339–1346. [Google Scholar] [CrossRef]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.-I.; Cheng, P.; Cho, H.-I.; Celis, E.; Quiceno, D.G.; Padhya, T. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef]
- Movahedi, K.; Laoui, D.; Gysemans, C.; Baeten, M.; Stange, G.; Van den Bossche, J.; Mack, M.; Pipeleers, D.; In't Veld, P.; De Baetselier, P.; et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010, 70, 5728–5739. [Google Scholar] [CrossRef]
- Doedens, A.L.; Stockmann, C.; Rubinstein, M.P.; Liao, D.; Zhang, N.; DeNardo, D.G.; Coussens, L.M.; Karin, M.; Goldrath, A.W.; Johnson, R.S. Macrophage expression of hypoxia-inducible factor-1α suppresses T-cell function and promotes tumor progression. Cancer Res. 2010, 70, 7465–7475. [Google Scholar] [CrossRef]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Clambey, E.T.; McNamee, E.N.; Westrich, J.A.; Glover, L.E.; Campbell, E.L.; Jedlicka, P.; de Zoeten, E.F.; Cambier, J.C.; Stenmark, K.R.; Colgan, S.P. Hypoxia-inducible factor-1 alpha–dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc. Natl. Acad. Sci. USA 2012, 109, 2784–2793. [Google Scholar] [CrossRef]
- Ben-Shoshan, J.; Maysel-Auslender, S.; Mor, A.; Keren, G.; George, J. Hypoxia controls CD4+ CD25+ regulatory T-cell homeostasis via hypoxia-inducible factor-1α. Eur. J. Immunol. 2008, 38, 2412–2418. [Google Scholar] [CrossRef] [PubMed]
- Lukashev, D.; Klebanov, B.; Kojima, H.; Grinberg, A.; Ohta, A.; Berenfeld, L.; Wenger, R.H.; Ohta, A.; Sitkovsky, M. Cutting edge: Hypoxia-inducible factor 1α and its activation-inducible short isoform I. 1 negatively regulate functions of CD4+ and CD8+ T lymphocytes. J. Immunol. 2006, 177, 4962–4965. [Google Scholar] [CrossRef] [PubMed]
- Krzywinska, E.; Stockmann, C. Hypoxia, Metabolism and Immune Cell Function. Biomedicines 2018, 6, e56. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Omori, K.; Murata, T. Role of prostaglandins in tumor microenvironment. Cancer Metastasis Rev. 2018, 37, 347–354. [Google Scholar] [CrossRef]
- Hanna, V.S.; Hafez, E.A.A. Synopsis of arachidonic acid metabolism: A review. J. Adv. Res. 2018, 11, 23–32. [Google Scholar] [CrossRef]
- Zelenay, S.; van der Veen, A.G.; Böttcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef]
- Miao, J.; Lu, X.; Hu, Y.; Piao, C.; Wu, X.; Liu, X.; Huang, C.; Wang, Y.; Li, D.; Liu, J. Prostaglandin E(2) and PD-1 mediated inhibition of antitumor CTL responses in the human tumor microenvironment. Oncotarget 2017, 8, 89802–89810. [Google Scholar] [CrossRef]
- Zhao, X.; Xu, Z.; Li, H. NSAIDs Use and Reduced Metastasis in Cancer Patients: Results from a meta-analysis. Sci. Rep. 2017, 7, e1875. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, H.; Chen, P.; Xie, H.; Tao, Y. Demystifying the manipulation of host immunity, metabolism, and extraintestinal tumors by the gut microbiome. Signal. Transduct. Target. Ther. 2019, 4, e41. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 2018, 359, e97. [Google Scholar] [CrossRef]
- Tanoue, T.; Morita, S.; Plichta, D.R.; Skelly, A.N.; Suda, W.; Sugiura, Y.; Narushima, S.; Vlamakis, H.; Motoo, I.; Sugita, K.; et al. A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature 2019, 565, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Riquelme, E.; Zhang, Y.; Zhang, L.; Montiel, M.; Zoltan, M.; Dong, W.; Quesada, P.; Sahin, I.; Chandra, V.; San Lucas, A.; et al. Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer Outcomes. Cell 2019, 178, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.M.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, e1079. [Google Scholar] [CrossRef] [PubMed]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Man Lei, Y.; Jabri, B.; Alegre, M.-L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti–PD-L1 efficacy. Science 2015, 350, e1084. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Carmagnani Pestana, R.; Hess, K.; Viola, G.M.; Subbiah, V. Impact of antibiotic use on survival in patients with advanced cancers treated on immune checkpoint inhibitor phase I clinical trials. Ann. Oncol. 2018, 29, 2396–2398. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; van Esch, B.C.A.M.; Wagenaar, G.T.M.; Garssen, J.; Folkerts, G.; Henricks, P.A.J. Pro- and anti-inflammatory effects of short chain fatty acids on immune and endothelial cells. Eur. J. Pharmacol. 2018, 831, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Luu, M.; Pautz, S.; Kohl, V.; Singh, R.; Romero, R.; Lucas, S.; Hofmann, J.; Raifer, H.; Vachharajani, N.; Carrascosa, L.C.; et al. The short-chain fatty acid pentanoate suppresses autoimmunity by modulating the metabolic-epigenetic crosstalk in lymphocytes. Nat. Commun. 2019, 10, e760. [Google Scholar] [CrossRef]
- David, C.J.; Chen, M.; Assanah, M.; Canoll, P.; Manley, J.L. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 2010, 463, 364–368. [Google Scholar] [CrossRef]
- Dong, G.; Mao, Q.; Xia, W.; Xu, Y.; Wang, J.; Xu, L.; Jiang, F. PKM2 and cancer: The function of PKM2 beyond glycolysis. Oncol. Lett. 2016, 11, 1980–1986. [Google Scholar] [CrossRef]
- Palsson-McDermott, E.M.; Dyck, L.; Zasłona, Z.; Menon, D.; McGettrick, A.F.; Mills, K.H.G.; O'Neill, L.A. Pyruvate Kinase M2 Is Required for the Expression of the Immune Checkpoint PD-L1 in Immune Cells and Tumors. Front. Immunol. 2017, 8, e1300. [Google Scholar] [CrossRef]
- Watanabe, R.; Shirai, T.; Namkoong, H.; Zhang, H.; Berry, G.J.; Wallis, B.B.; Schaefgen, B.; Harrison, D.G.; Tremmel, J.A.; Giacomini, J.C.; et al. Pyruvate controls the checkpoint inhibitor PD-L1 and suppresses T cell immunity. J. Clin. Investig. 2017, 127, 2725–2738. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-R.; Tian, M.-X.; Yang, L.-X.; Lin, Y.-L.; Jin, L.; Ding, Z.-B.; Shen, Y.-H.; Peng, Y.-F.; Gao, D.-M.; Zhou, J.; et al. PKM2 promotes metastasis by recruiting myeloid-derived suppressor cells and indicates poor prognosis for hepatocellular carcinoma. Oncotarget 2015, 6, 846–861. [Google Scholar] [CrossRef] [PubMed]
- Anastasiou, D.; Yu, Y.; Israelsen, W.J.; Jiang, J.-K.; Boxer, M.B.; Hong, B.S.; Tempel, W.; Dimov, S.; Shen, M.; Jha, A.; et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat. Chem. Biol. 2012, 8, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Imbert, C.; Montfort, A.; Fraisse, M.; Marcheteau, E.; Gilhodes, J.; Martin, E.; Bertrand, F.; Marcellin, M.; Burlet-Schiltz, O.; de Peredo, A.G.; et al. Resistance of melanoma to immune checkpoint inhibitors is overcome by targeting the sphingosine kinase-1. Nat. Commun. 2020, 11, 437. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Cabrera, T.; Aptsiauri, N. “Hard” and “soft” lesions underlying the HLA class I alterations in cancer cells: Implications for immunotherapy. Int. J. Cancer 2010, 127, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Luo, N.; Nixon, M.J.; Gonzalez-Ericsson, P.I.; Sanchez, V.; Opalenik, S.R.; Li, H.; Zahnow, C.A.; Nickels, M.L.; Liu, F.; Tantawy, M.N.; et al. DNA methyltransferase inhibition upregulates MHC-I to potentiate cytotoxic T lymphocyte responses in breast cancer. Nat. Commun. 2018, 9, e248. [Google Scholar] [CrossRef]
- Magner, W.J.; Kazim, A.L.; Stewart, C.; Romano, M.A.; Catalano, G.; Grande, C.; Keiser, N.; Santaniello, F.; Tomasi, T.B. Activation of MHC Class I, II, and CD40 Gene Expression by Histone Deacetylase Inhibitors. J. Immunol. 2000, 165, e7017. [Google Scholar] [CrossRef]
- Reid, M.A.; Dai, Z.; Locasale, J.W. The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat. Cell Biol. 2017, 19, 1298–1306. [Google Scholar] [CrossRef]
- Catalán, E.; Charni, S.; Jaime, P.; Aguiló, J.I.; Enríquez, J.A.; Naval, J.; Pardo, J.; Villalba, M.; Anel, A. MHC-I modulation due to changes in tumor cell metabolism regulates tumor sensitivity to CTL and NK cells. Oncoimmunology 2015, 4, 985924. [Google Scholar] [CrossRef]
- Marijt, K.A.; Sluijter, M.; Blijleven, L.; Tolmeijer, S.H.; Scheeren, F.A.; van der Burg, S.H.; van Hall, T. Metabolic stress in cancer cells induces immune escape through a PI3K-dependent blockade of IFNγ receptor signaling. J. Immunother. Cancer 2019, 7, e152. [Google Scholar] [CrossRef]
- Charni, S.; de Bettignies, G.; Rathore, M.G.; Aguiló, J.I.; van den Elsen, P.J.; Haouzi, D.; Hipskind, R.A.; Enriquez, J.A.; Sanchez-Beato, M.; Pardo, J.; et al. Oxidative Phosphorylation Induces De Novo Expression of the MHC Class I in Tumor Cells through the ERK5 Pathway. J. Immunol. 2010, 185, e3498. [Google Scholar] [CrossRef] [PubMed]
- Oliveras-Ferraros, C.; Cufí, S.; Vazquez-Martin, A.; Menendez, O.J.; Bosch-Barrera, J.; Martin-Castillo, B.; Joven, J.; Menendez, J.A. Metformin rescues cell surface major histocompatibility complex class I (MHC-I) deficiency caused by oncogenic transformation. Cell Cycle 2012, 11, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Turgeon, M.-O.; Perry, N.J.S.; Poulogiannis, G. DNA Damage, Repair, and Cancer Metabolism. Front. Oncol. 2018, 8, e15. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Kinnaird, A.; Zhao, S.; Wellen, K.E.; Michelakis, E.D. Metabolic control of epigenetics in cancer. Nat. Rev. Cancer 2016, 16, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.; Galeas, J.; Cheng, H. Tumor mutation burden in lung cancer: A new predictive biomarker for immunotherapy or too soon to tell? J. Thorac. Dis. 2018, 10, 3994–3998. [Google Scholar] [CrossRef]
- Friedman, J.M. Obesity: Causes and control of excess body fat. Nature 2009, 459, 340–342. [Google Scholar] [CrossRef]
- World Cancer Research Fund/American Institute for Cancer Research. Diet, nutrition, physical activity and cancer: A global perspective. Contin. Update Proj. Expert Rep. 2018. [Google Scholar]
- Coelho, M.; Oliveira, T.; Fernandes, R. Biochemistry of adipose tissue: An endocrine organ. Arch. Med. Sci 2013, 9, 191–200. [Google Scholar] [CrossRef]
- Abella, V.; Scotece, M.; Conde, J.; Lopez, V.; Lazzaro, V.; Pino, J.; Gomez-Reino, J.J.; Gualillo, O. Adipokines, metabolic syndrome and rheumatic diseases. J. Immunol. Res. 2014, 2014, e343746. [Google Scholar] [CrossRef]
- Cornier, M.A.; Dabelea, D.; Hernandez, T.L.; Lindstrom, R.C.; Steig, A.J.; Stob, N.R.; Van Pelt, R.E.; Wang, H.; Eckel, R.H. The metabolic syndrome. Endocr Rev. 2008, 29, 777–822. [Google Scholar] [CrossRef] [PubMed]
- Leal Vde, O.; Mafra, D. Adipokines in obesity. Clin. Chim. Acta. 2013, 419, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Ahima, R.S.; Osei, S.Y. Adipokines in obesity. Front. Horm. Res. 2008, 36, 182–197. [Google Scholar]
- Lihn, A.S.; Pedersen, S.B.; Richelsen, B. Adiponectin: Action, regulation and association to insulin sensitivity. Obes. Rev. 2005, 6, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Turer, A.T.; Scherer, P.E. Adiponectin: mechanistic insights and clinical implications. Diabetologia 2012, 55, 2319–2326. [Google Scholar] [CrossRef] [PubMed]
- Kelesidis, T.; Kelesidis, I.; Chou, S.; Mantzoros, C.S. Narrative review: The role of leptin in human physiology: emerging clinical applications. Ann. Intern. Med. 2010, 152, 93–100. [Google Scholar] [CrossRef]
- Pinto, S.; Roseberry, A.G.; Liu, H.; Diano, S.; Shanabrough, M.; Cai, X.; Friedman, J.M.; Horvath, T.L. Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science 2004, 304, 110–115. [Google Scholar] [CrossRef]
- Unger, R.H. The hyperleptinemia of obesity-regulator of caloric surpluses. Cell 2004, 117, 145–146. [Google Scholar] [CrossRef]
- Myers, M.G., Jr.; Leibel, R.L.; Seeley, R.J.; Schwartz, M.W. Obesity and leptin resistance: distinguishing cause from effect. Trends Endocrinol. Metab. 2010, 21, 643–651. [Google Scholar] [CrossRef]
- Quail, D.F.; Dannenberg, A.J. The obese adipose tissue microenvironment in cancer development and progression. Nat. Rev. Endocrinol. 2019, 15, 139–154. [Google Scholar] [CrossRef]
- Ellulu, M.S.; Patimah, I.; Khaza'ai, H.; Rahmat, A.; Abed, Y. Obesity and inflammation: the linking mechanism and the complications. Arch. Med. Sci. 2017, 13, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.E.; Kim, J.M.; Joung, K.H.; Lee, J.H.; You, B.R.; Choi, M.J.; Ryu, M.J.; Ko, Y.B.; Lee, M.A.; Lee, J.; et al. The Roles of Adipokines, Proinflammatory Cytokines, and Adipose Tissue Macrophages in Obesity-Associated Insulin Resistance in Modest Obesity and Early Metabolic Dysfunction. PLoS ONE 2016, 11, e0154003. [Google Scholar] [CrossRef] [PubMed]
- Caer, C.; Rouault, C.; Le Roy, T.; Poitou, C.; Aron-Wisnewsky, J.; Torcivia, A.; Bichet, J.C.; Clement, K.; Guerre-Millo, M.; Andre, S. Immune cell-derived cytokines contribute to obesity-related inflammation, fibrogenesis and metabolic deregulation in human adipose tissue. Sci Rep. 2017, 7, e3000. [Google Scholar] [CrossRef] [PubMed]
- DeFuria, J.; Belkina, A.C.; Jagannathan-Bogdan, M.; Snyder-Cappione, J.; Carr, J.D.; Nersesova, Y.R.; Markham, D.; Strissel, K.J.; Watkins, A.A.; Zhu, M.; et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc. Natl. Acad. Sci. USA 2013, 110, 5133–5138. [Google Scholar] [CrossRef]
- McArdle, M.A.; Finucane, O.M.; Connaughton, R.M.; McMorrow, A.M.; Roche, H.M. Mechanisms of obesity-induced inflammation and insulin resistance: Insights into the emerging role of nutritional strategies. Front. Endocrinol. (Lausanne) 2013, 4, e52. [Google Scholar] [CrossRef] [PubMed]
- Turbitt, W.J.; Collins, S.D.; Meng, H.; Rogers, C.J. Increased Adiposity Enhances the Accumulation of MDSCs in the Tumor Microenvironment and Adipose Tissue of Pancreatic Tumor-Bearing Mice and in Immune Organs of Tumor-Free Hosts. Nutrients 2019, 11, e3012. [Google Scholar] [CrossRef]
- Aguilar, E.G.; Murphy, W.J. Obesity induced T cell dysfunction and implications for cancer immunotherapy. Curr. Opin. Immunol. 2018, 51, 181–186. [Google Scholar] [CrossRef]
- Mirsoian, A.; Bouchlaka, M.N.; Sckisel, G.D.; Chen, M.; Pai, C.-C.S.; Maverakis, E.; Spencer, R.G.; Fishbein, K.W.; Siddiqui, S.; Monjazeb, A.M.; et al. Adiposity induces lethal cytokine storm after systemic administration of stimulatory immunotherapy regimens in aged mice. J. Exp. Med. 2014, 211, 2373–2383. [Google Scholar] [CrossRef]
- Wang, Z.; Aguilar, E.G.; Luna, J.I.; Dunai, C.; Khuat, L.T.; Le, C.T.; Mirsoian, A.; Minnar, C.M.; Stoffel, K.M.; Sturgill, I.R.; et al. Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat. Med. 2019, 25, 141–151. [Google Scholar] [CrossRef]
- McQuade, J.L.; Daniel, C.R.; Hess, K.R.; Mak, C.; Wang, D.Y.; Rai, R.R.; Park, J.J.; Haydu, L.E.; Spencer, C.; Wongchenko, M.; et al. Association of body-mass index and outcomes in patients with metastatic melanoma treated with targeted therapy, immunotherapy, or chemotherapy: a retrospective, multicohort analysis. Lancet Oncol. 2018, 19, 310–322. [Google Scholar] [CrossRef]
- Cortellini, A.; Bersanelli, M.; Buti, S.; Cannita, K.; Santini, D.; Perrone, F.; Giusti, R.; Tiseo, M.; Michiara, M.; Di Marino, P.; et al. A multicenter study of body mass index in cancer patients treated with anti-PD-1/PD-L1 immune checkpoint inhibitors: When overweight becomes favorable. J. Immunother. Cancer 2019, 7, e57. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, D.M.; Bajaj, S.; Zhong, J.; Weber, J.S.; Pavlick, A.C.; Balar, A.V.; Hsu, M.; Osman, I. The relationship between obesity and immunotherapy: It's complicated. J. Clin. Oncol. 2019, 37, 9562. [Google Scholar] [CrossRef]
- Procaccini, C.; De Rosa, V.; Galgani, M.; Carbone, F.; La Rocca, C.; Formisano, L.; Matarese, G. Role of adipokines signaling in the modulation of T cells function. Front. Immunol. 2013, 4, 332. [Google Scholar] [CrossRef] [PubMed]
- Clements, V.K.; Long, T.; Long, R.; Figley, C.; Smith, D.M.C.; Ostrand-Rosenberg, S. Frontline Science: High fat diet and leptin promote tumor progression by inducing myeloid-derived suppressor cells. J. Leukoc. Biol. 2018, 103, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.A.; James, B.R.; Sjaastad, F.V.; Kucaba, T.A.; Kim, H.; Brincks, E.L.; Chua, S.C., Jr.; Wilber, A.; Griffith, T.S. Cutting Edge: Elevated Leptin during Diet-Induced Obesity Reduces the Efficacy of Tumor Immunotherapy. J. Immunol. (Baltimore, Md.: 1950) 2018, 201, 1837–1841. [Google Scholar] [CrossRef] [PubMed]
- Rivadeneira, D.B.; DePeaux, K.; Wang, Y.; Kulkarni, A.; Tabib, T.; Menk, A.V.; Sampath, P.; Lafyatis, R.; Ferris, R.L.; Sarkar, S.N.; et al. Oncolytic Viruses Engineered to Enforce Leptin Expression Reprogram Tumor-Infiltrating T Cell Metabolism and Promote Tumor Clearance. Immunity 2019, 51, 548–560. [Google Scholar] [CrossRef]
- Saucillo, D.C.; Gerriets, V.A.; Sheng, J.; Rathmell, J.C.; Maciver, N.J. Leptin metabolically licenses T cells for activation to link nutrition and immunity. J. Immunol. (Baltimore, Md.: 1950) 2014, 192, 136–144. [Google Scholar] [CrossRef]
- Murphy, W.J.; Longo, D.L. The Surprisingly Positive Association Between Obesity and Cancer Immunotherapy Efficacy. JAMA 2019, 321, 1247–1248. [Google Scholar] [CrossRef]
- Sanchez, A.; Furberg, H.; Kuo, F.; Vuong, L.; Ged, Y.; Patil, S.; Ostrovnaya, I.; Petruzella, S.; Reising, A.; Patel, P.; et al. Transcriptomic signatures related to the obesity paradox in patients with clear cell renal cell carcinoma: a cohort study. Lancet Oncol. 2019, 21, 283–293. [Google Scholar] [CrossRef]
- Kichenadasse, G.; Miners, J.O.; Mangoni, A.A.; Rowland, A.; Hopkins, A.M.; Sorich, M.J. Association Between Body Mass Index and Overall Survival With Immune Checkpoint Inhibitor Therapy for Advanced Non-Small Cell Lung Cancer. JAMA Oncol. 2019. [Google Scholar] [CrossRef]
- Tannenbaum, A. The Initiation and Growth of Tumors: Introduction. I. Effects of Underfeeding. Am. J. Cancer 1940, 38, e335. [Google Scholar]
- JAMA Network. Available online: https://jamanetwork.com/journals/jama/fullarticle/277025 (accessed on 28 March 2020).
- Balasubramanian, P.; Howell, P.R.; Anderson, R.M. Aging and Caloric Restriction Research: A Biological Perspective With Translational Potential. EBioMedicine 2017, 21, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Wasinski, F.; Bacurau, R.F.P.; Moraes, M.R.; Haro, A.S.; Moraes-Vieira, P.M.M.; Estrela, G.R.; Paredes-Gamero, E.J.; Barros, C.C.; Almeida, S.S.; Câmara, N.O.; et al. Exercise and Caloric Restriction Alter the Immune System of Mice Submitted to a High-Fat Diet. Mediat. Inflamm. 2013, 2013, e8. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, T.; Das, S.K.; Golden, J.K.; Saltzman, E.; Roberts, S.B.; Meydani, S.N. Calorie restriction enhances T-cell-mediated immune response in adult overweight men and women. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 1107–1113. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Vanegas, S.M.; Du, X.; Noble, T.; Zingg, J.-M.A.; Meydani, M.; Meydani, S.N.; Wu, D. Caloric restriction favorably impacts metabolic and immune/inflammatory profiles in obese mice but curcumin/piperine consumption adds no further benefit. Nutr. Metab. 2013, 10, e29. [Google Scholar] [CrossRef] [PubMed]
- Turbitt, W.J.; Xu, Y.; Sosnoski, D.M.; Collins, S.D.; Meng, H.; Mastro, A.M.; Rogers, C.J. Physical Activity Plus Energy Restriction Prevents 4T1.2 Mammary Tumor Progression, MDSC Accumulation, and an Immunosuppressive Tumor Microenvironment. Cancer Prev. Res. 2019, 12, e493. [Google Scholar] [CrossRef]
- Tang, D.; Tao, S.; Chen, Z.; Koliesnik, I.O.; Calmes, P.G.; Hoerr, V.; Han, B.; Gebert, N.; Zörnig, M.; Löffler, B.; et al. Dietary restriction improves repopulation but impairs lymphoid differentiation capacity of hematopoietic stem cells in early aging. J. Exp. Med. 2016, 213, 535–553. [Google Scholar] [CrossRef]
- Messaoudi, I.; Warner, J.; Fischer, M.; Park, B.; Hill, B.; Mattison, J.; Lane, M.A.; Roth, G.S.; Ingram, D.K.; Picker, L.J.; et al. Delay of T cell senescence by caloric restriction in aged long-lived nonhuman primates. Proc. Natl. Acad. Sci. USA 2006, 103, 19448–19453. [Google Scholar] [CrossRef]
- Lee, J.-E.; Rayyan, M.; Liao, A.; Edery, I.; Pletcher, S.D. Acute Dietary Restriction Acts via TOR, PP2A, and Myc Signaling to Boost Innate Immunity in Drosophila. Cell Rep. 2017, 20, 479–490. [Google Scholar] [CrossRef]
- Ruby, C.E.; Weinberg, A.D. OX40-enhanced tumor rejection and effector T cell differentiation decreases with age. J. Immunol. (Baltimore, Md.: 1950) 2009, 182, 1481–1489. [Google Scholar] [CrossRef]
- Farazi, M.; Nguyen, J.; Goldufsky, J.; Linnane, S.; Lukaesko, L.; Weinberg, A.D.; Ruby, C.E. Caloric restriction maintains OX40 agonist-mediated tumor immunity and CD4 T cell priming during aging. Cancer Immunol. Immunother. 2014, 63, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Fabbiano, S.; Suárez-Zamorano, N.; Rigo, D.; Veyrat-Durebex, C.; Stevanovic Dokic, A.; Colin, D.J.; Trajkovski, M. Caloric Restriction Leads to Browning of White Adipose Tissue through Type 2 Immune Signaling. Cell Metab. 2016, 24, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Flint, T.R.; Janowitz, T.; Connell, C.M.; Roberts, E.W.; Denton, A.E.; Coll, A.P.; Jodrell, D.I.; Fearon, D.T. Tumor-Induced IL-6 Reprograms Host Metabolism to Suppress Anti-tumor Immunity. Cell Metab. 2016, 24, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Gudny Geirsdottir, O.; Thorsdottir, I. Nutritional status of cancer patients in chemotherapy; dietary intake, nitrogen balance and screening. Food Nutr. Res. 2008, 52, e1856. [Google Scholar] [CrossRef]
- O’Flanagan, C.H.; Smith, L.A.; McDonell, S.B.; Hursting, S.D. When less may be more: Calorie restriction and response to cancer therapy. BMC Med. 2017, 15, e106. [Google Scholar] [CrossRef]
- Gerriets, V.A.; MacIver, N.J. Role of T cells in malnutrition and obesity. Front. Immunol. 2014, 5, e379. [Google Scholar] [CrossRef]
- Nishioka, N.; Uchino, J.; Hirai, S.; Katayama, Y.; Yoshimura, A.; Okura, N.; Tanimura, K.; Harita, S.; Imabayashi, T.; Chihara, Y.; et al. Association of Sarcopenia with and Efficacy of Anti-PD-1/PD-L1 Therapy in Non-Small-Cell Lung Cancer. J. Clin. Med. 2019, 8, e450. [Google Scholar] [CrossRef]
- Shiroyama, T.; Nagatomo, I.; Koyama, S.; Hirata, H.; Nishida, S.; Miyake, K.; Fukushima, K.; Shirai, Y.; Mitsui, Y.; Takata, S.; et al. Impact of sarcopenia in patients with advanced non–small cell lung cancer treated with PD-1 inhibitors: A preliminary retrospective study. Sci. Rep. 2019, 9, e2447. [Google Scholar] [CrossRef]
- Turner, D.C.; Kondic, A.G.; Anderson, K.M.; Robinson, A.G.; Garon, E.B.; Riess, J.W.; Jain, L.; Mayawala, K.; Kang, J.; Ebbinghaus, S.W.; et al. Pembrolizumab Exposure–Response Assessments Challenged by Association of Cancer Cachexia and Catabolic Clearance. Clin. Cancer Res. 2018, 24, e5841. [Google Scholar] [CrossRef]
- Sun, P.; Wang, H.; He, Z.; Chen, X.; Wu, Q.; Chen, W.; Sun, Z.; Weng, M.; Zhu, M.; Ma, D.; et al. Fasting inhibits colorectal cancer growth by reducing M2 polarization of tumor-associated macrophages. Oncotarget 2017, 8, 74649–74660. [Google Scholar] [CrossRef]
- Di Biase, S.; Lee, C.; Brandhorst, S.; Manes, B.; Buono, R.; Cheng, C.-W.; Cacciottolo, M.; Martin-Montalvo, A.; de Cabo, R.; Wei, M.; et al. Fasting-Mimicking Diet Reduces HO-1 to Promote T Cell-Mediated Tumor Cytotoxicity. Cancer Cell 2016, 30, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Lévesque, S.; Le Naour, J.; Pietrocola, F.; Paillet, J.; Kremer, M.; Castoldi, F.; Baracco, E.E.; Wang, Y.; Vacchelli, E.; Stoll, G.; et al. A synergistic triad of chemotherapy, immune checkpoint inhibitors, and caloric restriction mimetics eradicates tumors in mice. OncoImmunology 2019, 8, e1657375. [Google Scholar]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coleman, M.F.; Cozzo, A.J.; Pfeil, A.J.; Etigunta, S.K.; Hursting, S.D. Cell Intrinsic and Systemic Metabolism in Tumor Immunity and Immunotherapy. Cancers 2020, 12, 852. https://doi.org/10.3390/cancers12040852
Coleman MF, Cozzo AJ, Pfeil AJ, Etigunta SK, Hursting SD. Cell Intrinsic and Systemic Metabolism in Tumor Immunity and Immunotherapy. Cancers. 2020; 12(4):852. https://doi.org/10.3390/cancers12040852
Chicago/Turabian StyleColeman, Michael F., Alyssa J. Cozzo, Alexander J. Pfeil, Suhas K. Etigunta, and Stephen D. Hursting. 2020. "Cell Intrinsic and Systemic Metabolism in Tumor Immunity and Immunotherapy" Cancers 12, no. 4: 852. https://doi.org/10.3390/cancers12040852
APA StyleColeman, M. F., Cozzo, A. J., Pfeil, A. J., Etigunta, S. K., & Hursting, S. D. (2020). Cell Intrinsic and Systemic Metabolism in Tumor Immunity and Immunotherapy. Cancers, 12(4), 852. https://doi.org/10.3390/cancers12040852