The Core-Clock Gene NR1D1 Impacts Cell Motility In Vitro and Invasiveness in a Zebrafish Xenograft Colon Cancer Model

 ,
,  , , ,
, , ,  and
and 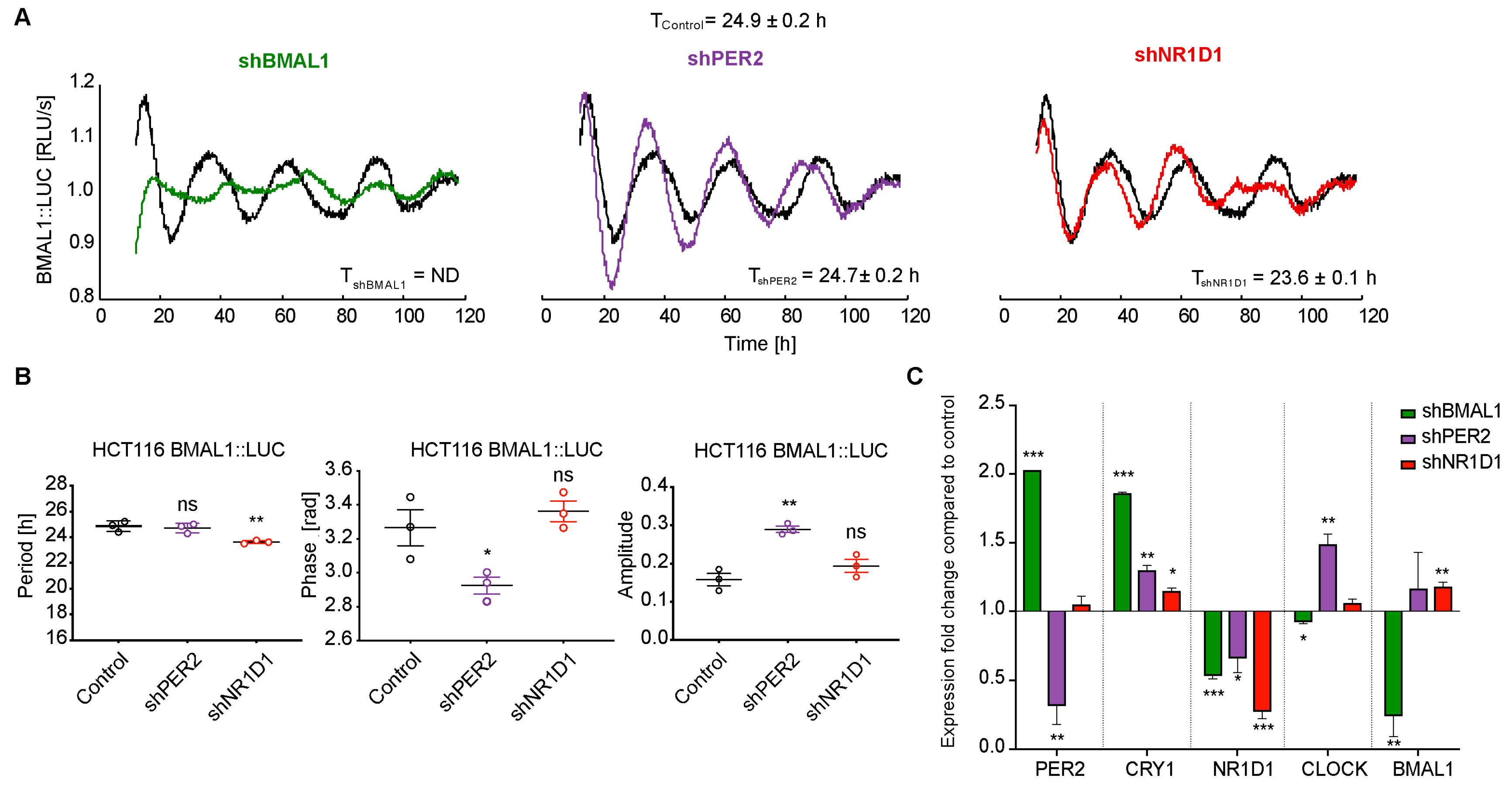{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. The Knockdown of Core-Clock Genes Affects the Oscillatory Phenotype of HCT116 Cells In Vitro
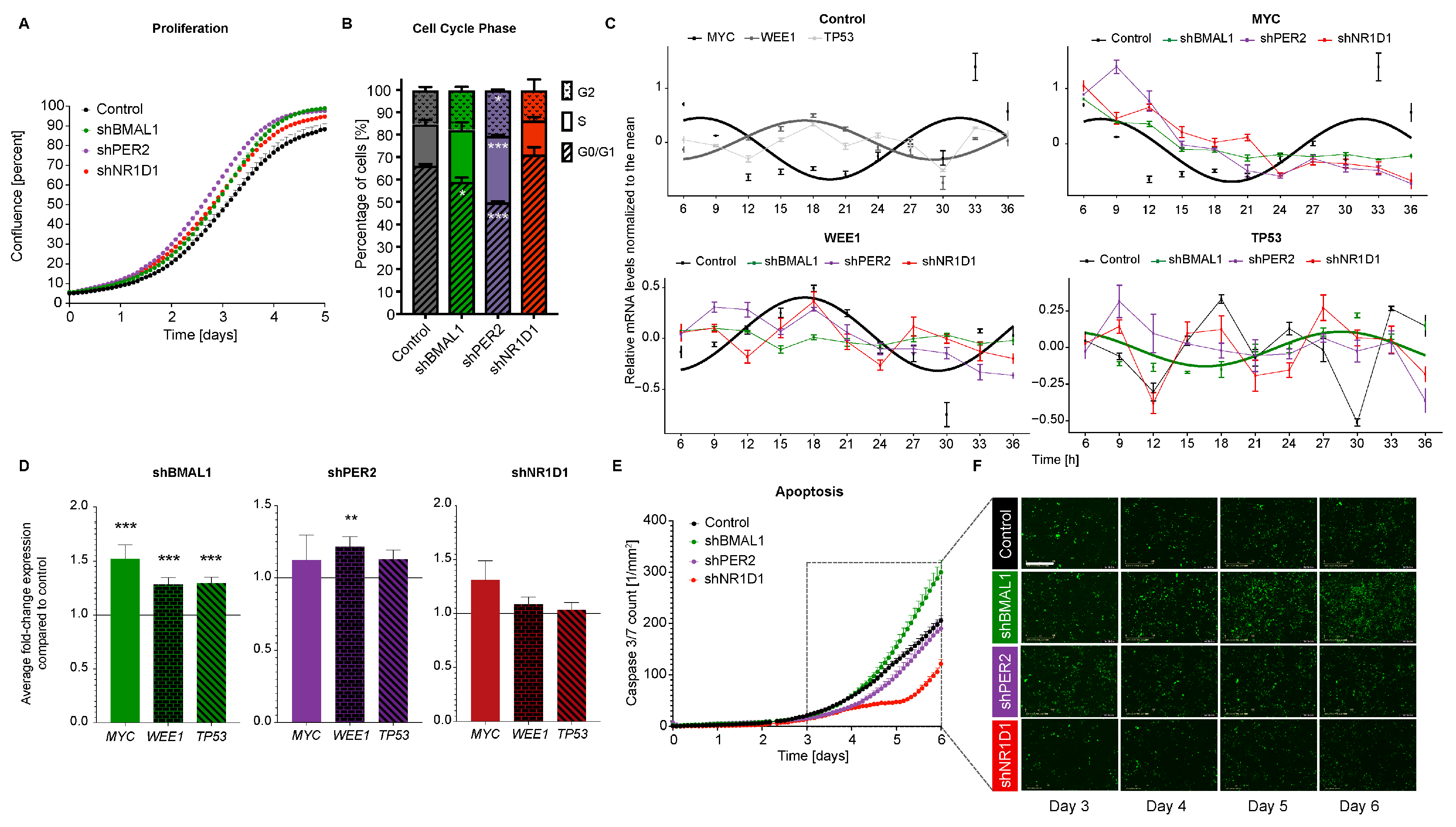
2.2. The Downregulation of Core-Clock Genes Impacts Cell Proliferation, Apoptosis and Migration in CRC Cell Lines
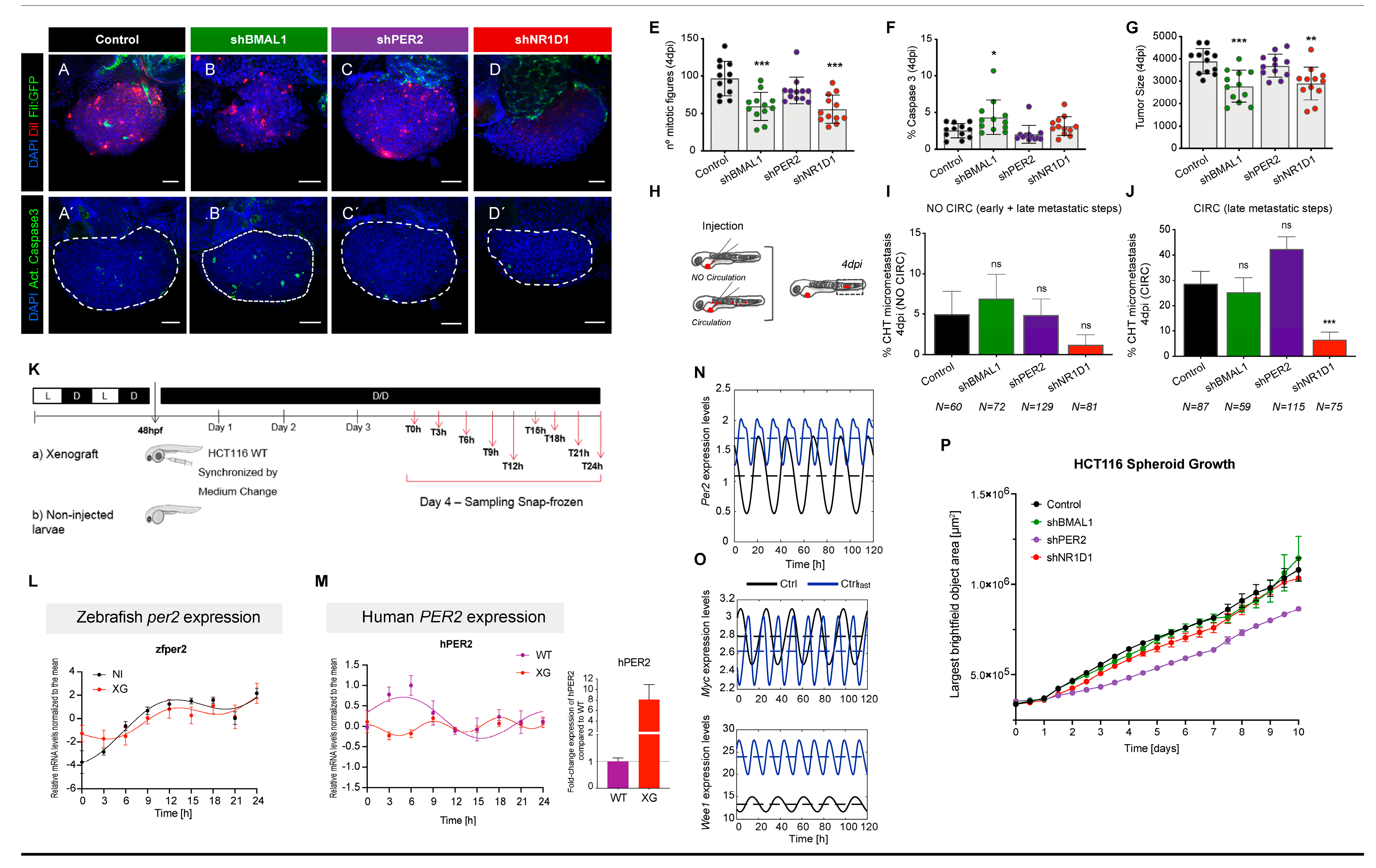
2.3. Downregulation of BMAL1 and NR1D1 Impairs Tumorigenesis In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Lentivirus Production
4.3. Transduction with Lentiviral Vectors
4.4. Bioluminescence Measurements
4.5. Animal Care and Handling
4.6. Cell Labelling
4.7. Zebrafish Xenograft Microinjection
4.8. Micrometastasis Assay
4.9. Immunofluorescence
4.10. Imaging Processing and Analysis
4.11. RNA Extraction, cDNA Synthesis (Reverse Transcription) and Quantitative Real-Time PCR (qPCR)
4.12. Cell Cycle Assay
4.13. Proliferation, Apoptosis, and Migration Measurements
4.13.1. Proliferation Assays
4.13.2. Apoptosis Assays
4.13.3. Migration Assays
4.13.4. 3D Spheroid Assay
4.14. Rhythmicity Analysis of Time-Course Data
4.14.1. Harmonic Regression Using R
4.14.2. Cosinor Analysis
4.15. Statistical Analysis
4.16. Mathematical Modelling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Cederroth, C.R.; Albrecht, U.; Bass, J.; Brown, S.A.; Dyhrfjeld-Johnsen, J.; Gachon, F.; Green, C.B.; Hastings, M.H.; Helfrich-Forster, C.; Hogenesch, J.B.; et al. Medicine in the Fourth Dimension. Cell Metab. 2019, 30, 238–250. [Google Scholar] [CrossRef]
- Lowrey, P.L.; Takahashi, J.S. Genetics of circadian rhythms in Mammalian model organisms. Adv. Genet. 2011, 74, 175–230. [Google Scholar] [CrossRef]
- Albrecht, U. Timing to perfection: The biology of central and peripheral circadian clocks. Neuron 2012, 74, 246–260. [Google Scholar] [CrossRef]
- Bass, J. Circadian topology of metabolism. Nature 2012, 491, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Kondratova, A.A.; Kondratov, R.V. The circadian clock and pathology of the ageing brain. Nat. Rev. Neurosci. 2012, 13, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, J.S.; Hong, H.-K.; Ko, C.H.; McDearmon, E.L. The genetics of mammalian circadian order and disorder: Implications for physiology and disease. Nat. Rev. Genet. 2008, 9, 764–775. [Google Scholar] [CrossRef]
- Levi, F.; Schibler, U. Circadian rhythms: Mechanisms and therapeutic implications. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 593–628. [Google Scholar] [CrossRef] [PubMed]
- Sulli, G.; Lam, M.T.Y.; Panda, S. Interplay between Circadian Clock and Cancer: New Frontiers for Cancer Treatment. Trends Cancer 2019, 5, 475–494. [Google Scholar] [CrossRef]
- Ballesta, A.; Innominato, P.F.; Dallmann, R.; Rand, D.A.; Levi, F.A. Systems Chronotherapeutics. Pharmacol. Rev. 2017, 69, 161–199. [Google Scholar] [CrossRef]
- Lehmann, R.; Childs, L.; Thomas, P.; Abreu, M.; Fuhr, L.; Herzel, H.; Leser, U.; Relogio, A. Assembly of a comprehensive regulatory network for the mammalian circadian clock: A bioinformatics approach. PLoS ONE 2015, 10, e0126283. [Google Scholar] [CrossRef]
- Deb, S.P.; Singh, S.; Deb, S. MDM2 overexpression, activation of signaling networks, and cell proliferation. Subcell. Biochem. 2014, 85, 215–234. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Mass, R.D.; Campa, C.; Kim, R. Targeting VEGF-A to treat cancer and age-related macular degeneration. Annu. Rev. Med. 2007, 58, 491–504. [Google Scholar] [CrossRef]
- Feuers, R.J.; Casciano, D.A.; Tsai, T.H.; Scheving, L.E. Regulation of the circadian rhythm of hepatic pyruvate kinase in mice. Prog. Clin. Biol. Res. 1987, 227A, 163–172. [Google Scholar]
- Fuhr, L.; El-Athman, R.; Scrima, R.; Cela, O.; Carbone, A.; Knoop, H.; Li, Y.; Hoffmann, K.; Laukkanen, M.O.; Corcione, F.; et al. The Circadian Clock Regulates Metabolic Phenotype Rewiring Via HKDC1 and Modulates Tumor Progression and Drug Response in Colorectal Cancer. EBioMedicine 2018, 33, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Fuhr, L.; Abreu, M.; Pett, P.; Relogio, A. Circadian systems biology: When time matters. Comput. Struct. Biotechnol. J. 2015, 13, 417–426. [Google Scholar] [CrossRef] [PubMed]
- El-Athman, R.; Genov, N.N.; Mazuch, J.; Zhang, K.; Yu, Y.; Fuhr, L.; Abreu, M.; Li, Y.; Wallach, T.; Kramer, A.; et al. The Ink4a/Arf locus operates as a regulator of the circadian clock modulating RAS activity. PLoS Biol. 2017, 15, e2002940. [Google Scholar] [CrossRef] [PubMed]
- Gaucher, J.; Montellier, E.; Sassone-Corsi, P. Molecular Cogs: Interplay between Circadian Clock and Cell Cycle. Trends Cell Biol. 2018, 28, 368–379. [Google Scholar] [CrossRef]
- Bieler, J.; Cannavo, R.; Gustafson, K.; Gobet, C.; Gatfield, D.; Naef, F. Robust synchronization of coupled circadian and cell cycle oscillators in single mammalian cells. Mol. Syst. Biol. 2014, 10, 739. [Google Scholar] [CrossRef]
- Sulli, G.; Rommel, A.; Wang, X.; Kolar, M.J.; Puca, F.; Saghatelian, A.; Plikus, M.V.; Verma, I.M.; Panda, S. Pharmacological activation of REV-ERBs is lethal in cancer and oncogene-induced senescence. Nature 2018, 553, 351–355. [Google Scholar] [CrossRef]
- Gery, S.; Komatsu, N.; Baldjyan, L.; Yu, A.; Koo, D.; Koeffler, H.P. The circadian gene per1 plays an important role in cell growth and DNA damage control in human cancer cells. Mol. Cell 2006, 22, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Miki, T.; Matsumoto, T.; Zhao, Z.; Lee, C.C. p53 regulates Period2 expression and the circadian clock. Nat. Commun. 2013, 4, 2444. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Fang, D. The Roles of SIRT1 in Cancer. Genes Cancer 2013, 4, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Fior, R.; Povoa, V.; Mendes, R.V.; Carvalho, T.; Gomes, A.; Figueiredo, N.; Ferreira, M.G. Single-cell functional and chemosensitive profiling of combinatorial colorectal therapy in zebrafish xenografts. Proc. Natl. Acad. Sci. USA 2017, 114, E8234–E8243. [Google Scholar] [CrossRef] [PubMed]
- Relogio, A.; Thomas, P.; Medina-Perez, P.; Reischl, S.; Bervoets, S.; Gloc, E.; Riemer, P.; Mang-Fatehi, S.; Maier, B.; Schafer, R.; et al. Ras-mediated deregulation of the circadian clock in cancer. PLoS Genet. 2014, 10, e1004338. [Google Scholar] [CrossRef] [PubMed]
- Levi, F. From circadian rhythms to cancer chronotherapeutics. Chronobiol. Int. 2002, 19, 1–19. [Google Scholar] [CrossRef]
- Dallmann, R.; Okyar, A.; Levi, F. Dosing-Time Makes the Poison: Circadian Regulation and Pharmacotherapy. Trends Mol. Med. 2016, 22, 430–445. [Google Scholar] [CrossRef]
- Sporl, F.; Schellenberg, K.; Blatt, T.; Wenck, H.; Wittern, K.P.; Schrader, A.; Kramer, A. A circadian clock in HaCaT keratinocytes. J. Investig. Dermatol. 2011, 131, 338–348. [Google Scholar] [CrossRef]
- Zeng, Z.L.; Luo, H.Y.; Yang, J.; Wu, W.J.; Chen, D.L.; Huang, P.; Xu, R.H. Overexpression of the circadian clock gene Bmal1 increases sensitivity to oxaliplatin in colorectal cancer. Clin. Cancer Res. 2014, 20, 1042–1052. [Google Scholar] [CrossRef]
- Wood, P.A.; Yang, X.; Taber, A.; Oh, E.Y.; Ansell, C.; Ayers, S.E.; Al-Assaad, Z.; Carnevale, K.; Berger, F.G.; Pena, M.M.; et al. Period 2 mutation accelerates ApcMin/+ tumorigenesis. Mol. Cancer Res. 2008, 6, 1786–1793. [Google Scholar] [CrossRef]
- Ray, S.; Valekunja, U.K.; Stangherlin, A.; Howell, S.A.; Snijders, A.P.; Damodaran, G.; Reddy, A.B. Circadian rhythms in the absence of the clock gene Bmal1. Science 2020, 367, 800–806. [Google Scholar] [CrossRef]
- Gotoh, T.; Kim, J.K.; Liu, J.; Vila-Caballer, M.; Stauffer, P.E.; Tyson, J.J.; Finkielstein, C.V. Model-driven experimental approach reveals the complex regulatory distribution of p53 by the circadian factor Period 2. Proc. Natl. Acad. Sci. USA 2016, 113, 13516–13521. [Google Scholar] [CrossRef] [PubMed]
- Vatine, G.; Vallone, D.; Gothilf, Y.; Foulkes, N.S. It’s time to swim! Zebrafish and the circadian clock. FEBS Lett. 2011, 585, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Shostak, A.; Ruppert, B.; Ha, N.; Bruns, P.; Toprak, U.H.; Project, I.M.-S.; Eils, R.; Schlesner, M.; Diernfellner, A.; Brunner, M. MYC/MIZ1-dependent gene repression inversely coordinates the circadian clock with cell cycle and proliferation. Nat. Commun. 2016, 7, 11807. [Google Scholar] [CrossRef]
- Fuhr, L.; Abreu, M.; Carbone, A.; El-Athman, R.; Bianchi, F.; Laukkanen, M.O.; Mazzoccoli, G.; Relogio, A. The Interplay between Colon Cancer Cells and Tumour-Associated Stromal Cells Impacts the Biological Clock and Enhances Malignant Phenotypes. Cancers 2019, 11, 988. [Google Scholar] [CrossRef] [PubMed]
- Luck, S.; Thurley, K.; Thaben, P.F.; Westermark, P.O. Rhythmic degradation explains and unifies circadian transcriptome and proteome data. Cell Rep. 2014, 9, 741–751. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basti, A.; Fior, R.; Yalҫin, M.; Póvoa, V.; Astaburuaga, R.; Li, Y.; Naderi, J.; Godinho Ferreira, M.; Relógio, A. The Core-Clock Gene NR1D1 Impacts Cell Motility In Vitro and Invasiveness in a Zebrafish Xenograft Colon Cancer Model. Cancers 2020, 12, 853. https://doi.org/10.3390/cancers12040853
Basti A, Fior R, Yalҫin M, Póvoa V, Astaburuaga R, Li Y, Naderi J, Godinho Ferreira M, Relógio A. The Core-Clock Gene NR1D1 Impacts Cell Motility In Vitro and Invasiveness in a Zebrafish Xenograft Colon Cancer Model. Cancers. 2020; 12(4):853. https://doi.org/10.3390/cancers12040853
Chicago/Turabian StyleBasti, Alireza, Rita Fior, Müge Yalҫin, Vanda Póvoa, Rosario Astaburuaga, Yin Li, Julian Naderi, Miguel Godinho Ferreira, and Angela Relógio. 2020. "The Core-Clock Gene NR1D1 Impacts Cell Motility In Vitro and Invasiveness in a Zebrafish Xenograft Colon Cancer Model" Cancers 12, no. 4: 853. https://doi.org/10.3390/cancers12040853
APA StyleBasti, A., Fior, R., Yalҫin, M., Póvoa, V., Astaburuaga, R., Li, Y., Naderi, J., Godinho Ferreira, M., & Relógio, A. (2020). The Core-Clock Gene NR1D1 Impacts Cell Motility In Vitro and Invasiveness in a Zebrafish Xenograft Colon Cancer Model. Cancers, 12(4), 853. https://doi.org/10.3390/cancers12040853