Exploring the Role of Mutations in Fanconi Anemia Genes in Hereditary Cancer Patients

 , , , ,
, , , ,  , , ,
, , , 
Abstract
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Patients and Controls
4.2. DNA Isolation
4.3. NGS Panel Testing
4.4. RNA Analysis
4.5. GnomAD Analysis
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mamrak, N.E.; Shimamura, A.; Howlett, N.G. Recent discoveries in the molecular pathogenesis of the inherited bone marrow failure syndrome Fanconi anemia. Blood Rev. 2017, 31, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Asur, R.S.; Kimble, D.C.; Lach, F.P.; Jung, M.; Donovan, F.X.; Kamat, A.; Noonan, R.J.; Thomas, J.W.; Park, M.; Chines, P.; et al. Somatic mosaicism of an intragenic FANCB duplication in both fibroblast and peripheral blood cells observed in a Fanconi anemia patient leads to milder phenotype. Mol. Genet. Genom. Med. 2018, 6, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Kottemann, M.C.; Smogorzewska, A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 2013, 493, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Bogliolo, M.; Surrallés, J. Fanconi anemia: A model disease for studies on human genetics and advanced therapeutics. Curr. Opin. Genet. Dev. 2015, 33, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Catucci, I.; Osorio, A.; Arver, B.; Neidhardt, G.; Bogliolo, M.; Zanardi, F.; Riboni, M.; Minardi, S.; Pujol, R.; Azzollini, J.; et al. Individuals with FANCM biallelic mutations do not develop Fanconi anemia, but show risk for breast cancer, chemotherapy toxicity and may display chromosome fragility. Genet. Med. 2018, 20, 452–457. [Google Scholar] [CrossRef]
- Daly, M.B.; Pilarski, R.; Berry, M.; Buys, S.S.; Farmer, M.; Friedman, S.; Garber, J.E.; Kauff, N.D.; Khan, S.; Klein, C.; et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast and Ovarian, Version 2. J. Natl. Compr. Cancer Netw. 2017, 15, 9–20. [Google Scholar] [CrossRef]
- Gracia-Aznarez, F.J.; Fernandez, V.; Pita, G.; Peterlongo, P.; Dominguez, O.; de la Hoya, M.; Duran, M.; Osorio, A.; Moreno, L.; Gonzalez-Neira, A.; et al. Whole exome sequencing suggests much of non-BRCA1/BRCA2 familial breast cancer is due to moderate and low penetrance susceptibility alleles. PLoS ONE 2013, 8, e55681. [Google Scholar] [CrossRef]
- Kiiski, J.I.; Pelttari, L.M.; Khan, S.; Freysteinsdottir, E.S.; Reynisdottir, I.; Hart, S.N.; Shimelis, H.; Vilske, S.; Kallioniemi, A.; Schleutker, J.; et al. Exome sequencing identifies FANCM as a susceptibility gene for triple-negative breast cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 15172–15177. [Google Scholar] [CrossRef]
- Peterlongo, P.; Catucci, I.; Colombo, M.; Caleca, L.; Mucaki, E.; Bogliolo, M.; Marin, M.; Damiola, F.; Bernard, L.; Pensotti, V.; et al. FANCM c.5791C > T nonsense mutation (rs144567652) induces exon skipping, affects DNA repair activity and is a familial breast cancer risk factor. Hum. Mol. Genet. 2015, 24, 5345–5355. [Google Scholar] [CrossRef]
- Neidhardt, G.; Hauke, J.; Ramser, J.; Groß, E.; Gehrig, A.; Müller, C.R.; Kahlert, A.K.; Hackmann, K.; Honisch, E.; Niederacher, D.; et al. Association Between Loss-of-Function Mutations Within the FANCM Gene and Early-Onset Familial Breast Cancer. JAMA Oncol. 2017, 3, 1245–1248. [Google Scholar] [CrossRef]
- Dicks, E.; Song, H.; Ramus, S.J.; Oudenhove, E.V.; Tyrer, J.P.; Intermaggio, M.P.; Kar, S.; Harrington, P.; Bowtell, D.D.; Group, A.S.; et al. Germline whole exome sequencing and large-scale replication identifies. Oncotarget 2017, 8, 50930–50940. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Dumont, T.; Myszka, A.; Karpinski, P.; Sasiadek, M.M.; Akopyan, H.; Hammet, F.; Tsimiklis, H.; Park, D.J.; Pope, B.J.; Slezak, R.; et al. FANCM and RECQL genetic variants and breast cancer susceptibility: Relevance to South Poland and West Ukraine. BMC Med. Genet. 2018, 19, 12. [Google Scholar] [CrossRef] [PubMed]
- Schubert, S.; van Luttikhuizen, J.L.; Auber, B.; Schmidt, G.; Hofmann, W.; Penkert, J.; Davenport, C.F.; Hille-Betz, U.; Wendeburg, L.; Bublitz, J.; et al. The identification of pathogenic variants in BRCA1/2 negative, high risk, hereditary breast and/or ovarian cancer patients: High frequency of FANCM pathogenic variants. Int. J. Cancer 2019, 144, 2683–2694. [Google Scholar] [CrossRef] [PubMed]
- Nurmi, A.; Muranen, T.A.; Pelttari, L.M.; Kiiski, J.I.; Heikkinen, T.; Lehto, S.; Kallioniemi, A.; Schleutker, J.; Bützow, R.; Blomqvist, C.; et al. Recurrent moderate-risk mutations in Finnish breast and ovarian cancer patients. Int. J. Cancer 2019, 145, 2692–2700. [Google Scholar] [CrossRef] [PubMed]
- Figlioli, G.; Bogliolo, M.; Catucci, I.; Caleca, L.; Lasheras, S.V.; Pujol, R.; Kiiski, J.I.; Muranen, T.A.; Barnes, D.R.; Dennis, J.; et al. The FANCM:p.Arg658* truncating variant is associated with risk of triple-negative breast cancer. NPJ Breast Cancer 2019, 5, 38. [Google Scholar] [CrossRef] [PubMed]
- Seal, S.; Barfoot, R.; Jayatilake, H.; Smith, P.; Renwick, A.; Bascombe, L.; McGuffog, L.; Evans, D.G.; Eccles, D.; Easton, D.F.; et al. Evaluation of Fanconi Anemia genes in familial breast cancer predisposition. Cancer Res. 2003, 63, 8596–8599. [Google Scholar]
- Haiman, C.A.; Hsu, C.; de Bakker, P.I.; Frasco, M.; Sheng, X.; Van Den Berg, D.; Casagrande, J.T.; Kolonel, L.N.; Le Marchand, L.; Hankinson, S.E.; et al. Comprehensive association testing of common genetic variation in DNA repair pathway genes in relationship with breast cancer risk in multiple populations. Hum. Mol. Genet. 2008, 17, 825–834. [Google Scholar] [CrossRef]
- Solyom, S.; Winqvist, R.; Nikkilä, J.; Rapakko, K.; Hirvikoski, P.; Kokkonen, H.; Pylkäs, K. Screening for large genomic rearrangements in the FANCA gene reveals extensive deletion in a Finnish breast cancer family. Cancer Lett. 2011, 302, 113–118. [Google Scholar] [CrossRef]
- Abbasi, S.; Rasouli, M. A rare FANCA gene variation as a breast cancer susceptibility allele in an Iranian population. Mol. Med. Rep. 2017, 15, 3983–3988. [Google Scholar] [CrossRef]
- van der Heijden, M.S.; Yeo, C.J.; Hruban, R.H.; Kern, S.E. Fanconi anemia gene mutations in young-onset pancreatic cancer. Cancer Res. 2003, 63, 2585–2588. [Google Scholar]
- Couch, F.J.; Johnson, M.R.; Rabe, K.; Boardman, L.; McWilliams, R.; de Andrade, M.; Petersen, G. Germ line Fanconi anemia complementation group C mutations and pancreatic cancer. Cancer Res. 2005, 65, 383–386. [Google Scholar] [PubMed]
- Berwick, M.; Satagopan, J.M.; Ben-Porat, L.; Carlson, A.; Mah, K.; Henry, R.; Diotti, R.; Milton, K.; Pujara, K.; Landers, T.; et al. Genetic heterogeneity among Fanconi anemia heterozygotes and risk of cancer. Cancer Res. 2007, 67, 9591–9596. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.R.; Doyle, M.A.; Ryland, G.L.; Rowley, S.M.; Choong, D.Y.; Tothill, R.W.; Thorne, H.; Barnes, D.R.; Li, J.; Ellul, J.; et al. Exome sequencing identifies rare deleterious mutations in DNA repair genes FANCC and BLM as potential breast cancer susceptibility alleles. PLoS Genet. 2012, 8, e1002894. [Google Scholar] [CrossRef] [PubMed]
- Dörk, T.; Peterlongo, P.; Mannermaa, A.; Bolla, M.K.; Wang, Q.; Dennis, J.; Ahearn, T.; Andrulis, I.L.; Anton-Culver, H.; Arndt, V.; et al. Two truncating variants in FANCC and breast cancer risk. Sci. Rep. 2019, 9, 12524. [Google Scholar] [CrossRef] [PubMed]
- Bakker, J.L.; van Mil, S.E.; Crossan, G.; Sabbaghian, N.; De Leeneer, K.; Poppe, B.; Adank, M.; Gille, H.; Verheul, H.; Meijers-Heijboer, H.; et al. Analysis of the novel fanconi anemia gene SLX4/FANCP in familial breast cancer cases. Hum. Mutat. 2013, 34, 70–73. [Google Scholar] [CrossRef]
- de Garibay, G.R.; Díaz, A.; Gaviña, B.; Romero, A.; Garre, P.; Vega, A.; Blanco, A.; Tosar, A.; Díez, O.; Pérez-Segura, P.; et al. Low prevalence of SLX4 loss-of-function mutations in non-BRCA1/2 breast and/or ovarian cancer families. Eur. J. Hum. Genet. 2013, 21, 883–886. [Google Scholar] [CrossRef][Green Version]
- Shah, S.; Kim, Y.; Ostrovnaya, I.; Murali, R.; Schrader, K.A.; Lach, F.P.; Sarrel, K.; Rau-Murthy, R.; Hansen, N.; Zhang, L.; et al. Assessment of SLX4 Mutations in Hereditary Breast Cancers. PLoS ONE 2013, 8, e66961. [Google Scholar] [CrossRef]
- Park, D.J.; Lesueur, F.; Nguyen-Dumont, T.; Pertesi, M.; Odefrey, F.; Hammet, F.; Neuhausen, S.L.; John, E.M.; Andrulis, I.L.; Terry, M.B.; et al. Rare mutations in XRCC2 increase the risk of breast cancer. Am. J. Hum. Genet. 2012, 90, 734–739. [Google Scholar] [CrossRef]
- Hilbers, F.S.; Wijnen, J.T.; Hoogerbrugge, N.; Oosterwijk, J.C.; Collee, M.J.; Peterlongo, P.; Radice, P.; Manoukian, S.; Feroce, I.; Capra, F.; et al. Rare variants in XRCC2 as breast cancer susceptibility alleles. J. Med. Genet. 2012, 49, 618–620. [Google Scholar] [CrossRef]
- Kluźniak, W.; Wokołorczyk, D.; Rusak, B.; Huzarski, T.; Gronwald, J.; Stempa, K.; Rudnicka, H.; Kashyap, A.; Dębniak, T.; Jakubowska, A.; et al. Inherited variants in XRCC2 and the risk of breast cancer. Breast Cancer Res. Treat. 2019, 178, 657–663. [Google Scholar] [CrossRef]
- Castellanos, E.; Gel, B.; Rosas, I.; Tornero, E.; Santín, S.; Pluvinet, R.; Velasco, J.; Sumoy, L.; Del Valle, J.; Perucho, M.; et al. A comprehensive custom panel design for routine hereditary cancer testing: Preserving control, improving diagnostics and revealing a complex variation landscape. Sci. Rep. 2017, 7, 39348. [Google Scholar] [CrossRef] [PubMed]
- Feliubadalo, L.; Lopez-Fernandez, A.; Pineda, M.; Diez, O.; Del Valle, J.; Gutierrez-Enriquez, S.; Teule, A.; Gonzalez, S.; Stjepanovic, N.; Salinas, M.; et al. Opportunistic testing of BRCA1, BRCA2 and mismatch repair genes improves the yield of phenotype driven hereditary cancer gene panels. Int. J. Cancer 2019, 145, 2682–2691. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.M.; Kirby, M.; Jansen, M.; Lach, F.P.; Schulte, J.; Singh, T.R.; Batish, S.D.; Auerbach, A.D.; Williams, D.A.; Meetei, A.R. Identification and characterization of mutations in FANCL gene: A second case of Fanconi anemia belonging to FA-L complementation group. Hum. Mutat. 2009, 30, E761–E770. [Google Scholar] [CrossRef] [PubMed]
- Figlioli, G.; Kvist, A.; Tham, E.; Soukupova, J.; Kleiblova, P.; Muranen, T.A.; Andrieu, N.; Azzollini, J.; Balmaña, J.; Barroso, A.; et al. The Spectrum of FANCM Protein Truncating Variants in European Breast Cancer Cases. Cancers (Basel) 2020, 12, 292. [Google Scholar] [CrossRef] [PubMed]
- Obón-Santacana, M.; Vilardell, M.; Carreras, A.; Duran, X.; Velasco, J.; Galván-Femenía, I.; Alonso, T.; Puig, L.; Sumoy, L.; Duell, E.J.; et al. GCAT|Genomes for life: A prospective cohort study of the genomes of Catalonia. BMJ Open 2018, 8, e018324. [Google Scholar] [CrossRef]
- Fowler, A.; Mahamdallie, S.; Ruark, E.; Seal, S.; Ramsay, E.; Clarke, M.; Uddin, I.; Wylie, H.; Strydom, A.; Lunter, G.; et al. Accurate clinical detection of exon copy number variants in a targeted NGS panel using DECoN. Wellcome Open Res. 2016, 1, 20. [Google Scholar] [CrossRef]


{kind=link}
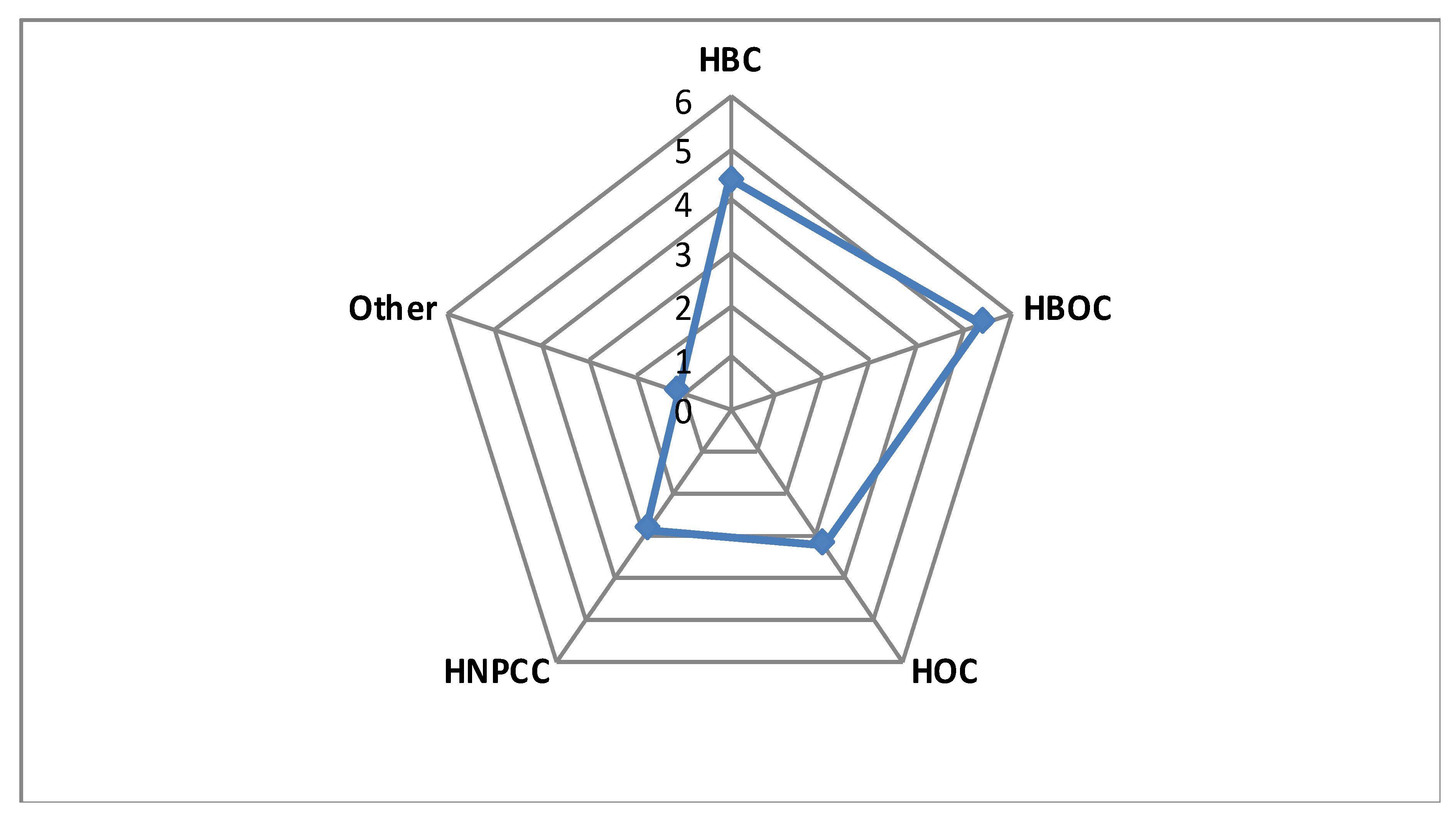
| Clinical Suspicion | GCAT Women Cohort (n = 100) | GnomAD European >23,000 women β | Study Cohort Versus NFE γ, Non-Cancer GnomAD (OR/95%CI/p-Value) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Genes | Pathogenic Variants | Breast (HBC) | Ovary (HOC) | Breast + Ovary (HBOC) | HNPCC α | Other | All Patients | HBC + HOC + HBOC | HBC + HBOC | ||
| FANCA | 10 | 7 | 0 | 2 | 1 | 0 | 3 | 147 | 1.94/0.91–3.7/0.047 | 2.34/1.04–4.59/0.02 | 3.14/1.4–6.17/0.003* |
| FANCL | 8 | 3 | 1 | 3 | 1 | 0 | 1 | 187 | 1.22/0.52–2.46/0.549 | 1.42/0.56–3/0.356 | 1.63/0.59–3.64/0.283 |
| FANCM | 6 | 2 | 3 | 0 | 1 | 0 | 0 | 159 | 1.07/0.38–2.39/0.828 | 1.19/0.38–2.85/0.618 | 0.63/0.08–2.34/0.774 |
| FANCI | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 25 | 1.14/0.03–7/0.593 | 1.12/0,04–9.29/0.492 | 2.02/0.05–12.4/0.399 |
| FANCE | 2 | 1 | 0 | 0 | 1 | 0 | 0 | 17 | 1.14/0.03–6.97/0.593 | 1.52/0.04–9.3/0.492 | 2.03/0.05–12.4/0.399 |
| FANCC | 2 | 1 | 1 | 0 | 0 | 0 | 0 | 44 | 1.29/0.15–4.97/0.67 | 1.72/0.2–6.63/0.332 | 1.15/0.03–6.78/0.586 |
| FANCF | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 26 | 1.09/0.02–6.6/0.608 | 1.45/0.04–8.88/0.506 | 1.94/0.05–11.87/0.412 |
| RAD51 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 4 | 7.12/0.14–72/0.159 | 9.49/0.19–96/0.122 | 12.7/ 0.26–128/0.093 |
| SLX4 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 36 | NA | NA | NA |
| ERCC4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 22 | NA | NA | NA |
| FANCB | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NA | NA | NA |
| FANCD2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 21 | NA | NA | NA |
| FANCG | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 43 | NA | NA | NA |
| XRCC2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 22 | NA | NA | NA |
| TOTAL | 31 | 17 | 5 | 5 | 4 | 0 | 5 | 753 | |||
| Clinical Suspicion | Number of Patients (Women) |
|---|---|
| Hereditary breast cancer, HBC | 385 (370) |
| Hereditary non-polyposis colon cancer, HNPCC | 210 (130) |
| Hereditary ovarian cancer, HOC | 154 (154) |
| Other hereditary cancer conditions | 102 (55) |
| Hereditary breast and ovarian cancer, HBOC | 93 (90) |
| Familial (attenuated) adenomatous polyposis, FAP/AFAP | 77 (19) |
| Total | 1021 (818) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
del Valle, J.; Rofes, P.; Moreno-Cabrera, J.M.; López-Dóriga, A.; Belhadj, S.; Vargas-Parra, G.; Teulé, À.; Cuesta, R.; Muñoz, X.; Campos, O.; et al. Exploring the Role of Mutations in Fanconi Anemia Genes in Hereditary Cancer Patients. Cancers 2020, 12, 829. https://doi.org/10.3390/cancers12040829
del Valle J, Rofes P, Moreno-Cabrera JM, López-Dóriga A, Belhadj S, Vargas-Parra G, Teulé À, Cuesta R, Muñoz X, Campos O, et al. Exploring the Role of Mutations in Fanconi Anemia Genes in Hereditary Cancer Patients. Cancers. 2020; 12(4):829. https://doi.org/10.3390/cancers12040829
Chicago/Turabian Styledel Valle, Jesús, Paula Rofes, José Marcos Moreno-Cabrera, Adriana López-Dóriga, Sami Belhadj, Gardenia Vargas-Parra, Àlex Teulé, Raquel Cuesta, Xavier Muñoz, Olga Campos, and et al. 2020. "Exploring the Role of Mutations in Fanconi Anemia Genes in Hereditary Cancer Patients" Cancers 12, no. 4: 829. https://doi.org/10.3390/cancers12040829
APA Styledel Valle, J., Rofes, P., Moreno-Cabrera, J. M., López-Dóriga, A., Belhadj, S., Vargas-Parra, G., Teulé, À., Cuesta, R., Muñoz, X., Campos, O., Salinas, M., de Cid, R., Brunet, J., González, S., Capellá, G., Pineda, M., Feliubadaló, L., & Lázaro, C. (2020). Exploring the Role of Mutations in Fanconi Anemia Genes in Hereditary Cancer Patients. Cancers, 12(4), 829. https://doi.org/10.3390/cancers12040829