Current Strategies and Future Perspectives for Precision Medicine in Pancreatic Cancer

Abstract
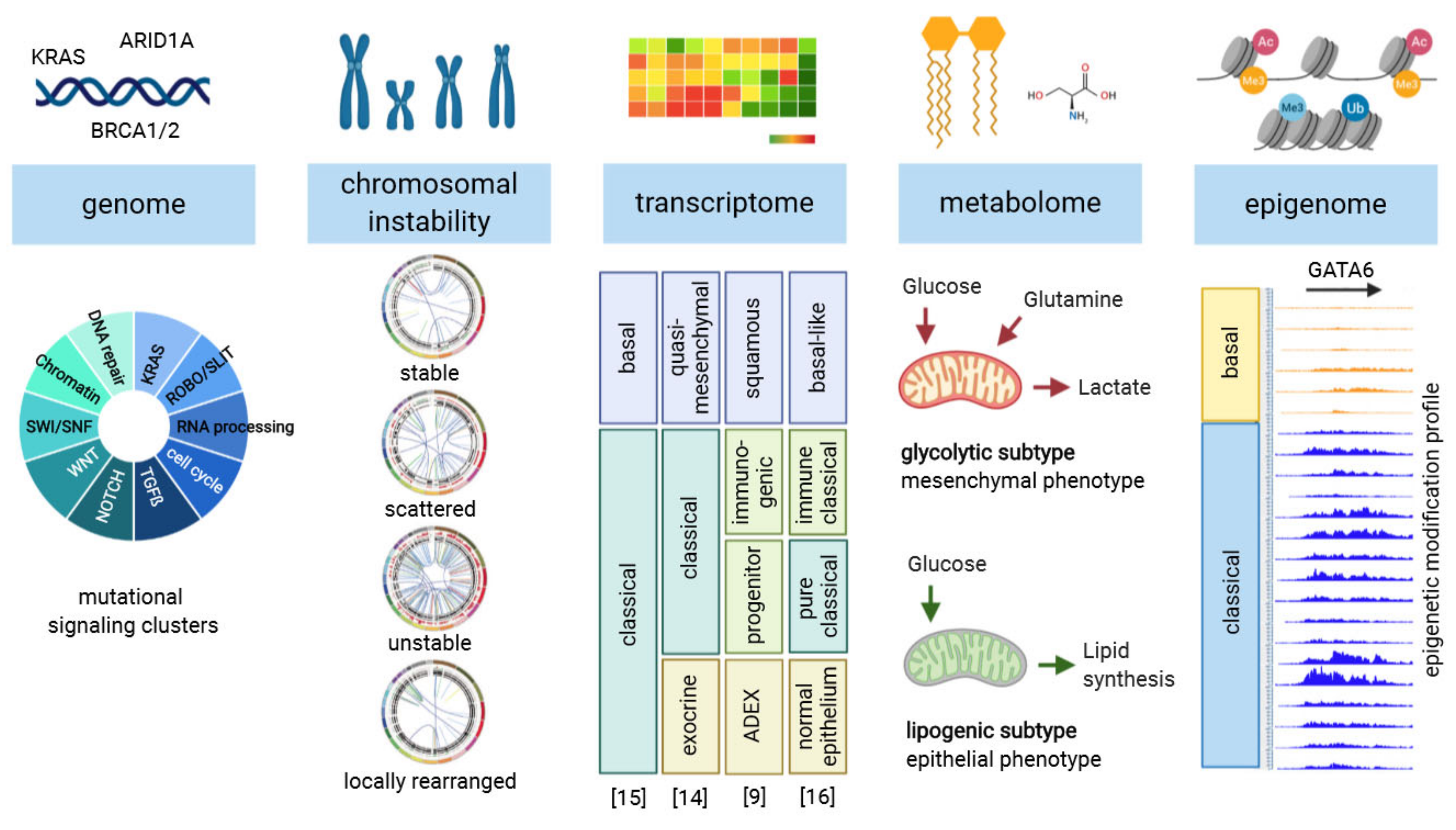
1. Introduction
2. Selected Mutant Gene Profiles and Cancer Subtypes are Eligible for Targeted Therapy
3. Precision Medicine Initiatives
4. Novel Preclinical Strategies for Precision Medicine
4.1. Opportunities of Metabolic Profiles to Develop Targeted Therapies
4.2. Epigenetic Therapies for Defined Subtypes
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, L.R.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Neoptolemos, J.P.; Kleeff, J.; Michl, P.; Costello, E.; Greenhalf, W.; Palmer, D.H. Therapeutic developments in pancreatic cancer: Current and future perspectives. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.; Schwarz, L.; Borbath, I.; Henry, A.; Van Laethem, J.-L.; Malka, D.; Ducreux, M.; Conroy, T. An update on treatment options for pancreatic adenocarcinoma. Ther. Adv. Med Oncol. 2019, 11, 1758835919875568. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. New Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef]
- Biankin, A.V.; Initiative, A.P.C.G.; Waddell, N.; Kassahn, K.; Gingras, M.-C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.-M.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef]
- Bailey, P.; Initiative, A.P.C.G.; Chang, D.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christophi, C.; Bruxner, T.J.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Waddell, N.; Initiative, A.P.C.G.; Pajic, M.; Patch, A.-M.; Chang, D.; Kassahn, K.; Bailey, P.; Johns, A.L.; Miller, D.K.; Nones, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef]
- Schutte, M.; Hruban, R.H.; Geradts, J.; Maynard, R.; Hilgers, W.; Rabindran, S.K.; Moskaluk, CA.; Hahn, S.A.; Schwarte-Waldhoff, I.; Schmiegel, W.; et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997, 57, 3126–3130. [Google Scholar] [PubMed]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Puleo, F.; Nicolle, R.; Blum, Y.; Cros, J.; Marisa, L.; Demetter, P.; Quertinmont, E.; Svrcek, M.; Elarouci, N.; Iovanna, J.L.; et al. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology 2018, 155, 1999–2013.e3. [Google Scholar] [CrossRef]
- Daemen, A.; Peterson, D.; Sahu, N.; Mccord, R.; Du, X.; Liu, B.; Kowanetz, K.; Hong, R.; Moffat, J.; Gao, M.; et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc. Natl. Acad. Sci. USA 2015, 112, E4410–E4417. [Google Scholar] [CrossRef]
- Lomberk, G.; Blum, Y.; Nicolle, R.; Nair, A.; Gaonkar, K.S.; Marisa, L.; Mathison, A.; Sun, Z.; Yan, H.; Elarouci, N.; et al. Distinct epigenetic landscapes underlie the pathobiology of pancreatic cancer subtypes. Nat. Commun. 2018, 9, 1978. [Google Scholar] [CrossRef]
- Pishvaian, M.J.; Bender, R.J.; Halverson, D.; Rahib, L.; Hendifar, A.E.; Mikhail, S.; Chung, V.; Picozzi, V.J.; Sohal, D.; Blais, E.M.; et al. Molecular Profiling of Patients with Pancreatic Cancer: Initial Results from the Know Your Tumor Initiative. Clin. Cancer Res. 2018, 24, 5018–5027. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Lord, C.; Serra, V.; Tutt, A.; Balmaña, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.; De Bono, J.S. A decade of clinical development of PARP inhibitors in perspective. Ann. Oncol. 2019, 30, 1437–1447. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, -Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Nevala-Plagemann, C.; Hidalgo, M.; Garrido-Laguna, I. From state-of-the-art treatments to novel therapies for advanced-stage pancreatic cancer. Nat. Rev. Clin. Oncol. 2019, 17, 108–123. [Google Scholar] [CrossRef] [PubMed]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef] [PubMed]
- Tiriac, H.; Belleau, P.; Engle, D.D.; Plenker, D.; Deschênes, A.; Somerville, T.D.D.; Froeling, F.E.M.; Burkhart, R.A.; Denroche, R.E.; Jang, G.H.; et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov. 2018, 8, 1112–1129. [Google Scholar] [CrossRef]
- Collisson, E.A.; Bailey, P.; Chang, D.K.; Biankin, A.V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 207–220. [Google Scholar] [CrossRef]
- Dreyer, S.; Jamieson, N.; Cooke, S.; Valle, J.; McKay, C.; Biankin, A.; Chang, D. PRECISION-Panc: The Next Generation Therapeutic Development Platform for Pancreatic Cancer. Clin. Oncol. 2020, 32, 1–4. [Google Scholar] [CrossRef]
- Dreyer, S.; Jamieson, N.; Morton, J.P.; Sansom, O.; Biankin, A.; Chang, D. Pancreatic Cancer: From Genome Discovery to PRECISION-Panc. Clin. Oncol. 2019, 32, 5–8. [Google Scholar] [CrossRef]
- Aung, K.L.; Fischer, S.E.; Denroche, R.E.; Jang, G.-H.; Dodd, A.; Creighton, S.; Southwood, B.; Liang, S.-B.; Chadwick, D.; Zhang, A.; et al. Genomics-Driven Precision Medicine for Advanced Pancreatic Cancer: Early Results from the COMPASS Trial. Clin. Cancer Res. 2017, 24, 1344–1354. [Google Scholar] [CrossRef]
- Law, H.C.-H.; Lagundžin, D.; Clement, E.J.; Qiao, F.; Wagner, Z.S.; Krieger, K.L.; Costanzo-Garvey, D.; Caffrey, T.C.; Grem, J.L.; DiMaio, D.J.; et al. The Proteomic Landscape of Pancreatic Ductal Adenocarcinoma Liver Metastases Identifies Molecular Subtypes and Associations with Clinical Response. Clin. Cancer Res 2020, 26, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Storz, P.; Crawford, H.C. Carcinogenesis of Pancreatic Ductal Adenocarcinoma. Gastroenterology 2020. [Google Scholar] [CrossRef] [PubMed]
- Christenson, E.S.; Jaffee, E.; Azad, N.S. Current and emerging therapies for patients with advanced pancreatic ductal adenocarcinoma: A bright future. Lancet Oncol. 2020, 21, e135–e145. [Google Scholar] [CrossRef]
- Mahajan, U.M.; Langhoff, E.; Goni, E.; Costello, E.; Greenhalf, W.; Halloran, C.; Ormanns, S.; Kruger, S.; Boeck, S.; Ribback, S.; et al. Immune Cell and Stromal Signature Associated With Progression-Free Survival of Patients With Resected Pancreatic Ductal Adenocarcinoma. Gastroenterology 2018, 155, 1625–1639.e2. [Google Scholar] [CrossRef] [PubMed]
- Wartenberg, M.; Cibin, S.; Zlobec, I.; Vassella, E.; Eppenberger-Castori, S.M.; Terracciano, L.; Eichmann, M.; Worni, M.; Gloor, B.; Perren, A.; et al. Integrated Genomic and Immunophenotypic Classification of Pancreatic Cancer Reveals Three Distinct Subtypes with Prognostic/Predictive Significance. Clin. Cancer Res. 2018, 24, 4444–4454. [Google Scholar] [CrossRef]
- Armitage, E.; Barbas, C. Metabolomics in cancer biomarker discovery: Current trends and future perspectives. J. Pharm. Biomed. Anal. 2014, 87, 1–11. [Google Scholar] [CrossRef]
- Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 2001, 69, 89–95. [Google Scholar]
- Bennett, R.M.; Devarajan, P. Characteristics of an Ideal Biomarker of Kidney Diseases. In Biomarkers of Kidney Disease; Elsevier: Amsterdam, The Netherlands, 2011; pp. 1–24. [Google Scholar]
- Davis, V.W.; Bathe, O.F.; Schiller, D.; Slupsky, C.M.; Sawyer, M.B. Metabolomics and surgical oncology: Potential role for small molecule biomarkers. J. Surg. Oncol. 2010, 103, 451–459. [Google Scholar] [CrossRef]
- Monteiro, M.; Carvalho, M.; Bastos, M.L.; De Pinho, P.G. Metabolomics Analysis for Biomarker Discovery: Advances and Challenges. Curr. Med. Chem. 2013, 20, 257–271. [Google Scholar] [CrossRef]
- Fiehn, O.; Kristal, B.; Van Ommen, B.; Sumner, L.W.; Sansone, S.-A.; Taylor, C.; Hardy, N.; Kaddurah-Daouk, R. Establishing Reporting Standards for Metabolomic and Metabonomic Studies: A Call for Participation. OMICS A J. Integr. Boil. 2006, 10, 158–163. [Google Scholar] [CrossRef]
- Griffin, J.L. Metabolic profiles to define the genome: Can we hear the phenotypes? Philos. Trans. R. Soc. B Boil. Sci. 2004, 359, 857–871. [Google Scholar] [CrossRef]
- Koen, N.; Du Preez, I. Metabolomics and personalized medicine. In Advances in Protein Chemistry and Structural Biology; Elsevier: Amsterdam, The Netherlands, 2016; pp. 53–78. [Google Scholar]
- Daemen, A.; Liu, B.; Song, K.; Kwong, M.; Gao, M.; Hong, R.; Nannini, M.; Peterson, D.; Liederer, B.M.; De La Cruz, C.; et al. Pan-Cancer Metabolic Signature Predicts Co-Dependency on Glutaminase and De Novo Glutathione Synthesis Linked to a High-Mesenchymal Cell State. Cell Metab. 2018, 28, 383–399.e9. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Nilsson, R.; Sharma, S.; Madhusudhan, N.; Kitami, T.; Souza, A.L.; Kafri, R.; Kirschner, M.W.; Clish, C.; Mootha, V.K. Metabolite Profiling Identifies a Key Role for Glycine in Rapid Cancer Cell Proliferation. Science 2012, 336, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M. Pancreatic cancer. N. Engl. J. Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Baine, M.J.; Jain, M.; Sasson, A.R.; Batra, S.K. Early diagnosis of pancreatic cancer: Challenges and new developments. Biomarkers Med. 2012, 6, 597–612. [Google Scholar] [CrossRef] [PubMed]
- Locker, G.Y.; Hamilton, S.; Harris, J.; Jessup, J.M.; Kemeny, N.; Macdonald, J.S.; Somerfield, M.R.; Hayes, D.F.; Bast, R.C. ASCO 2006 Update of Recommendations for the Use of Tumor Markers in Gastrointestinal Cancer. J. Clin. Oncol. 2006, 24, 5313–5327. [Google Scholar] [CrossRef]
- Goggins, M. Molecular Markers of Early Pancreatic Cancer. J. Clin. Oncol. 2005, 23, 4524–4531. [Google Scholar] [CrossRef]
- Goonetilleke, K.; Siriwardena, A.K. Systematic review of carbohydrate antigen (CA 19-9) as a biochemical marker in the diagnosis of pancreatic cancer. Eur. J. Surg. Oncol. (EJSO) 2007, 33, 266–270. [Google Scholar] [CrossRef]
- Kunovský, L.; Tesarikova, P.; Kala, Z.; Kroupa, R.; Kysela, P.; Dolina, J.; Trna, J. The Use of Biomarkers in Early Diagnostics of Pancreatic Cancer. Can. J. Gastroenterol. Hepatol. 2018, 2018, 1–10. [Google Scholar] [CrossRef]
- Mayerle, J.; Kalthoff, H.; Reszka, R.; Kamlage, B.; Peter, E.; Schniewind, B.; Maldonado, S.G.; Pilarsky, C.; Heidecke, C.-D.; Schatz, P.; et al. Metabolic biomarker signature to differentiate pancreatic ductal adenocarcinoma from chronic pancreatitis. Gut 2017, 67, 128–137. [Google Scholar] [CrossRef]
- Costello, E. A metabolomics-based biomarker signature discriminates pancreatic cancer from chronic pancreatitis. Gut 2017, 67, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Phua, L.C.; Goh, S.; Tai, D.W.M.; Leow, W.Q.; Alkaff, S.M.F.; Chan, C.Y.; Kam, J.H.; Lim, T.K.H.; Chan, E.C.Y. Metabolomic prediction of treatment outcome in pancreatic ductal adenocarcinoma patients receiving gemcitabine. Cancer Chemother. Pharmacol. 2017, 81, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Schuettengruber, B.; Martinez, A.-M.; Iovino, N.; Cavalli, G. Trithorax group proteins: Switching genes on and keeping them active. Nat. Rev. Mol. Cell Boil. 2011, 12, 799–814. [Google Scholar] [CrossRef] [PubMed]
- Benitz, S.; Straub, T.; Mahajan, U.M.; Mutter, J.; Czemmel, S.; Unruh, T.; Wingerath, B.; Deubler, S.; Fahr, L.; Cheng, T.; et al. Ring1b-dependent epigenetic remodelling is an essential prerequisite for pancreatic carcinogenesis. Gut 2019, 68, 2007–2018. [Google Scholar] [CrossRef] [PubMed]
- Hessmann, E.; Johnsen, S.A.; Siveke, J.T.; Ellenrieder, V. Epigenetic treatment of pancreatic cancer: Is there a therapeutic perspective on the horizon? Gut 2016, 66, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Diaferia, G.R.; Balestrieri, C.; Prosperini, E.; Nicoli, P.; Spaggiari, P.; Zerbi, A.; Natoli, G. Dissection of transcriptional and cis -regulatory control of differentiation in human pancreatic cancer. EMBO J. 2016, 35, 595–617. [Google Scholar] [CrossRef]


{kind=link}
| Pathway | Incidence Multiple Genes | Incidence Single Genes | ||
|---|---|---|---|---|
| Epigenetic regulators | ~35% (134/383) | ARID1A, KMT2C, KMT2D, KDM6A, SMARCA4, SETD2, ARID2, PBRM1, HDAC1, CREBBP, SETDB1, SETD1B, EP300, JARID2, KMT2A, SMARCA1, SMARCA2, KDM5C, SETBP1, KDM2B, ARID3C, DNMT3B, DNMT1, ARID4A, KDM5A, SMARCB1, SMARCD1, SETD1A, KAT6B | 8% | ARID1A |
| 5% | KMT2C | |||
| 2.9% | SMARCA4 | |||
| 2.3% | SETD2 | |||
| DDR pathway components | ~9% (34/383) | BRCA1, BRCA2, PALB2, ATM, ATR, MLH1, MSH2, MSH6, RPA1, STK11, FANCA, FANCC | 4% | ATM |
| 2.1% | BRCA2 | |||
| 1.3% | BRCA1 | |||
| 0.5% | PALB2 | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Regel, I.; Mayerle, J.; Ujjwal Mukund, M. Current Strategies and Future Perspectives for Precision Medicine in Pancreatic Cancer. Cancers 2020, 12, 1024. https://doi.org/10.3390/cancers12041024
Regel I, Mayerle J, Ujjwal Mukund M. Current Strategies and Future Perspectives for Precision Medicine in Pancreatic Cancer. Cancers. 2020; 12(4):1024. https://doi.org/10.3390/cancers12041024
Chicago/Turabian StyleRegel, Ivonne, Julia Mayerle, and Mahajan Ujjwal Mukund. 2020. "Current Strategies and Future Perspectives for Precision Medicine in Pancreatic Cancer" Cancers 12, no. 4: 1024. https://doi.org/10.3390/cancers12041024
APA StyleRegel, I., Mayerle, J., & Ujjwal Mukund, M. (2020). Current Strategies and Future Perspectives for Precision Medicine in Pancreatic Cancer. Cancers, 12(4), 1024. https://doi.org/10.3390/cancers12041024