1. Introduction
UTX (ubiquitously transcribed tetratricopeptide repeat, X chromosome) is encoded by the gene
KDM6A, located on the X chromosome.
KDM6A is frequently affected by deleterious mutations in urothelial carcinoma (UC) and other cancers. UTX is therefore considered a tumor suppressor [
1]. Its mode of action is not fully understood and may differ between cancer types [
2,
3]. UTX has several molecular functions, including, prominently, a specific histone demethylase activity towards dimethylated or trimethylated lysine 27 of histone H3 (H3K27me2/3) [
4,
5]. UTX participates in the MLL2/3 complex (also known as COMPASS-like), which catalyzes H3K4 methylation, and in interactions with the chromatin remodeling SWI/SNF complex and the histone acetyltransferase CBP [
1]. During fetal development, UTX modulates stem cell differentiation and HOX gene regulation [
5,
6]. It is therefore plausible to assume that UTX inactivation in urothelial carcinoma might promote cancer development via aberrant urothelial differentiation. This idea is supported by observations in other cancer types. For instance, loss of UTX in myeloid leukemia leads to dysregulation of transcription factor programs steering the differentiation of hematopoietic cells [
7,
8]. Similarly, in the pancreas, UTX deficiency results in squamous metaplasia and cancer by deregulation of tissue-specific enhancer activities [
9]. However,
KDM6A mutations are found across all molecular subtypes of invasive UC [
10] and are even frequent in well-differentiated papillary UC [
11], as reviewed in [
2]. To date, there is no direct evidence on whether and to which extent urothelial differentiation is disturbed by UTX loss of function.
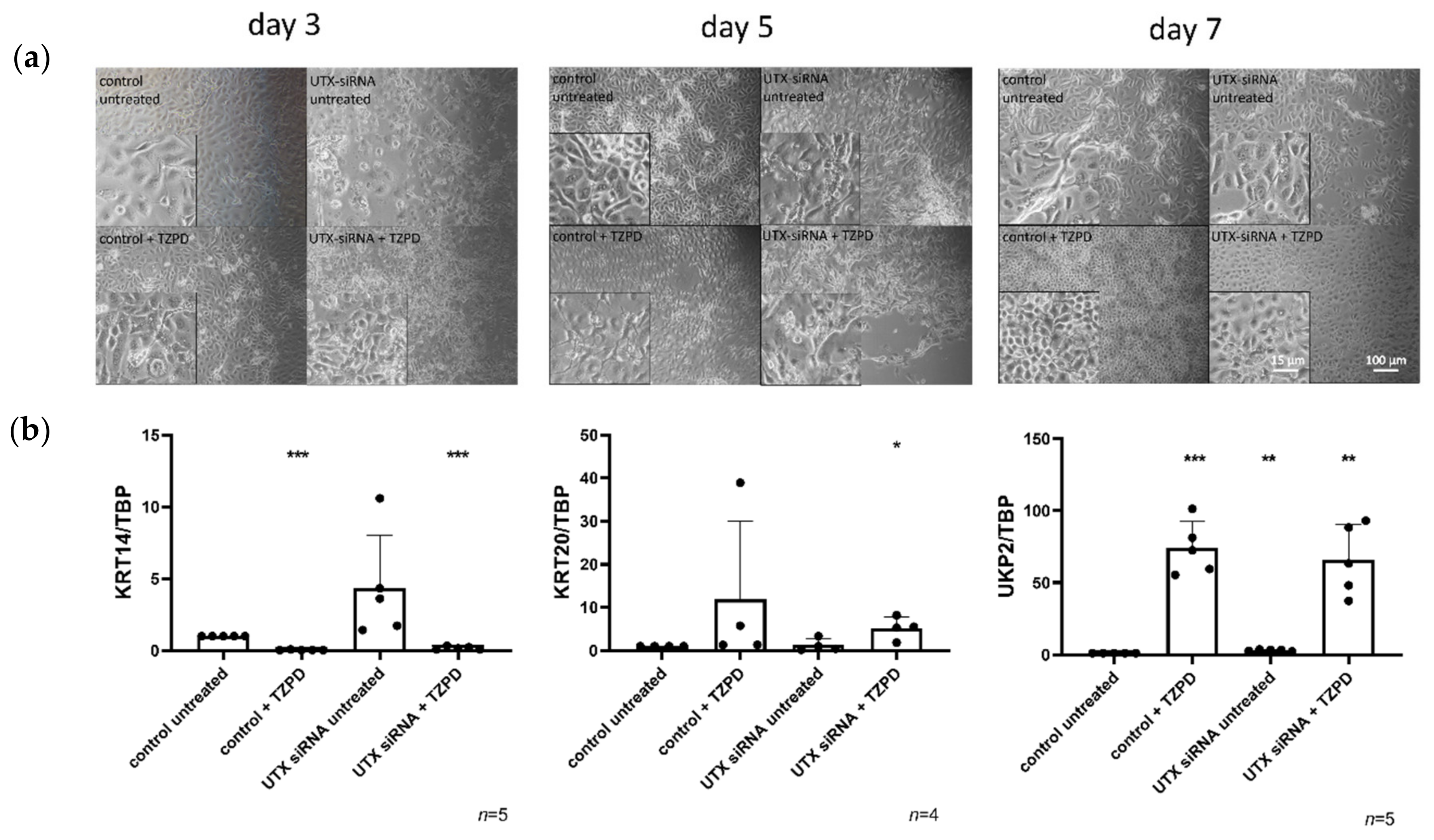
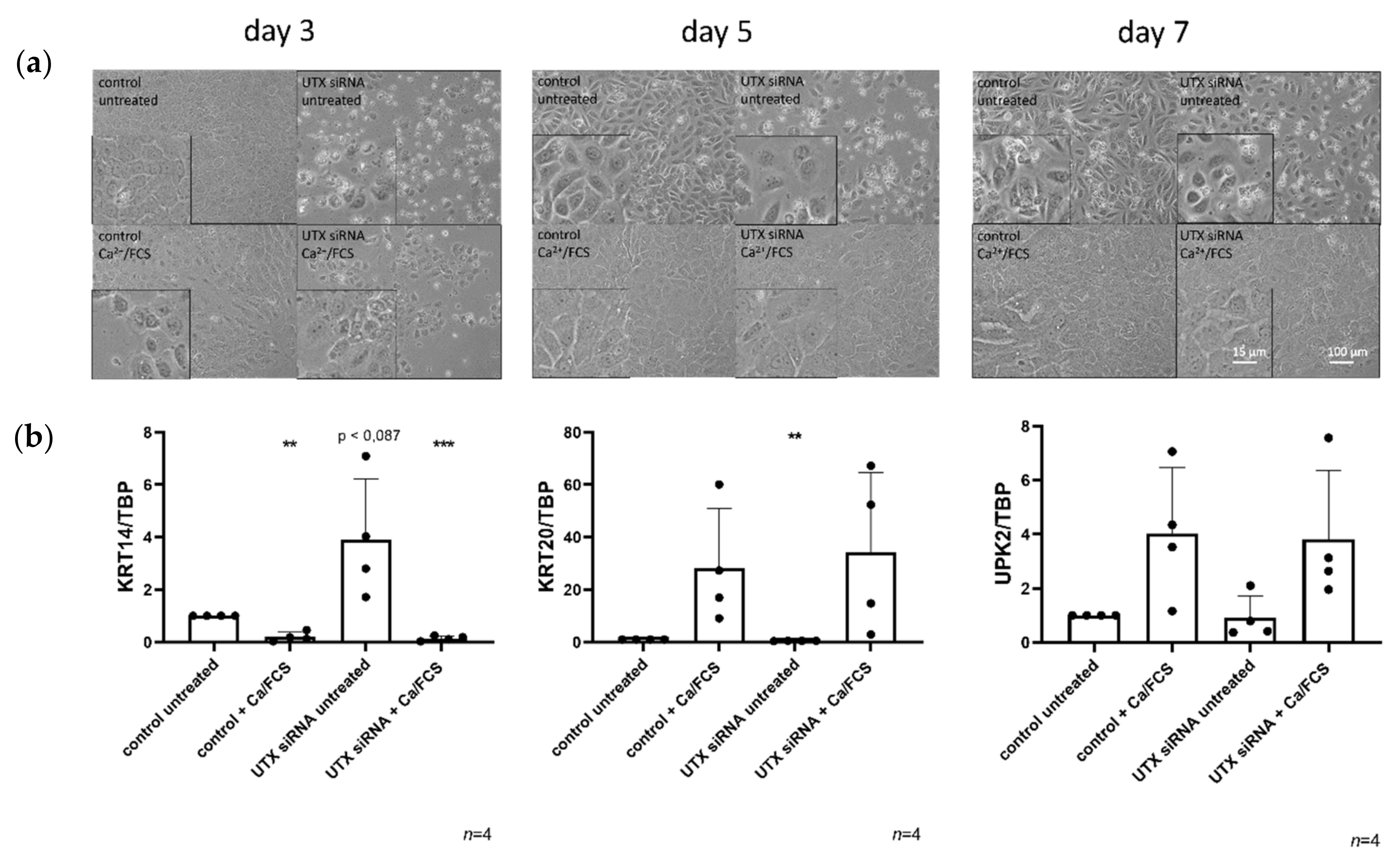
To address this question, we used two models of urothelial differentiation. First, primary cultures of normal urothelial cells (UECs) derived from ureters of nephrectomy patients consist mainly of cells with a basal phenotype (KRT14-/KRT5+/KRT20-) and a variable proportion of KRT14+/KRT5+/KRT20- cells, which are regarded as stem cells in the urothelium [
12,
13,
14,
15,
16,
17]. Treatment with a PPARγ agonist (troglitazone) and the EGF receptor inhibitor PD153035 (TZ/PD protocol) induces biochemical markers of urothelial differentiation, such as KRT20 and uroplakins, e.g., UPK2, while decreasing KRT14 and KRT5 expression [
18]. Alternatively, urothelial differentiation can be elicited by increasing the Ca
2+ concentration in the culture medium and adding calf serum (Ca/FCS protocol) [
19]. The spontaneous immortalized urothelial cell line HBLAK provides a more conveniently available model than primary urothelial cultures, but in these cells the Ca/FCS protocol is more efficacious than the TZ/PD protocol [
20]. Like UEC cultures, HBLAK contains a subpopulation of KRT14+/KRT5+/KRT20− cells (hereafter KRT14
high cells), and upon Ca/FCS treatment yields a high percentage of cells expressing KRT20 and UPK2, whereas KRT14
high cells decrease in proportion.
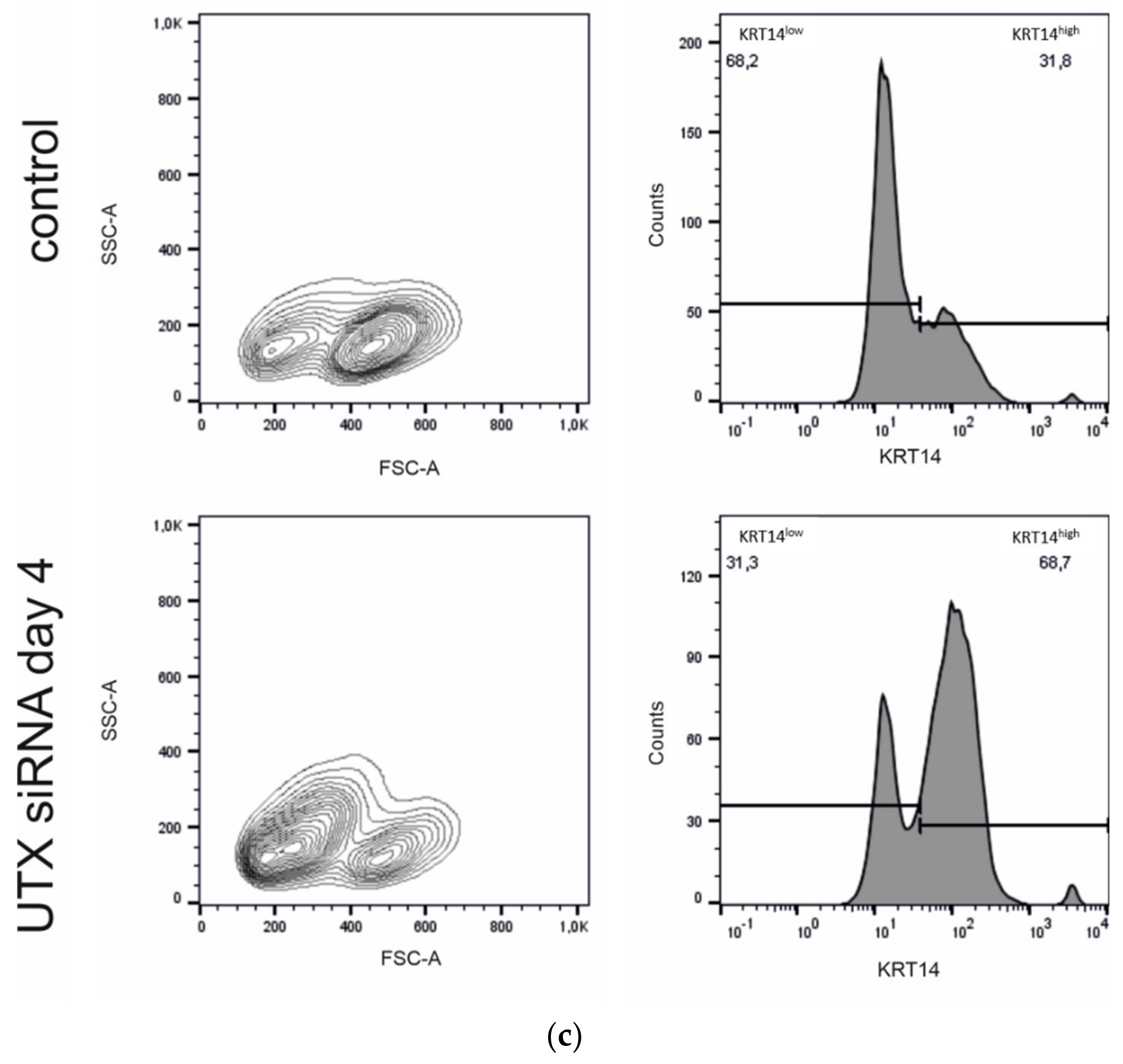
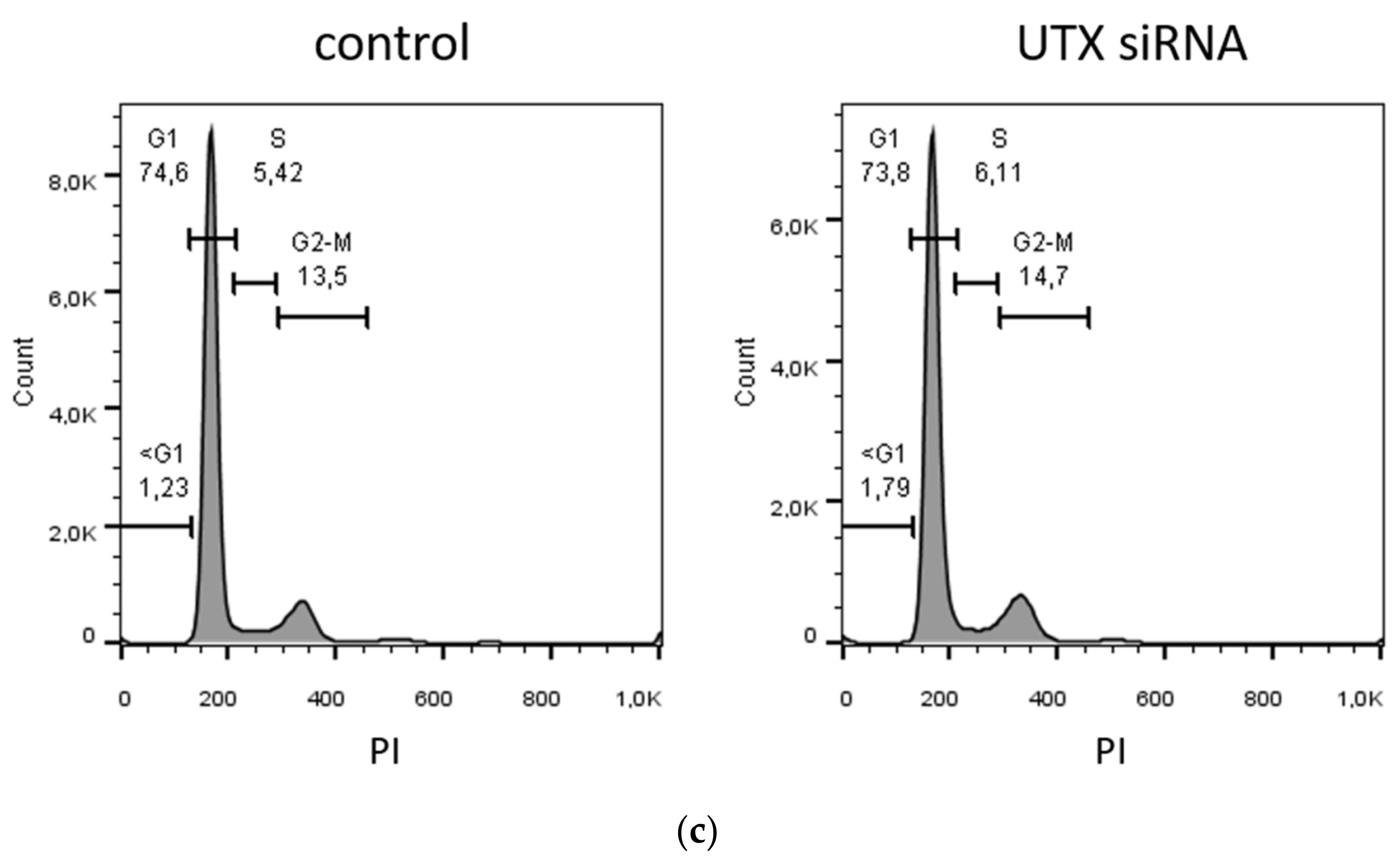
Here, we studied the effect of efficient UTX siRNA-mediated knockdown on TZ/PD-induced differentiation of UECs and on Ca/FCS-induced differentiation of HBLAK cells. Unexpectedly, we did not observe a major effect on differentiation in either cell model, but increased apoptotic cell death prior to and independent of differentiation induction, which was partly mediated by p53 activation. Interestingly, cell death resulted in an increased ratio of KRT14high over KRT14low cells. Therefore, we characterized these two populations in more detail in the HBLAK cell line. Finally, we observed an analogous effect of UTX knockdown in the BFTC-905 urothelial carcinoma cell line, which also contains KRT14high and KRT14low cells.
3. Discussion
Mutations inactivating UTX are found across all stages of urothelial carcinoma (UC), albeit more commonly in lower stage tumors [
11], and intriguingly, across all molecular subtypes of muscle-invasive bladder cancers (MIBC) [
2]. In particular, according to the large TCGA study [
10], there is no significant difference in their frequency between cancers with a basal-squamous (BASQ) phenotype, which present with markers of basal urothelial cells, and cancers with luminal phenotypes exhibiting markers of intermediate and luminal differentiated urothelial cells. Most current evidence indicates that basal cells are precursors of intermediate and luminal cells [
12,
13,
14,
16,
17]. Therefore, it may seem a priori unlikely that UTX inactivation contributes to urothelial carcinogenesis by blocking the differentiation of basal to luminal cells, since in that case UTX mutations should be more prevalent in BASQ UC. This assumption is borne out by our finding that UTX knockdown in models of urothelial differentiation, wherein cells with a basal phenotype differentiate into luminal cells, did not significantly inhibit differentiation. Our conclusion should, however, be considered with caution in so far, as in vitro differentiation models of urothelial cells do not in every respect fully recapitulate in vivo biochemical differentiation and stratification.
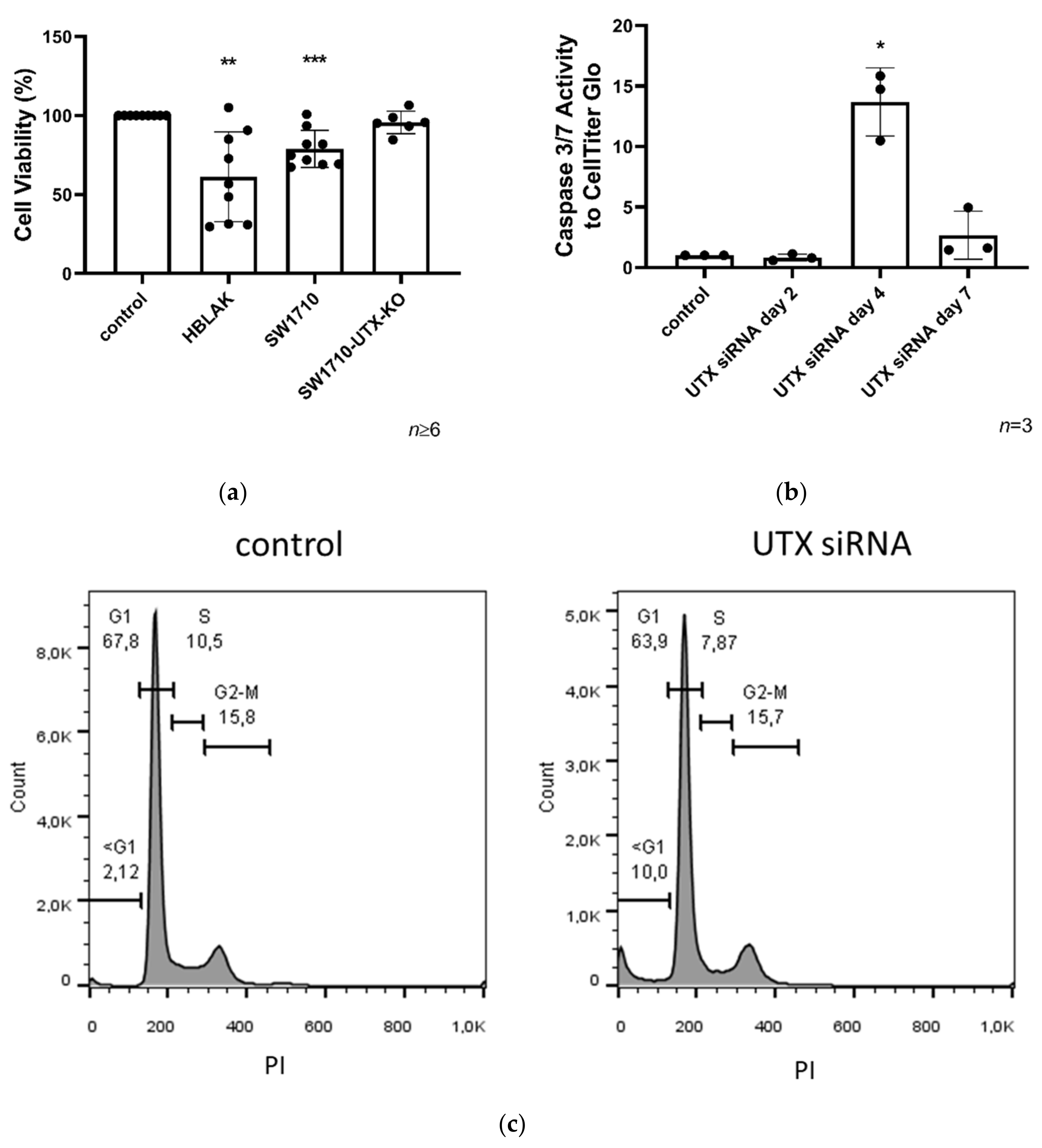
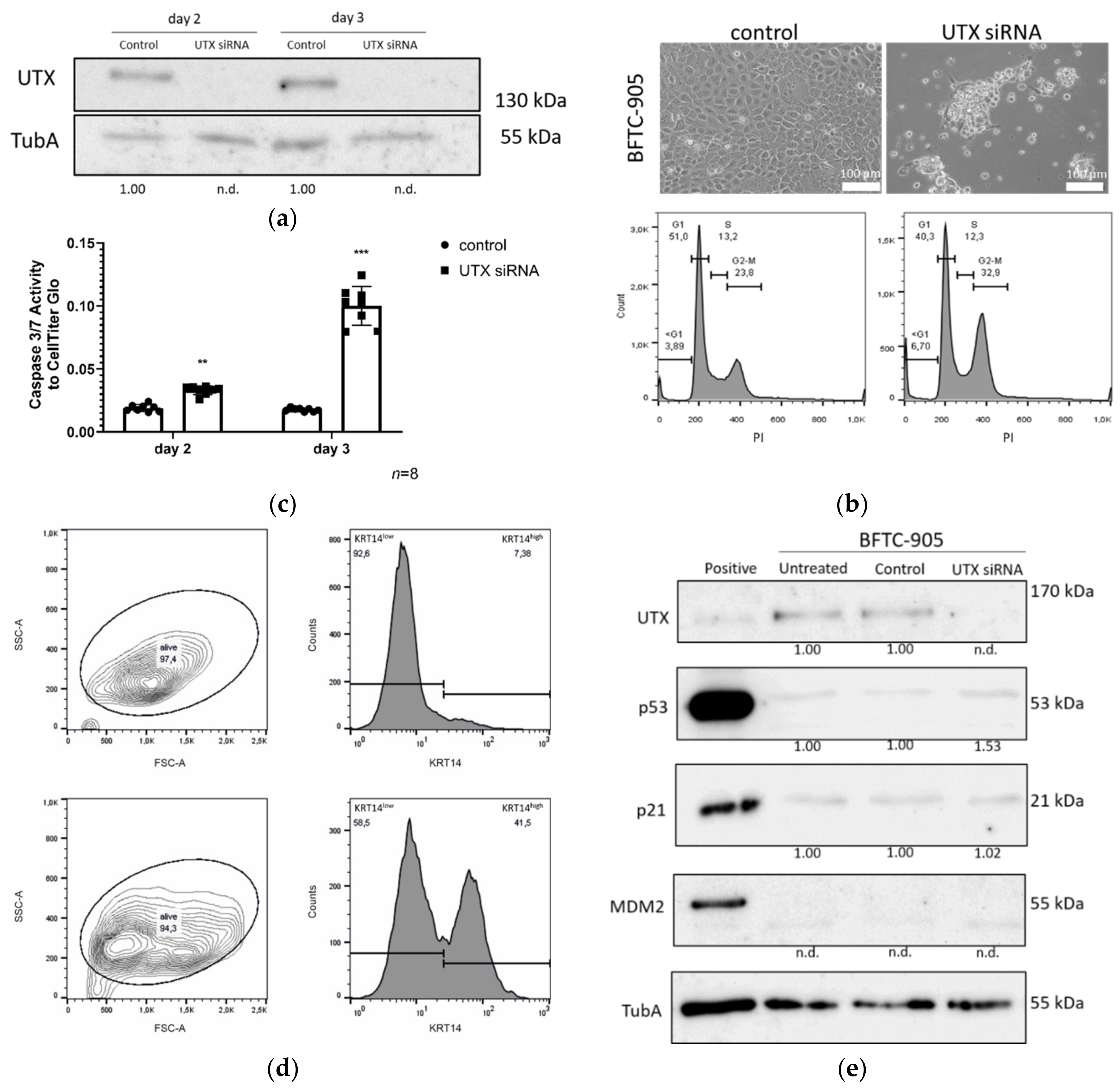
Our investigation focused on an unexpected observation; namely, that the UTX knockdown elicited significant cell death by apoptosis. This occurred independent of differentiation treatment after 2–3 days in cultured normal urothelial cells (UECs) and immortalized HBLAK cells. A similar effect was elicited in the BFTC-905 UC cell line, which like the other two cell types has a basal phenotype [
27,
29]. All three models moreover share the properties of being wild-type for p53 and all COMPASS components and containing a fraction of cells with high expression of KRT14, in addition to the ubiquitously expressed basal cell cytokeratin KRT5. KRT14 is generally considered a marker of urothelial stem cells [
12,
13,
14,
15,
16,
17]. In the urothelium between 1% and 14% of basal urothelial cells are estimated as KRT14
high, depending on developmental stage, physiological state and species [
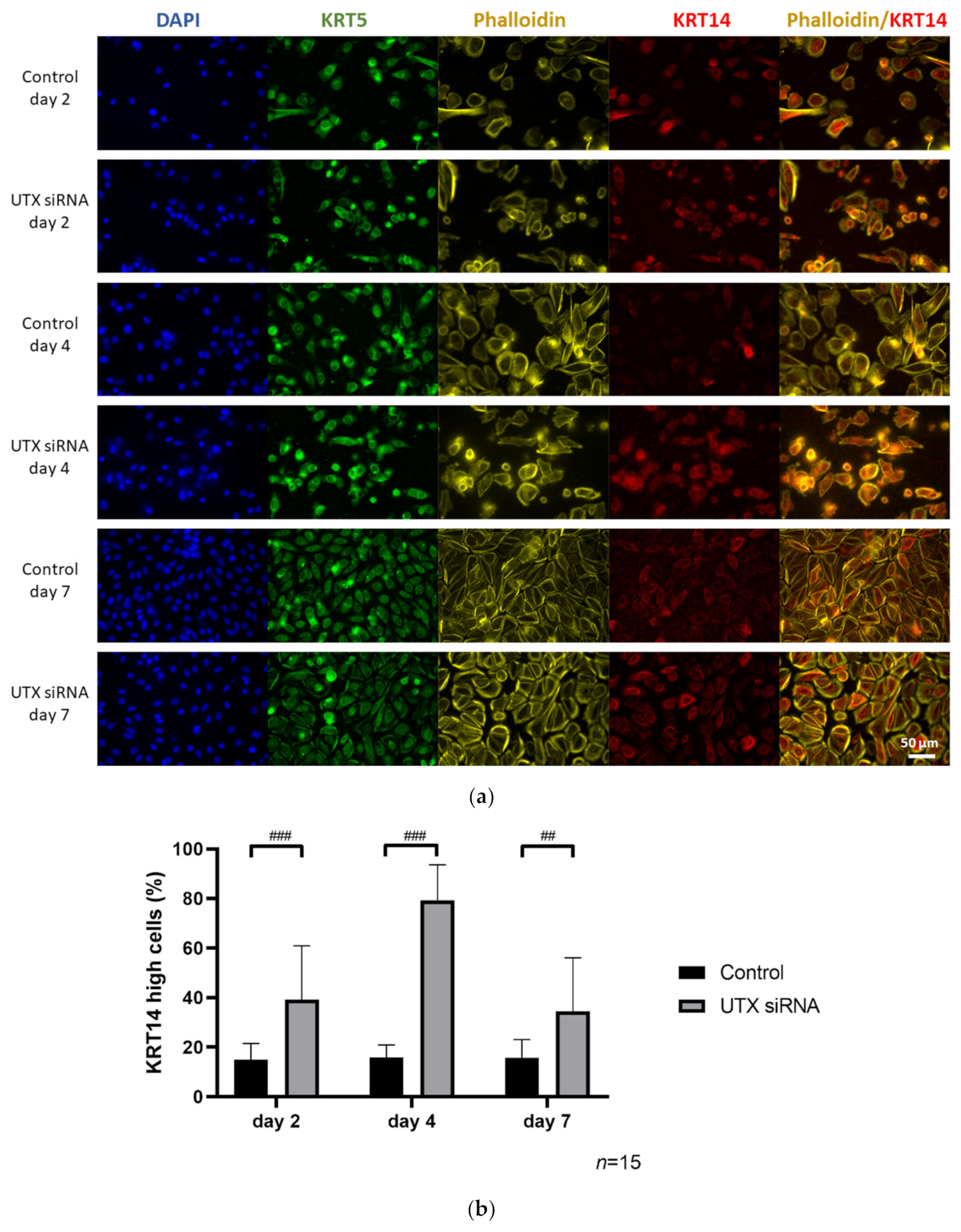
17]. Following UTX knockdown, we observed a relative increase of KRT14 expression in the urothelial culture models, which upon closer analysis turned out to reflect an increased fraction of KRT14
high cells due to apoptotic death of KRT14
low cells.
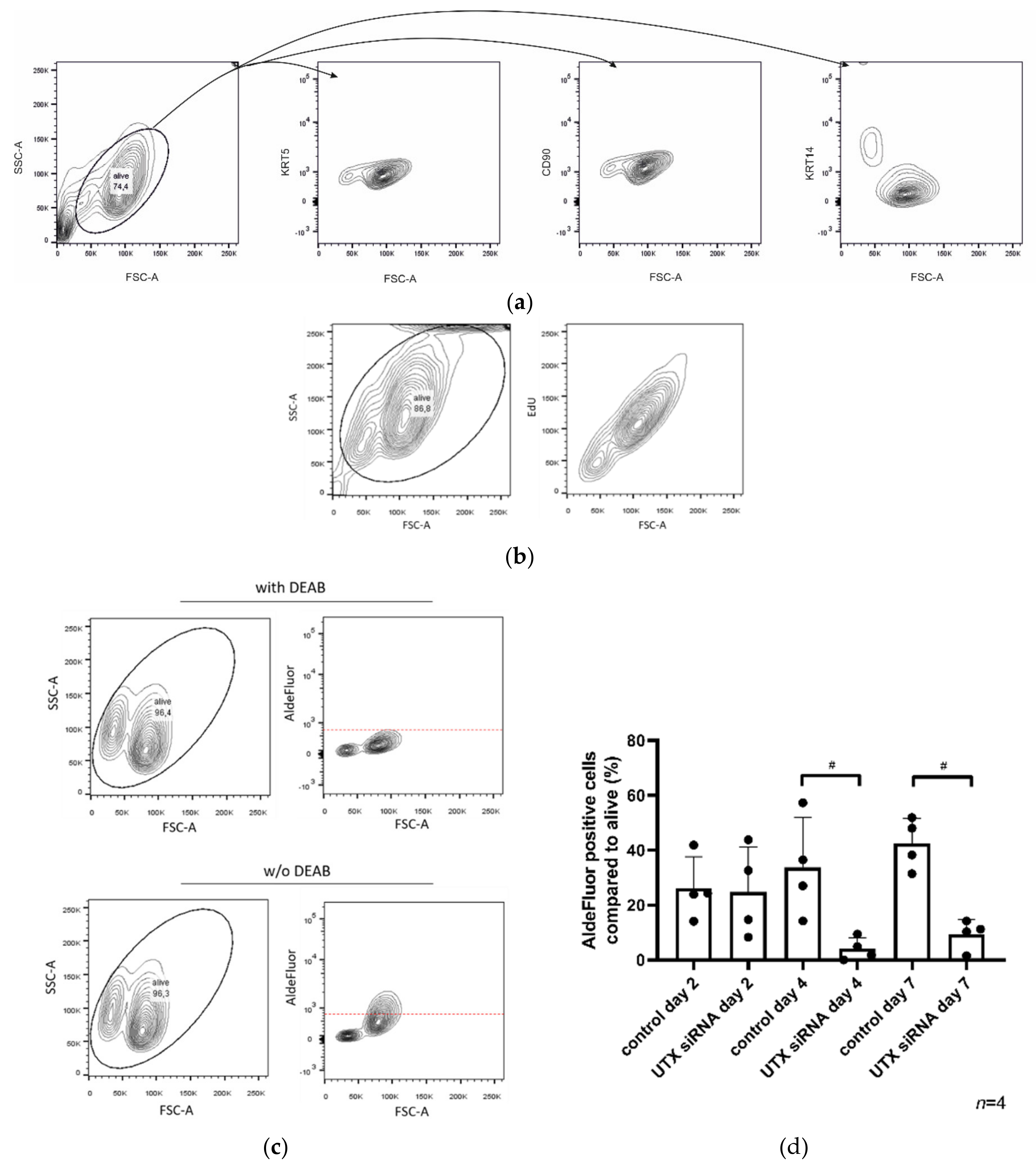
Since UECs are not consistently available and can be highly variable between individual cultures, we investigated this phenomenon in depth in HBLAK, and subsequently, in BFTC-905 cells. The previously reported KRT14
high fraction in HBLAK turned out to differ from KRT14
low cells by further properties; namely, cell size, proliferative activity and AldeFluor-assay activity. The observations that KRT14
high cells are smaller and less proliferative than KRT14
low cells are in accordance with expectations for a stem cell population. AldeFluor-assay activity would instead be expected to be higher in stem cells, oppositely to our observations. In fact, AldeFluor-assay activity reflects the activity of various aldehyde dehydrogenases [
30], whose expression does not necessarily have to be localized in the stem cell population of a tissue. AldeFluor activity was linked to tumor-initiating cells in bladder cancer in a previous publication, but the authors did not investigate the association with KRT14 expression [
31]. In a survey of three different UC cell lines (T24, TCCSUP and 5637), however, AldeFluor activity was not consistently associated with stem cell properties [
32]. Likewise, the lack of association between AldeFluor-assay activity and subpopulations in BFTC-905 argues against a strict link between stem cell properties and AldeFluor activity in urothelial cancer. Our results, rather, suggest that high ALDH activity may characterize KRT5-positive basal cells in the urothelium, possibly due to the most prominently expressed ALDH1A3 isoenzyme. Regardless, UTX knockdown enriched for the small cell, KRT14
high, weakly proliferative and AldeFluor
low cell population in HBLAK cells.
Collectively, these findings suggest that loss of UTX favors the survival of urothelial KRT14
high stem cells over more differentiated KRT14
low/KRT5+ basal cells. In vivo, loss of UTX could therefore over time lead to an expansion of the stem cell population with an increased likelihood of transformation. Notably, a function of UTX in the regulation of the ratio of stem cells to more differentiated basal cells rather than during further urothelial differentiation would account for the prevalence of UTX mutations throughout all UC subtypes. This argument also suggests that UTX inactivation might constitute an early event in urothelial carcinogenesis. Additional mutations in p53 or growth factor receptors would then lead to cancer. For instance, activation of STAT3 in a mouse model of urothelial carcinoma was associated with expansion of KRT14
high cells [
33]. Urothelial cancer is notorious for its multifocality and high recurrence rate after local surgery, which points towards a pronounced field effect. Recent studies have indeed observed clonal expansion of mutant cells in morphologically normal urothelium. In one study on four patients, intriguingly,
KMT2D, another COMPASS component, was frequently mutated in morphologically normal urothelial tissue from cancer-carrying bladders [
34]. An analogous scenario is established in the development of acute myeloid leukemia, which is often preceded by clonal hematopoiesis elicited by mutations in various genes, most often encoding epigenetic regulators like DNMT3A and TET2 [
35], which shift the balance between stem cells and differentiated progeny and displace normal with stem cells with mutants. Similarly, mutations inactivating
KMT2D appear to increase the B-cell population at risk to develop lymphomas by further genetic alterations [
36].
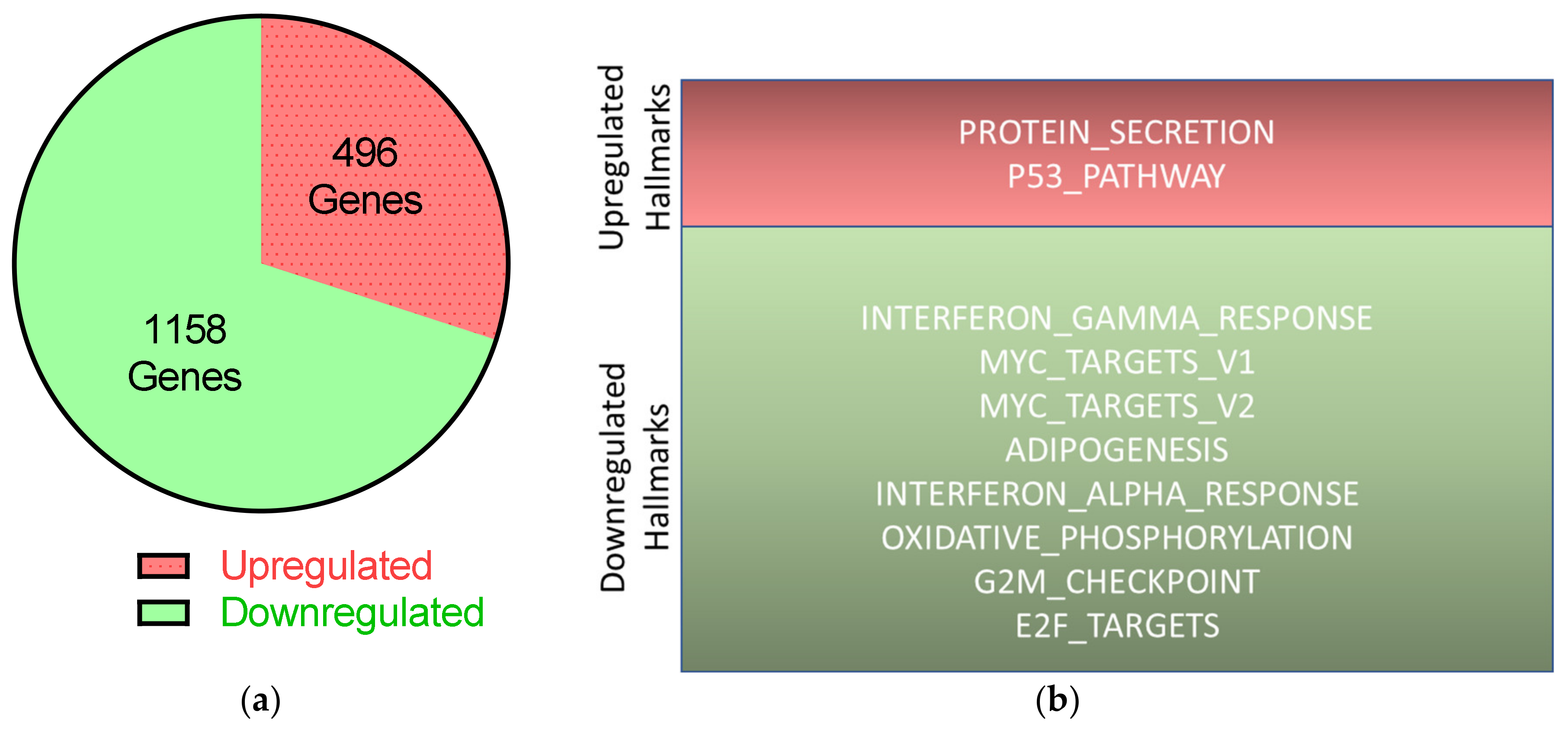
Cell death after UTX knockdown in HBLAK was partly mediated by p53, as suggested by the induction of canonical p53 response genes and by the decrease in cell death in HBLAK cells expressing a dominant-negative p53 protein. In the context of urothelial carcinogenesis, this may be relevant, as p53 function is obliterated or impeded in most MIBC. The cells most affected by UTX knockdown were the proliferating KRT14
low cells. Our findings might therefore predict that in vivo UTX-mutant cells might be able to proliferate identically or better if p53 is also inactivated. In other words, UTX mutations may select for additional genetic changes that inactivate p53. However, expression of a dominant-negative p53 protein only diminished, but did not obliterate cell death in HBLAK cells treated with UTX-siRNA, indicating the involvement of additional factors. Deeper analysis of the RNA-seq data suggests activation of FOXO transcription factors, which can also induce apoptosis, as a second possible factor. It is instructive to compare this situation to the regenerating liver, where, likewise, quiescent cells re-enter a proliferative state. Here, fine-tuning of the activity of p53 and further transcription factors by several pathways is required to avoid induction of apoptosis during proliferation and ensure genetic stability and correct lineage choice [
37]. In the urothelium, another tissue with high regenerative potential, such mechanisms have not yet been sufficiently studied. It should also be mentioned that cell death in BFTC-905 cells did not appear to be associated with p53 activation at all. Another difference in the BFTC-905 UC cell line compared to HBLAK cells was a clearly increased fraction of G2/M-phase cells. This further indicates that the response to UTX knockdown likewise lead to an enrichment of KRT14
high cells, but differed somewhat from that in HBLAK. These differences underline the conclusion that additional pathways beyond p53 activation are involved in the response to UTX knockdown.
Our findings raise the question of why UTX is required for survival of proliferating KRT14
low cells. The most likely answer is that these cells require UTX to set up a new stable epigenetic state. In support of this idea, the UTX antagonist EZH2 has been shown to be required for proper regeneration of the urothelium following damage by uropathogenic bacteria [
38]. The function of UTX during urothelial regeneration, which involves repletion of intermediate luminal, and especially umbrella cells, from the lower epithelial layers, should therefore be studied. Since interactions of UTX with a broad range of lineage-specific transcription factors have been described (reviewed in [
2]), another not necessarily exclusive explanation is that UTX may be required as a co-activator for specific transcription factors establishing the epigenetic state of KRT14
low/KRT5+ cells. These issues will require further experimental investigation.
4. Materials and Methods
4.1. Cell Culture
HBLAK cells were obtained from CELLnTEC Advanced Cell Systems (Bern, Switzerland) and were routinely cultured in CnT-Prime (CnT-PR), as described in [
20]. UECs were cultured in KSFM supplemented with epidermal growth factor and bovine pituitary extract, as described in [
39]. Both were passaged using accutase (Sigma Aldrich, Munich, Germany). Culture and use of normal urothelial cells from ureters of patients undergoing nephrectomy was permitted by the ethical committee of the HHU medical faculty (#1788). UC cell lines were cultured in Dulbecco’s modified eagle’s medium (DMEM, ThermoFisher Scientific, Langenselbold, Germany) supplemented with 10% fetal calf serum as previously described [
21]. Passaging was performed using trypsin (Sigma Aldrich, Munich). All cells were cultured at 37 °C with 5% CO
2. All cells were regularly authenticated by STR profiling and checked for mycoplasm contamination.
The SW1710-UTX-KO cell line was generated using UTX CRISPR/Cas9 KO Plasmids (Santa Cruz Biotechnology, Dallas, TX, USA, sc-402761-NIC and sc-402761-NIC-2) and clonal selection with 0.5 µg/mL puromycin (Invivogen, Toulouse, France) for 5 days as described [
21].
To generate HBLAK-p53DD cells, HBLAK cells were infected with the retroviral vector pBABE-hygro-p53DD (Addgene plasmid #9058, kindly provided by Prof. R. Weinberg, Boston, MA, USA). This vector expresses p53DD, a dominant-negative p53 mutant, consisting of the initial 14 amino acids and the oligomerization and COOH-domains of p53 but lacking the intermediate 288 (15–301) amino acids [
40]. Transduced cells were then selected with hygromycin.
4.2. Induction of Urothelial Differentiation
Two days after siRNA transfection, HBLAK cells were switched to CnT-PR-D medium (CELLnTEC) and treated with 2 mM CaCl2 and 5% fetal calf serum (Ca/FCS) and UECs were treated with 1 µM troglitazone and 1 µM PD153035 (TZ/PD) and further cultured for up to 10 days.
4.3. siRNA Transfection
All siRNAs were transfected at a final concentration of 10 nM using Lipofectamine™ RNAiMAX Transfection Reagent (ThermoFisher Scientific, catalogue number 13778150). The following siRNAs were purchased from ThermoFisher Scientific: ON-TARGETplus Human KDM6A siRNA (SMARTpool, Dharmacon, L-014140-01-0005, siRNA 01) with ON-TARGETplus Non-targeting Pool (Dharmacon, D-001810-10-05, siRNA 20) as a control, or Silencer® Select siRNA (4392420, Ambion, KDM6A, s14735) with Silencer™ Select Negative Control No. 1 siRNA (Invitrogen, 4390843, Waltham, MA, USA).
4.4. RNA isolation and RT-qPCR
Cells were lysed with TRIzol™ Reagent (Invitrogen, 15596026). Following addition of chloroform, RNA, DNA and protein were separated via centrifugation at 11,000 g at 4 °C for 10 min. RNA was then isolated by adding one volume of EtOH (70%) and further purified using the RNeasy Mini Kit (Qiagen, Hilden, Germany, catalogue number 74106). For reverse transcription 1 µg RNA was copied into cDNA using the QuantiTect Reverse Transcription Kit (Qiagen, catalogue number 205313). qPCR was performed with the QuantiTect SYBR Green PCR (Qiagen catalogue number 204145). The following primers were used:
KRT14 (forward: GCG CAC CAT GCA GAA CCT G reverse: CCT CCA CGC TGC CAA TCA TC, at 55 °C)
KRT20 (forward: GAA GTC CTC AGC AGC CAG TT, reverse: GGT CGC GAC TAC AGT GCA TA, at 60 °C)
UPK2 (forward: GAC AGC CAC TGA GTC CAG CAG, reverse: AGC ACC GTG ATG ACC ACC ATG, at 60 °C),
UTX (forward: CGA AAA ACA AGC GGA AAC T, reverse: TAT CAA GAT GAG GCG GAT G, at 55 °C),
TBP (forward: ACA ACA GCC TGC CAC CTT A, reverse: GAA TAG GCT GTG GGG TCA GT, at 60 °C)
Standard curves were carried in each experiment to calculate relative expression, and TBP was used as a reference gene.
4.5. Immunocytochemistry (ICC)
Cells were fixed and permeabilized using a solution of 1% paraformaldehyde and 0.02% Triton X-100 in PBS. All antibodies were diluted in the blocking solution (1% BSA, 0.1% saponine, 0.1% NaN3, dissolved in PBS). Used antibodies were: KRT5 (Abcam, Cambridge, UK, ab53121, at a 1:250 dilution) and KRT14 (Abcam, ab7800, 1:250) with secondary antibodies goat-anti-mouse IgG Alexa Fluor 633 (ThermoFischer Scientific, A-21052) and goat-anti-rabbit IgG Alexa Fluor 488 (ThermoFischer Scientific, A-11008). Phalloidin (Acti-stain 535 Phalloidin, tebu-bio, PHDR1, 1:1,000) was used to stain actin filaments and 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI, Roche, Mannheim, Germany, 10236276001) to stain DNA. Cover slips were mounted with Dako Fluorescence Mounting Medium (Dako, S3023). Detection was performed using ZEISS Axio Observer.Z1/7; Plan-Apochromat 40× /1.4 Oil DIC (UV) VIS-IR M27; 90 HE DAPI/ GFP/ Cy3 /Cy5; LED-module "wavelength" nm (Colibri 7); ZEISS Axiocam 512 mono.
Pictures were analyzed with ImageJ (version 1.52b) and a specific approach-based macro. The core adjustments were: DAPI-Channel (threshold: "Default"; "Watershed"); KRT14-Channel (threshold: "Huang"; "Watershed"). Regions of interest were counted by using "Extended Particle Analyzer" with a pixel ratio of 1:100,000.
4.6. Western Blot Analysis
Cells were scraped off, washed once with PBS and collected by centrifugation at 500 g for 5 min at RT. The cell pellet was then lysed with NP-40 lysis buffer (150 mM NaCl, 1% NP-40, 0.1% SDS, 1 mM EDTA, 50 mM Tris-HCl, pH 7.6, with protease inhibitor (Sigma Aldrich, P8340)). Lysates were cleared by centrifugation at 11,000 g for 20 min at 4 °C. Protein concentration was measured by the Pierce™ BCA Protein Assay Kit (Sigma Aldrich, 23225). Western blotting was performed using 15–50 µg protein in 10% or 12% SDS-PAGE gels in TGS-buffer (Bio-Rad, Feldkirchen, Germany, 1610732) at 20 mA for 60 min followed by transfer (100 V for 40 min) to a PVDF membrane with blotting buffer (125 mM Tris, 960 mM glycine, 10% methanol). The membrane was blocked with 5% milk in TBS-Tween (0.1%). The following antibodies were applied in TBS-Tween (0.1%); TubA (Abcam, #ab4074, at a dilution of 1:10,000); UTX (Cell Signal Technology, Frankfurt, Germany, 33510, 1:500); p53 (Merck Millipore, #OP43, at a dilution of 1:500); p21 (Merck Millipore; Darmstadt, Germany #OP64, at a dilution of 1:500); MDM2 (Oncogene Science, Uniondale, USA, #OP46, at a dilution of 1:500); goat anti-rabbit IgG H&L (HRP, Abcam, ab6721, 1:10,000); H3K27me2/3 (Active Motif, La Hulpe, Belgium, #39535); H3 (Cell Signaling Technology, #4499); goat anti-rabbit IgG H&L (HRP, Dako, Glostrup, Denmark, #P0448, 1:1,000); rabbit anti-mouse IgG H&L (HRP, Dako, #P0260, 1:1,000); IRDye
® 800CW goat anti-rabbit IgG (LI-COR Biosciences, Lincoln, NE, USA, 925-32211, 1:20,000). Membranes were developed by Clarity Western ECL Substrate (Bio-Rad #170-5061), and detection was performed on a Chemidoc Imagine System (Bio-Rad) or an Odyssey Classic with Image Studio Software (version 4.0, LI-COR Biosciences, Lincoln, NE, USA). Quantification as indicated below each blot figure was performed relative to tubulin α or histone H3 using the respective instrument or ImageJ software [
41].
4.7. Flow Cytometry
Cells were cultured on six-well plates and harvested by accutase or trypsin. After one washing, step cells were fixed and permeabilized using a solution of 1% paraformaldehyde and 0.02% triton X-100 in PBS. Antibodies against KRT5 (Sigma Aldrich, FCMAB291F, dilution 1:200), CD90 (Miltenyi Biotech, Bergisch-Gladbach, Germany, 120-007-297, dilution 1:100) and KRT14 (Novus Biologicals, Abingdon, UK, NBP2-34403APC, dilution 1:100), were diluted in blocking solution (1% BSA, 0.1% saponin, 0.1% NaN3 in PBS). Negative controls were performed with mouse Anti-IgG1-APC (Miltenyi Biotech, 130-117-058), mouse Anti-IgG1-PE (Miltenyi Biotech, 130-117-057) and mouse Anti-IgG1-FITC (Miltenyi Biotech, 130-095-897).
4.8. Viability Assay
Two-thousand cells in 100 µL cell culture media were seeded per well in 96-well-plates. After one day of culture cells were transfected with UTX or control siRNA. Two days later, viable cells were quantified by addition of 10 µL MTT for one or four (HBLAK) hours. After incubation at cell culture conditions, the supernatant was removed and cells were lysed in 50 µL DMSO. Absorbance was measured at 505 nm.
4.9. EdU-labeling
Cells were cultured and treated on six-well plates. On the day of measurement, the cells were incubated with 10 μM EdU for 4 h. Fixation and further steps were performed as described by the manufacturer (Sigma Aldrich, BCK-FC647-100). Detection was performed using a Miltenyi MACSQuant® Analyzer (Milteny Biotec, Bergisch-Gladbach, Germany) and evaluated using FlowJo (BD, Version 10.4, Franklin Lakes, NJ, USA).
4.10. AldeFluor-Assay
Cells were cultured and transfected on six-well plates. After harvesting and washing once with PBS, cells were resuspended in the AldeFluor reaction solution, as described by the manufacturer (STEMCELL Technologies, Cologne, Germany, catalogue number #01700). The detection was performed on a Miltenyi MACSQuant® Analyzer (Milteny Biotec) and evaluated using FlowJo (BD, Version 10.4, Franklin Lakes, NJ, USA).
4.11. Cell Cycle Analysis by Flow Cytometry
Cell cycle analyses were performed as previously described [
42]. Soluble and floating cells collected from the supernatant were stained with Nicoletti-buffer (50 µg/µL propidium iodide (PI), 0.1% sodium citrate and 0.1% Triton X-100), and profiles were established using a Miltenyi MACSQuant
® Analyzer (Milteny Biotec) and evaluated using FlowJo (BD, Version 10.4 Franklin Lakes, NJ, USA).
4.12. Caspase and CellTiter-Glo Assay
After one day of culture following siRNA transfection in 6-well plates, 2000 cells in 50 µL cell culture media were seeded on 96-well plates. After further two days of culture, 50 µL of Caspase-Glo® Reagent (Promega, G8090, Madison, WI, USA) or CellTiter-Glo® (Promega, G7570) was added to each well. After 30 min of incubation at 37 °C, signals were detected as described by the manufacturer.
4.13. RNA-Seq
Total RNA samples used for transcriptome analyses were quantified (Qubit RNA HS Assay, Thermo Fisher Scientific) and quality measured by capillary electrophoresis using the Fragment Analyzer and the Total RNA Standard Sensitivity Assay (Agilent Technologies, Inc. Santa Clara, CA, USA). All samples showed high quality RNA quality numbers (≥10). The library preparation was performed according to the manufacturer’s protocol using the “TruSeq Stranded mRNA Library Prep Kit” from Illumina®. Briefly, 300 ng total RNA were used for mRNA capturing, fragmentation, the synthesis of cDNA, adapter ligation and library amplification. Bead purified libraries were normalized and finally sequenced on the HiSeq 3000 system (Illumina Inc. San Diego, CA, USA) with a read setup of 1 × 150 bp. The bcl2fastq tool was used to convert the bcl files to fastq files as well for adapter trimming and demultiplexing.
Data analyses on fastq files were conducted with CLC Genomics Workbench (version 10.1.1, QIAGEN, Venlo, The Netherlands). The reads of all probes were adapter trimmed (Illumina TruSeq) and quality trimmed (using the default parameters: bases below Q13 were trimmed from the end of the reads, ambiguous nucleotides maximal 2). Mapping was done against the Homo sapiens (hg38) (Mai 25, 2017) genome sequence. After grouping of samples (three biological replicates each), a pairwise comparison was made and statistically determined using the Empirical Analysis of DGE (version 1.1, cutoff = 5). The Resulting
p values were corrected for multiple testing by FDR and Bonferroni-correction. A
p value of ≤0.05 was considered significant. RNA-seq data were further analyzed via the GSEA Software (GSEA v4.0.1 for Windows) [
43], the String (
https://string-db.org/) [
24] and DAVID databases [
25,
26]. The RNA-seq and the single cell RNA-seq data are available through the GEO database.
4.14. Single Cell RNA-Seq
HBLAK cells were harvested by accutase after routine cultivation in T25 flasks for two days. Cell viability and cell number analysis were performed via trypan blue staining in a Neubauer counting chamber. A total of 20,000 cells were used as input for the single-cell droplet libraries generation on the 10× Chromium Controller system utilizing the Chromium Single Cell 3 Reagent Kit v3 according to manufacturer’s instructions. Sequencing was carried out on a HiSeq 3000 system (Illumina Inc. San Diego, CA, USA) with a mean sequencing depth of ~40,000 reads/cell. Raw sequencing data were processed using the 10× Genomics CellRanger software (v3.1). Raw BCL-files were demultiplexed and processed to Fastq-files using the CellRanger mkfastq pipeline. Alignment of reads to the mm10 genome and UMI counting was performed via the CellRanger count pipeline to generate a gene-barcode matrix.
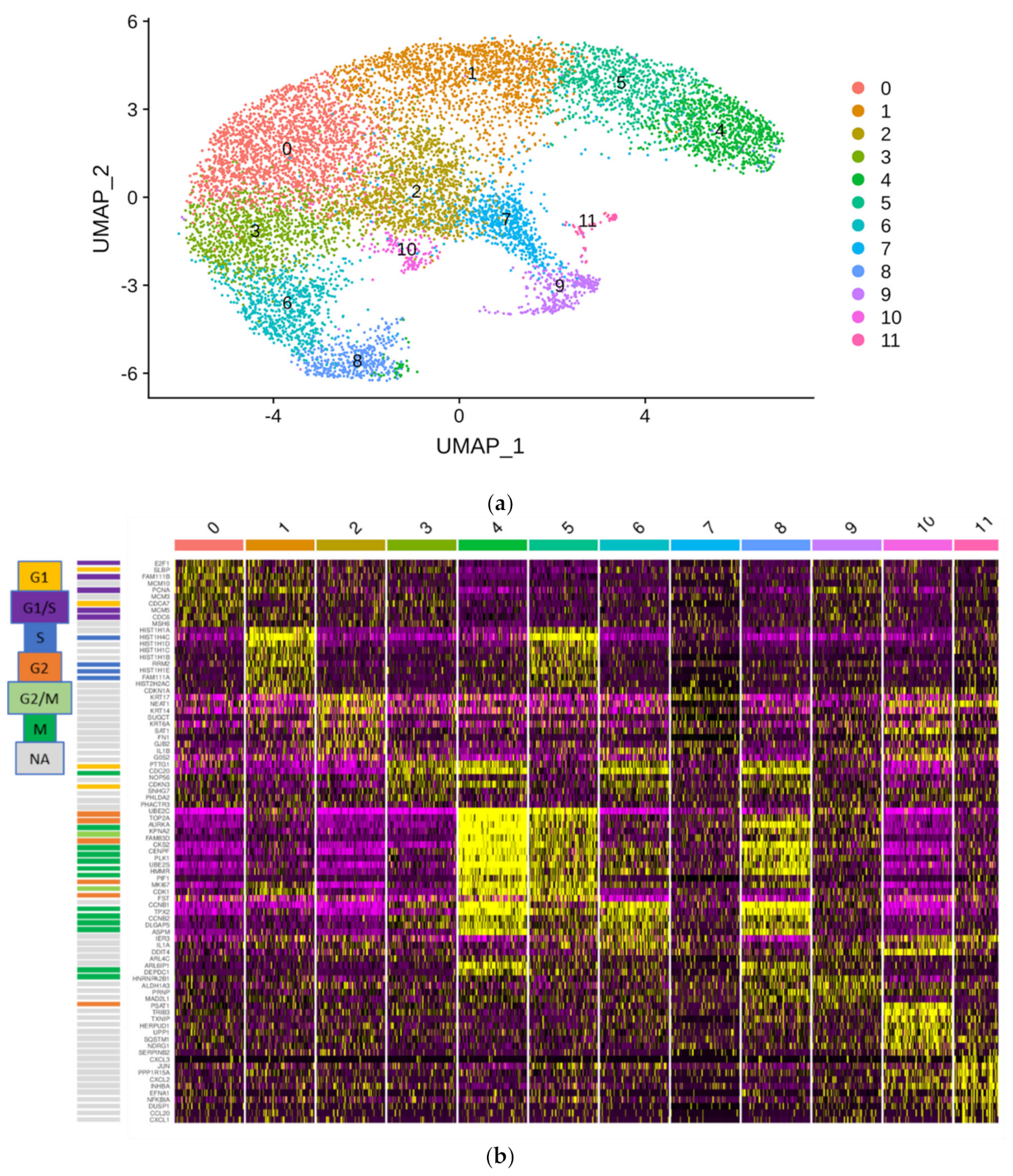
Further analyses were carried out with the Seurat v3.0 R package. Initial quality control consisted of removal of cells with fewer than 200 detected genes and removal of genes expressed in fewer than 3 cells. Furthermore, cells with a mapping rate of >10% to the mitochondrial genome were removed, under the assumption that they represent dead or damaged cells. Normalization was carried out utilizing SCTransform. Dimensional reduction of the data set was achieved by principal component analysis (PCA) based on identified variable genes and subsequent UMAP embedding. The number of meaningful principal components (PC) was selected by ranking them according to the percentage of variance explained by each PC, plotting them in an “elbow plot” and manually determining the number of PCs that represent the majority of variance in the data set. Cells were clustered using the graph-based clustering approach implemented in Seurat v3.0. Markers defining each cluster and differential gene expression between different clusters were calculated using a Wilcoxon Rank Sum test which was implemented in Seurat.
4.15. Statistics
All data were analyzed using the IBM SPSS Statistics v26.001 for Windows (2019) program. Results with p ≤ 0.05 were considered significant. Differences in mean values between two groups were statistically confirmed by means of the t test for independent samples. In all parametric tests used, the respective requirements were checked, and appropriate consequences were drawn for events of gross injuries. Unless indicated otherwise, the Tukey test was used as a post-hoc test to identify group differences in variance analysis.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










