Ex Vivo Organoid Cultures Reveal the Importance of the Tumor Microenvironment for Maintenance of Colorectal Cancer Stem Cells

 ,
, 
Abstract
1. Introduction
2. Results
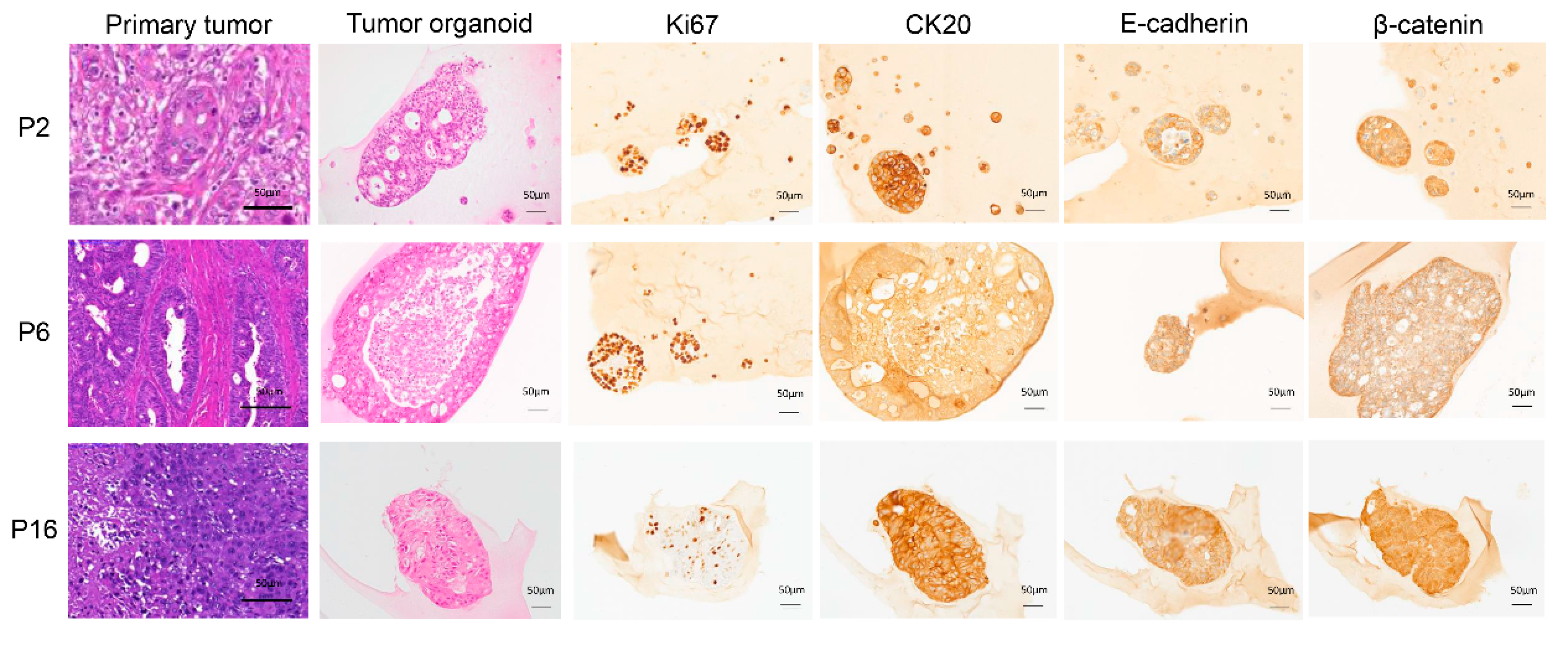
2.1. Establishment and Characterization of Long-Term CRC Organoid Cultures
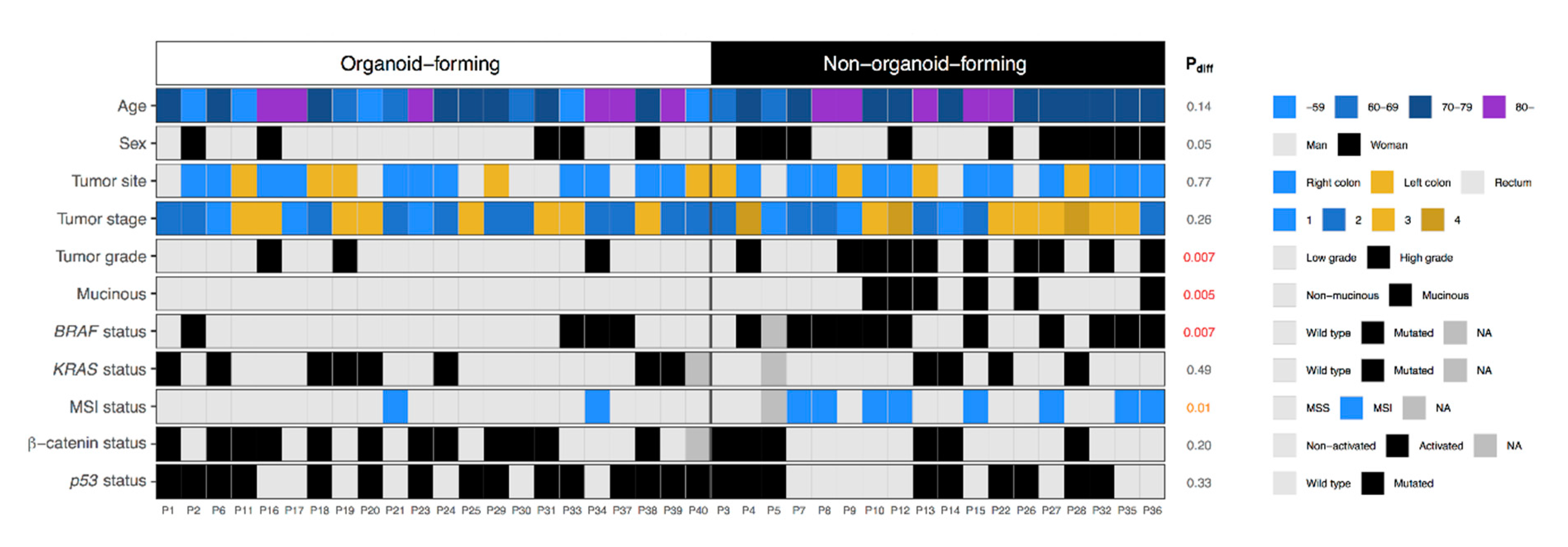
2.2. Establishment of Organoid Cultures in Relation to Clinicopathological Characteristics and Molecular Subtypes
2.3. Comparison of the Organoid-Forming and Non-Organoid-Forming Tumors Using RNA Sequencing Analysis
2.4. Survival Analysis of Organoid Establishment Status in the Cancer Genome Atlas (TCGA) Database
3. Discussion
4. Materials and Methods
4.1. Study Population of Patients with CRC
4.2. Tumor Cell Isolation, Organoid Culture, and Storage
4.3. Immunohistochemistry
4.4. DNA and RNA Extraction
4.5. Microsatellite Instability (MSI) Assessment
4.6. BRAFV600E Mutational Status
4.7. KRAS Sequencing
4.8. RNA Sequencing and Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020. [Google Scholar] [CrossRef] [PubMed]
- Popat, S.; Hubner, R.; Houlston, R.S. Systematic review of microsatellite instability and colorectal cancer prognosis. J. Clin. Oncol. 2005, 23, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Toyota, M.; Kondo, Y.; Lin, E.; Zhang, L.; Guo, Y.; Hernandez, N.S.; Chen, X.; Ahmed, S.; Konishi, K.; et al. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 18654–18659. [Google Scholar] [CrossRef]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.P. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 8681–8686. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- Fujii, M.; Shimokawa, M.; Date, S.; Takano, A.; Matano, M.; Nanki, K.; Ohta, Y.; Toshimitsu, K.; Nakazato, Y.; Kawasaki, K.; et al. A colorectal tumor organoid library demonstrates progressive loss of niche factor requirements during tumorigenesis. Cell Stem Cell 2016, 18, 827–838. [Google Scholar] [CrossRef]
- Korinek, V.; Barker, N.; Moerer, P.; van Donselaar, E.; Huls, G.; Peters, P.J.; Clevers, H. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat. Genet. 1998, 19, 379–383. [Google Scholar] [CrossRef]
- Van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef]
- Drost, J.; van Jaarsveld, R.H.; Ponsioen, B.; Zimberlin, C.; van Boxtel, R.; Buijs, A.; Sachs, N.; Overmeer, R.M.; Offerhaus, G.J.; Begthel, H.; et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 2015, 521, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Weeber, F.; van de Wetering, M.; Hoogstraat, M.; Dijkstra, K.K.; Krijgsman, O.; Kuilman, T.; Gadellaa-van Hooijdonk, C.G.M.; van der Velden, D.L.; Peeper, D.S.; Cuppen, E.P.J.G.; et al. Preserved genetic diversity in organoids cultured from biopsies of human colorectal cancer metastases. Proc. Natl. Acad. Sci. USA 2015, 112, 13308–13311. [Google Scholar] [CrossRef] [PubMed]
- Jung, P.; Sato, T.; Merlos-Suarez, A.; Barriga, F.M.; Iglesias, M.; Rossell, D.; Auer, H.; Gallardo, M.; Blasco, M.A.; Sancho, E.; et al. Isolation and in vitro expansion of human colonic stem cells. Nat. Med. 2011, 17, 1225–1227. [Google Scholar] [CrossRef] [PubMed]
- Clarke, C.N.; Kopetz, E.S. BRAF mutant colorectal cancer as a distinct subset of colorectal cancer: Clinical characteristics, clinical behavior, and response to targeted therapies. J. Gastrointest. Oncol. 2015, 6, 660–667. [Google Scholar] [CrossRef]
- Bylesjo, M.; Rantalainen, M.; Cloarec, O.; Nicholson, J.K.; Holmes, E.; Trygg, J. OPLS discriminant analysis: Combining the strengths of PLS-DA and SIMCA classification. J. Chemometr. 2006, 20, 341–351. [Google Scholar] [CrossRef]
- Barker, N.; Clevers, H. Leucine-rich repeat-containing G-protein-coupled receptors as markers of adult stem cells. Gastroenterology 2010, 138, 1681–1696. [Google Scholar] [CrossRef]
- Snippert, H.J.; Haegebarth, A.; Kasper, M.; Jaks, V.; van Es, J.H.; Barker, N.; van de Wetering, M.; van den Born, M.; Begthel, H.; Vries, R.G.; et al. Lgr6 marks stem cells in the hair follicle that generate all cell lineages of the skin. Science 2010, 327, 1385–1389. [Google Scholar] [CrossRef]
- De Lau, W.; Barker, N.; Low, T.Y.; Koo, B.K.; Li, V.S.W.; Teunissen, H.; Kujala, P.; Haegebarth, A.; Peters, P.J.; van de Wetering, M.; et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 2011, 476, 293–297. [Google Scholar] [CrossRef]
- Gong, X.; Carmon, K.S.; Lin, Q.S.; Thomas, A.; Yi, J.; Liu, Q.Y. LGR6 Is a high affinity receptor of R-spondins and potentially functions as a tumor suppressor. PLoS ONE 2012, 7, e37137. [Google Scholar] [CrossRef]
- Manieri, N.A.; Drylewicz, M.R.; Miyoshi, H.; Stappenbeck, T.S. Igf2bp1 is required for full induction of Ptgs2 mRNA in colonic mesenchymal stem cells in mice. Gastroenterology 2012, 143, 110–121. [Google Scholar] [CrossRef]
- Vikesaa, J.; Hansen, T.V.O.; Jonson, L.; Borup, R.; Wewer, U.M.; Christiansen, J.; Nielsen, F.C. RNA-binding IMPs promote cell adhesion and invadopodia formation. EMBO J. 2006, 25, 1456–1468. [Google Scholar] [CrossRef]
- Dalerba, P.; Dylla, S.J.; Park, I.K.; Liu, R.; Wang, X.H.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M.; et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.M.; Martinez, N.J.; Thornton, J.E.; Hagan, J.P.; Nguyen, K.D.; Gregory, R.I. Trim71 cooperates with microRNAs to repress Cdkn1a expression and promote embryonic stem cell proliferation. Nat. Commun. 2012, 3, 923. [Google Scholar] [CrossRef] [PubMed]
- Semenov, M.V.; Zhang, X.; He, X. DKK1 antagonizes Wnt signaling without promotion of LRP6 internalization and degradation. J. Biol. Chem. 2008, 283, 21427–21432. [Google Scholar] [CrossRef] [PubMed]
- Schueler, M.; Braun, D.A.; Chandrasekar, G.; Gee, H.Y.; Klasson, T.D.; Halbritter, J.; Bieder, A.; Porath, J.D.; Airik, R.; Zhou, W.B.; et al. DCDC2 mutations cause a renal-hepatic ciliopathy by disrupting wnt signaling. Am. J. Hum. Genet. 2015, 96, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Mikels, A.J.; Nusse, R. Purified Wnt5a protein activates or inhibits beta-catenin-TCF signaling depending on receptor context. PLoS Biol. 2006, 4, 570–582. [Google Scholar] [CrossRef]
- Gurney, A.; Axelrod, F.; Bond, C.J.; Cain, J.; Chartier, C.; Donigan, L.; Fischer, M.; Chaudhari, A.; Ji, M.; Kapoun, A.M.; et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 11717–11722. [Google Scholar] [CrossRef]
- Thibodeau, S.N.; Bren, G.; Schaid, D. Microsatellite instability in cancer of the proximal colon. Science 1993, 260, 816–819. [Google Scholar] [CrossRef]
- Ionov, Y.; Peinado, M.A.; Malkhosyan, S.; Shibata, D.; Perucho, M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993, 363, 558–561. [Google Scholar] [CrossRef]
- Geiersbach, K.B.; Samowitz, W.S. Microsatellite instability and colorectal cancer. Arch. Pathol. Lab. Med. 2011, 135, 1269–1277. [Google Scholar] [CrossRef]
- De Roock, W.; Claes, B.; Bernasconi, D.; De Schutter, J.; Biesmans, B.; Fountzilas, G.; Kalogeras, K.T.; Kotoula, V.; Papamichael, D.; Laurent-Puig, P.; et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol. 2010, 11, 753–762. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Kohne, C.H.; Lang, I.; Folprecht, G.; Nowacki, M.P.; Cascinu, S.; Shchepotin, I.; Maurel, J.; Cunningham, D.; Tejpar, S.; et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: Updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J. Clin. Oncol. 2011, 29, 2011–2019. [Google Scholar] [CrossRef] [PubMed]
- Todaro, M.; Alea, M.P.; Di Stefano, A.B.; Cammareri, P.; Vermeulen, L.; Lovino, F.; Tripodo, C.; Russo, A.; Gulotta, G.; Medema, J.P.; et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell 2007, 1, 389–402. [Google Scholar] [CrossRef]
- Benfey, P.N. Defining the path from stem cells to differentiated tissue. In Current Topics in Developmental Biology; Wassarman, P.M., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 116, pp. 35–44. [Google Scholar]
- Sadanandam, A.; Lyssiotis, C.A.; Homicsko, K.; Collisson, E.A.; Gibb, W.J.; Wullschleger, S.; Ostos, L.C.G.; Lannon, W.A.; Grotzinger, C.; Del Rio, M.; et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat. Med. 2013, 19, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Jorissen, R.N.; Lipton, L.; Gibbs, P.; Chapman, M.; Desai, J.; Jones, I.T.; Yeatman, T.J.; East, P.; Tomlinson, I.P.M.; Verspaget, H.W.; et al. DNA copy-number alterations underlie gene expression differences between microsatellite stable and unstable colorectal cancers. Clin. Cancer Res. 2008, 14, 8061–8069. [Google Scholar] [CrossRef] [PubMed]
- Leman, J.K.H.; Sandford, S.K.; Rhodes, J.L.; Kemp, R.A. Multiparametric analysis of colorectal cancer immune responses. World J. Gastroenterol. 2018, 24, 2995–3005. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.; In, J.; Zachos, N.; Huso, D.; Maegawa, S.; Issa, J.P.; Easwaran, H.; Baylin, S.B. Oncogenic BRAFV600E drives stem cell niche factors-independent growth and tumorigenic transformation in colon organoids. Cancer Res. 2016, 76, Am2016–Am4273. [Google Scholar]
- Eklof, V.; Wikberg, M.L.; Edin, S.; Dahlin, A.M.; Jonsson, B.A.; Oberg, A.; Rutegard, J.; Palmqvist, R. The prognostic role of KRAS, BRAF, PIK3CA and PTEN in colorectal cancer. Br. J. Cancer 2013, 108, 2153–2163. [Google Scholar] [CrossRef]
- Li, W.Q.; Kawakami, K.; Ruszkiewicz, A.; Bennett, G.; Moore, J.; Iacopetta, B. BRAF mutations are associated with distinctive clinical, pathological and molecular features of colorectal cancer independently of microsatellite instability status. Mol. Cancer. 2006, 5. [Google Scholar] [CrossRef]
- Samowitz, W.S.; Sweeney, C.; Herrick, J.; Albertsen, H.; Levin, T.R.; Murtaugh, M.A.; Wolff, R.K.; Slattery, M.L. Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer Res. 2005, 65, 6063–6070. [Google Scholar] [CrossRef]
- Seppala, T.T.; Bohm, J.P.; Friman, M.; Lahtinen, L.; Vayrynen, V.M.J.; Liipo, T.K.E.; Ristimaki, A.P.; Kairaluoma, M.V.J.; Kellokumpu, I.H.; Kuopio, T.H.I.; et al. Combination of microsatellite instability and BRAF mutation status for subtyping colorectal cancer. Br. J. Cancer 2015, 112, 1966–1975. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Brahmandam, M.; Cantor, M.; Namgyal, C.; Kawasaki, T.; Kirkner, G.; Meyerhardt, J.A.; Loda, M.; Fuchs, C.S. Distinct molecular features of colorectal carcinoma with signet ring cell component and colorectal carcinoma with mucinous component. Mod. Pathol. 2006, 19, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, K.; Clevers, H. Wnt/beta-catenin signaling in adult mammalian epithelial stem cells. Dev. Biol. 2017, 428, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Loh, K.M.; Nusse, R. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 2014, 346, 1248012. [Google Scholar] [CrossRef]
- Schatoff, E.M.; Leach, B.I.; Dow, L.E. WNT signaling and colorectal cancer. Curr. Colorectal Cancer Rep. 2017, 13, 101–110. [Google Scholar] [CrossRef]
- Tilg, H.; Adolph, T.E.; Gerner, R.R.; Moschen, A.R. The intestinal microbiota in colorectal cancer. Cancer Cell 2018, 33, 954–964. [Google Scholar] [CrossRef]
- Brennan, C.A.; Garrett, W.S. Gut microbiota, inflammation, and colorectal cancer. Annu. Rev. Microbiol. 2016, 70, 395–411. [Google Scholar] [CrossRef]
- Choi, C.R.; Al Bakir, I.; Ding, N.J.; Lee, G.H.; Askari, A.; Warusavitarne, J.; Moorghen, M.; Humphries, A.; Ignjatovic-Wilson, A.; Thomas-Gibson, S.; et al. Cumulative burden of inflammation predicts colorectal neoplasia risk in ulcerative colitis: A large single-centre study. Gut 2019, 68, 414–422. [Google Scholar] [CrossRef]
- Tlaskalova-Hogenova, H.; Vannucci, L.; Klimesova, K.; Stepankova, R.; Krizan, J.; Kverka, M. Microbiome and colorectal carcinoma: Insights from germ-free and conventional animal models. Cancer J. 2014, 20, 217–224. [Google Scholar] [CrossRef]
- Abu-Shakra, M.; Buskila, D.; Ehrenfeld, M.; Conrad, K.; Shoenfeld, Y. Cancer and autoimmunity: Autoimmune and rheumatic features in patients with malignancies. Ann. Rheum. Dis. 2001, 60, 433–440. [Google Scholar] [CrossRef]
- Zornig, I.; Halama, N.; Bermejo, J.L.; Ziegelmeier, C.; Dickes, E.; Migdoll, A.; Kaiser, I.; Waterboer, T.; Pawlita, M.; Grabe, N.; et al. Prognostic significance of spontaneous antibody responses against tumor-associated antigens in malignant melanoma patients. Int. J. Cancer 2015, 136, 138–151. [Google Scholar] [CrossRef] [PubMed]
- Barbera-Guillem, E.; May, K.F., Jr.; Nyhus, J.K.; Nelson, M.B. Promotion of tumor invasion by cooperation of granulocytes and macrophages activated by anti-tumor antibodies. Neoplasia 1999, 1, 453–460. [Google Scholar] [CrossRef] [PubMed]
- De Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef]
- Afshar-Kharghan, V. The role of the complement system in cancer. J. Clin. Investig. 2017, 127, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Ytting, H.; Jensenius, J.C.; Christensen, I.J.; Thiel, S.; Nielsen, H.J. Increased activity of the mannan-binding lectin complement activation pathway in patients with colorectal cancer. Scand. J. Gastroenterol. 2004, 39, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Ytting, H.; Christensen, I.J.; Thiel, S.; Jensenius, J.C.; Nielsen, H.J. Serum mannan-binding lectin-associated serine protease 2 levels in colorectal cancer: Relation to recurrence and mortality. Clin. Cancer Res. 2005, 11, 1441–1446. [Google Scholar] [CrossRef][Green Version]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Maimela, N.R.; Liu, S.S.; Zhang, Y. Fates of CD8+ T cells in tumor microenvironment. Comput. Struct. Biotechnol. 2019, 17, 1–13. [Google Scholar] [CrossRef]
- Glimelius, B.; Melin, B.; Enblad, G.; Alafuzoff, I.; Beskow, A.; Ahlstrom, H.; Bill-Axelson, A.; Birgisson, H.; Bjor, O.; Edqvist, P.H.; et al. U-CAN: A prospective longitudinal collection of biomaterials and clinical information from adult cancer patients in Sweden. Acta Oncol. 2018, 57, 187–194. [Google Scholar] [CrossRef]
- Bettington, M.L.; Walker, N.I.; Rosty, C.; Brown, I.S.; Clouston, A.D.; McKeone, D.M.; Pearson, S.A.; Klein, K.; Leggett, B.A.; Whitehall, V.L. A clinicopathological and molecular analysis of 200 traditional serrated adenomas. Mod. Pathol. 2015, 28, 414–427. [Google Scholar] [CrossRef]
- Takayama, T.; Miyanishi, K.; Hayashi, T.; Sato, Y.; Niitsu, Y. Colorectal cancer: Genetics of development and metastasis. J. Gastroenterol. 2006, 41, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Kobel, M.; Piskorz, A.M.; Lee, S.; Lui, S.H.; LePage, C.; Marass, F.; Rosenfeld, N.; Masson, A.M.M.; Brenton, J.D. Optimized p53 immunohistochemistry is an accurate predictor of TP53 mutation in ovarian carcinoma. J. Pathol. Clin. Res. 2016, 2, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; San Luis, B.; Lane, D.P. Intratumour heterogeneity of p53 expression; causes and consequences. J. Pathol. 2019, 249, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Bacher, J.W.; Flanagan, L.A.; Smalley, R.L.; Nassif, N.A.; Burgart, L.J.; Halberg, R.B.; Megid, W.M.; Thibodeau, S.N. Development of a fluorescent multiplex assay for detection of MSI-High tumors. Dis. Markers 2004, 20, 237–250. [Google Scholar] [CrossRef]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hiddessen, A.L.; Legler, T.C.; et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef]
- Pinheiro, L.B.; Coleman, V.A.; Hindson, C.M.; Herrmann, J.; Hindson, B.J.; Bhat, S.; Emslie, K.R. Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal. Chem. 2012, 84, 1003–1011. [Google Scholar] [CrossRef]
- Benlloch, S.; Paya, A.; Alenda, C.; Bessa, X.; Andreu, M.; Jover, R.; Castells, A.; Llor, X.; Aranda, F.I.; Massuti, B. Detection of BRAF V600E mutation in colorectal cancer: Comparison of automatic sequencing and real-time chemistry methodology. J. Mol. Diagn. 2006, 8, 540–543. [Google Scholar] [CrossRef]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef]
- Kim, D.; Landmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Racle, J.; de Jonge, K.; Baumgaertner, P.; Speiser, D.E.; Gfeller, D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. eLife 2017, 6, e26476. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Annotated Functions | p-Value | Number of Genes |
|---|---|---|
| Complement activation | <0.001 | 19 |
| Complement activation, classical pathway | <0.001 | 19 |
| Fc-gamma receptor signaling pathway involved in phagocytosis | <0.001 | 18 |
| Receptor-mediated endocytosis | <0.001 | 20 |
| Regulation of immune response | <0.001 | 19 |
| Immune response | <0.001 | 25 |
| Fc-epsilon receptor signaling pathway | <0.001 | 17 |
| Proteolysis | <0.001 | 24 |
| O-glycan processing | 0.006 | 7 |
| Maintenance of gastrointestinal epithelium | 0.027 | 4 |
| Inflammatory response | 0.029 | 14 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Larsson, P.; Ljuslinder, I.; Öhlund, D.; Myte, R.; Löfgren-Burström, A.; Zingmark, C.; Ling, A.; Edin, S.; Palmqvist, R. Ex Vivo Organoid Cultures Reveal the Importance of the Tumor Microenvironment for Maintenance of Colorectal Cancer Stem Cells. Cancers 2020, 12, 923. https://doi.org/10.3390/cancers12040923
Li X, Larsson P, Ljuslinder I, Öhlund D, Myte R, Löfgren-Burström A, Zingmark C, Ling A, Edin S, Palmqvist R. Ex Vivo Organoid Cultures Reveal the Importance of the Tumor Microenvironment for Maintenance of Colorectal Cancer Stem Cells. Cancers. 2020; 12(4):923. https://doi.org/10.3390/cancers12040923
Chicago/Turabian StyleLi, Xingru, Pär Larsson, Ingrid Ljuslinder, Daniel Öhlund, Robin Myte, Anna Löfgren-Burström, Carl Zingmark, Agnes Ling, Sofia Edin, and Richard Palmqvist. 2020. "Ex Vivo Organoid Cultures Reveal the Importance of the Tumor Microenvironment for Maintenance of Colorectal Cancer Stem Cells" Cancers 12, no. 4: 923. https://doi.org/10.3390/cancers12040923
APA StyleLi, X., Larsson, P., Ljuslinder, I., Öhlund, D., Myte, R., Löfgren-Burström, A., Zingmark, C., Ling, A., Edin, S., & Palmqvist, R. (2020). Ex Vivo Organoid Cultures Reveal the Importance of the Tumor Microenvironment for Maintenance of Colorectal Cancer Stem Cells. Cancers, 12(4), 923. https://doi.org/10.3390/cancers12040923