Molecular Heterogeneity and Cellular Diversity: Implications for Precision Treatment in Medulloblastoma

Abstract
1. Introduction
2. Molecular Heterogeneity in MB
2.1. Molecular Stratifications of MB
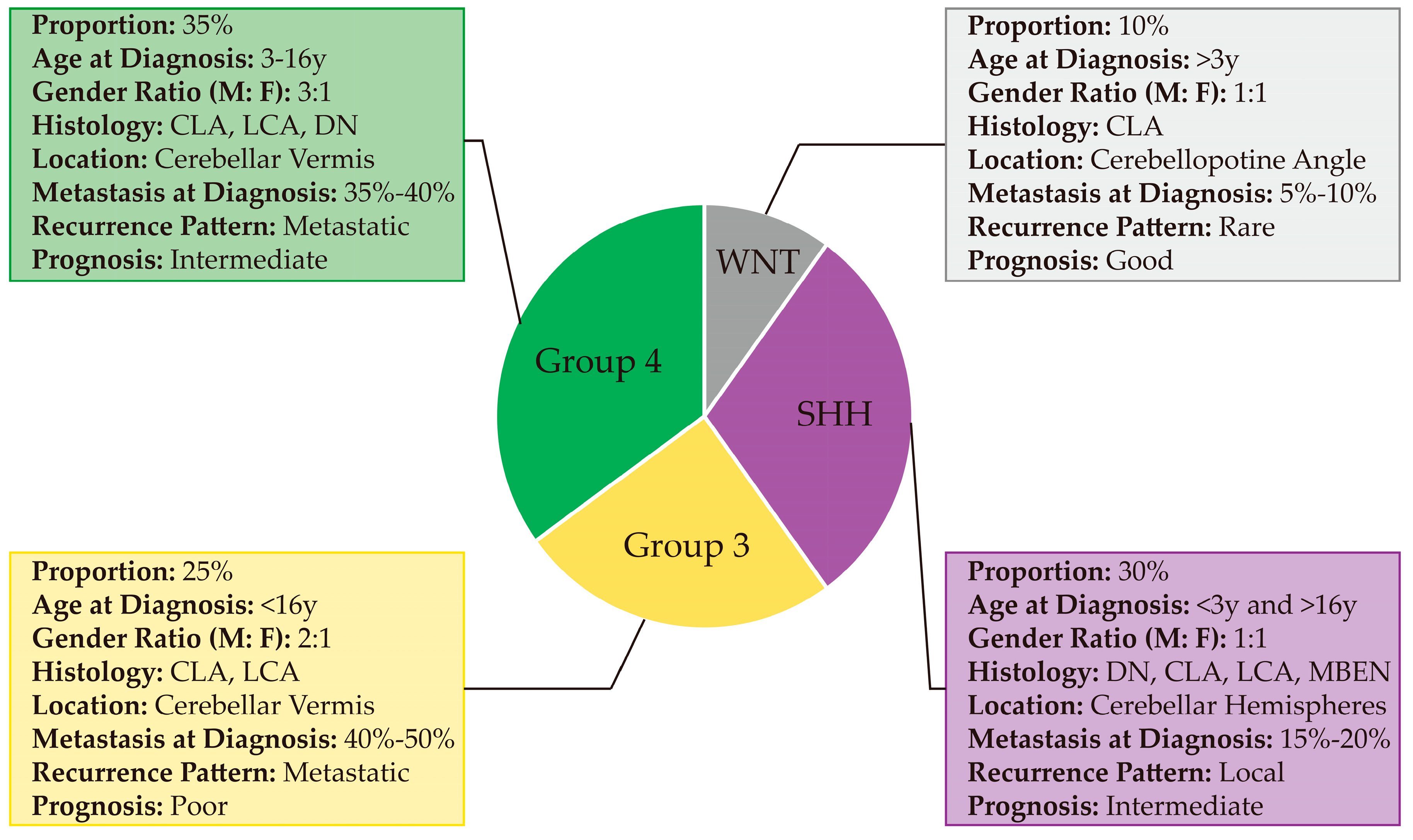
2.1.1. WNT
2.1.2. SHH
2.1.3. Group 3
2.1.4. Group 4
2.2. Epigenetic Regulation in MB Subgroups
2.2.1. DNA Methylation
2.2.2. Histone Modifications
2.2.3. ATP-Dependent Chromatin Remodeling
2.2.4. Genomic Structural Variations
2.3. Proteomics in MB Subgroups
3. Cellular Heterogeneity in MB
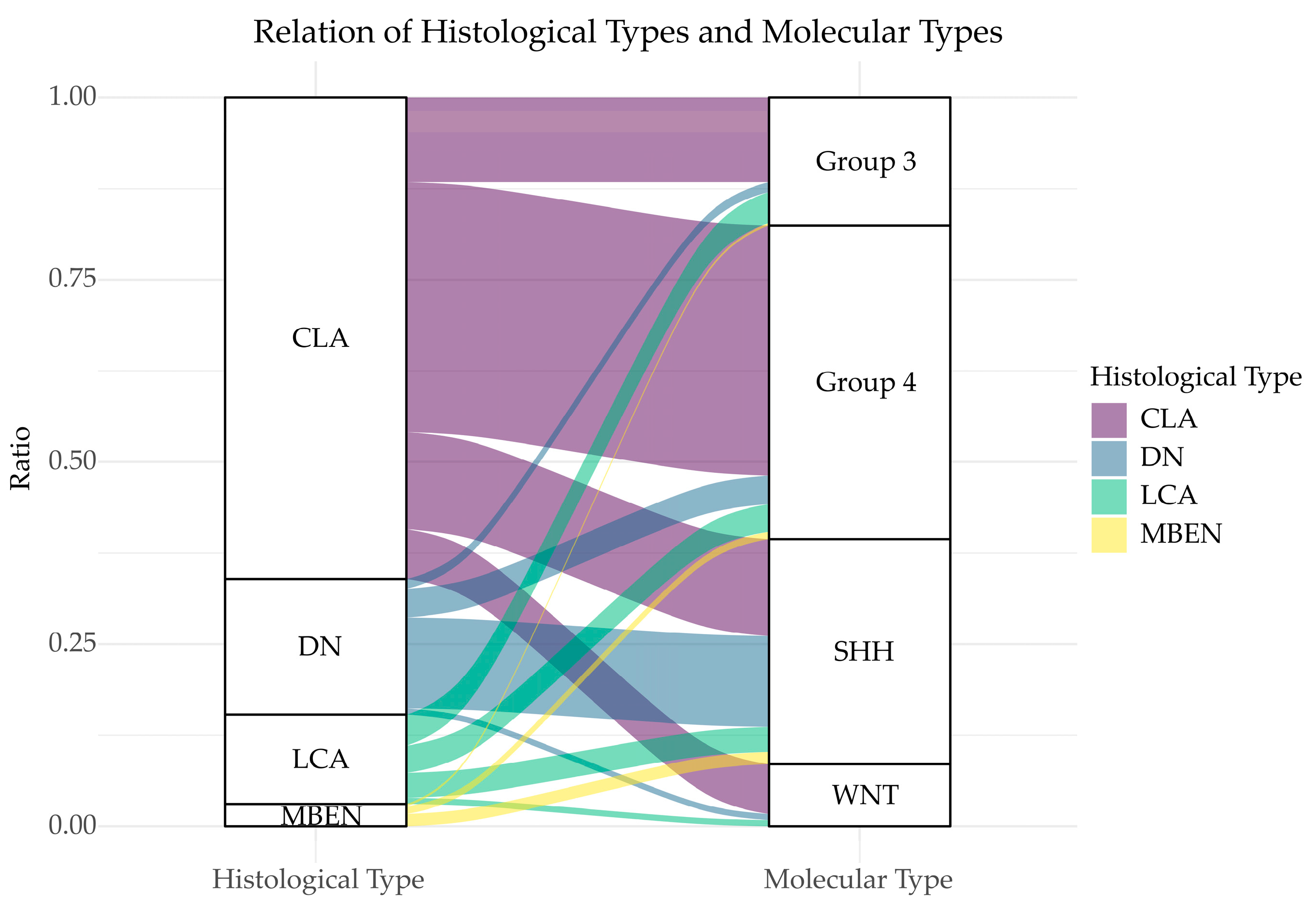
3.1. Histological Diversity of MB
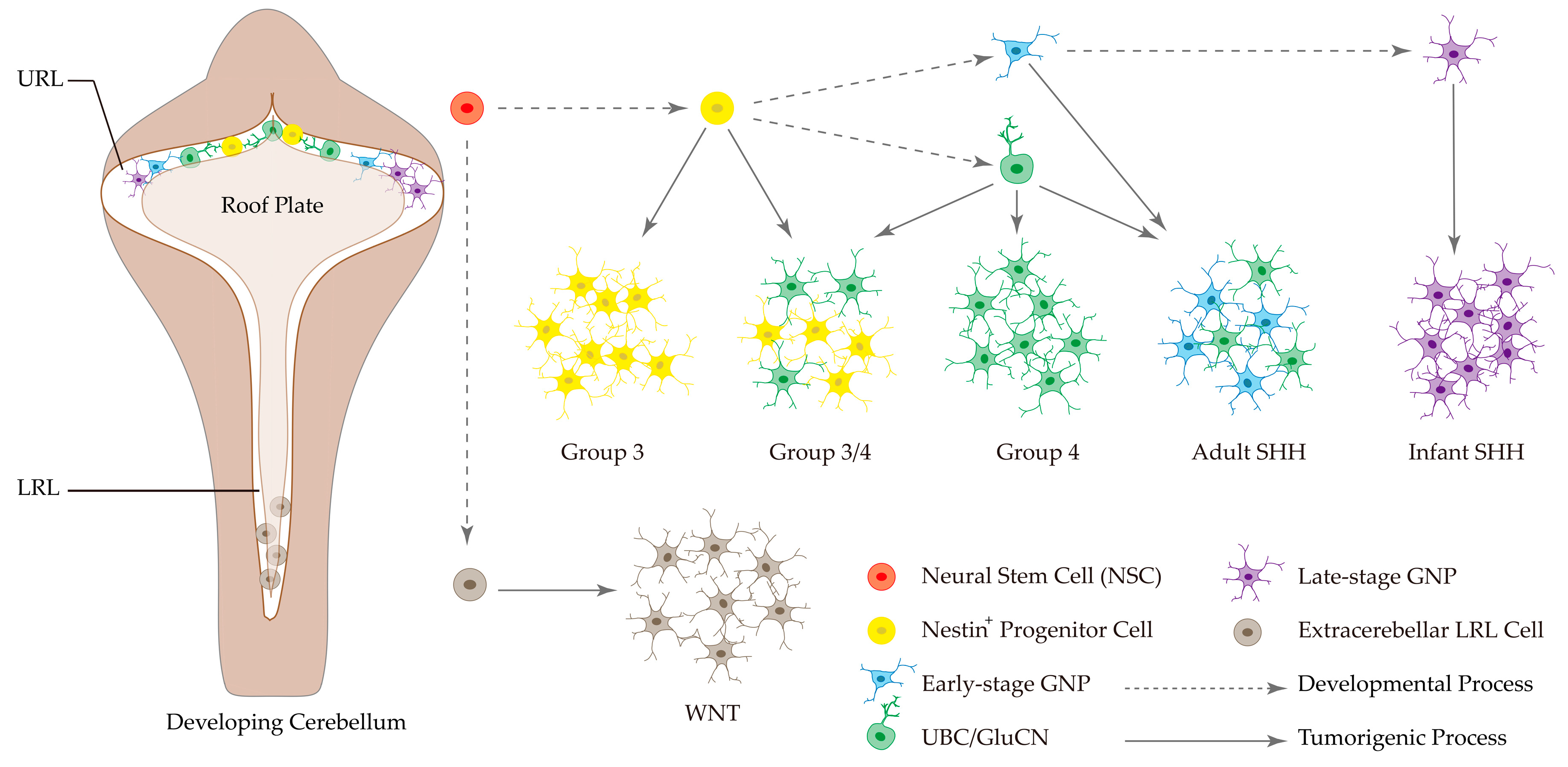
3.2. Cell of Origins in MB Subgroups
3.3. Diversity of Tumor Microenvironment in MB
4. Diagnosis, Current Therapies and Clinical Trials for MB Subgroups
4.1. Diagnosis of MB Subgroups
4.2. Current Therapies
4.3. Clinical Trials
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Khanna, V.; Achey, R.L.; Ostrom, Q.T.; Block-Beach, H.; Kruchko, C.; Barnholtz-Sloan, J.S.; de Blank, P.M. Incidence and survival trends for medulloblastomas in the United States from 2001 to 2013. J. Neurooncol. 2017, 135, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Ellison, D.W. Childhood medulloblastoma: Novel approaches to the classification of a heterogeneous disease. Acta Neuropathol. 2010, 120, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Eberhart, C.G.; Kepner, J.L.; Goldthwaite, P.T.; Kun, L.E.; Duffner, P.K.; Friedman, H.S.; Strother, D.R.; Burger, P.C. Histopathologic grading of medulloblastomas: A pediatric oncology group study. Cancer 2002, 94, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Albright, A.L.; Wisoff, J.H.; Zeltzer, P.M.; Boyett, J.M.; Rorke, L.B.; Stanley, P. Effects of medulloblastoma resections on outcome in children: A report from the Children’s Cancer Group. Neurosurgery 1996, 38, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.J.; Tsherniak, A.; Tamayo, P.; Santagata, S.; Ligon, A.; Greulich, H.; Berhoukim, R.; Amani, V.; Goumnerova, L.; Eberhart, C.G.; et al. Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J. Clin. Oncol. 2011, 29, 1424–1430. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Koster, J.; Bunt, J.; Hasselt, N.E.; Lakeman, A.; van Sluis, P.; Troost, D.; Meeteren, N.S.; Caron, H.N.; Cloos, J.; et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS ONE 2008, 3, e3088. [Google Scholar] [CrossRef]
- Northcott, P.A.; Korshunov, A.; Witt, H.; Hielscher, T.; Eberhart, C.G.; Mack, S.; Bouffet, E.; Clifford, S.C.; Hawkins, C.E.; French, P.; et al. Medulloblastoma comprises four distinct molecular variants. J. Clin. Oncol. 2011, 29, 1408–1414. [Google Scholar] [CrossRef]
- Remke, M.; Hielscher, T.; Northcott, P.A.; Witt, H.; Ryzhova, M.; Wittmann, A.; Benner, A.; von Deimling, A.; Scheurlen, W.; Perry, A.; et al. Adult medulloblastoma comprises three major molecular variants. J. Clin. Oncol. 2011, 29, 2717–2723. [Google Scholar] [CrossRef]
- Thompson, M.C.; Fuller, C.; Hogg, T.L.; Dalton, J.; Finkelstein, D.; Lau, C.C.; Chintagumpala, M.; Adesina, A.; Ashley, D.M.; Kellie, S.J.; et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J. Clin. Oncol. 2006, 24, 1924–1931. [Google Scholar] [CrossRef]
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol 2012, 123, 465–472. [Google Scholar] [CrossRef]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Bailey, S.; Clifford, S.C.; Doz, F.; Kool, M.; Dufour, C.; Vassal, G.; Milde, T.; et al. Risk stratification of childhood medulloblastoma in the molecular era: The current consensus. Acta Neuropathol. 2016, 131, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, F.M.G.; Remke, M.; Rampasek, L.; Peacock, J.; Shih, D.J.H.; Luu, B.; Garzia, L.; Torchia, J.; Nor, C.; Morrissy, A.S.; et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2017, 31, 737–754. [Google Scholar] [CrossRef] [PubMed]
- Ris, M.D.; Packer, R.; Goldwein, J.; Jones-Wallace, D.; Boyett, J.M. Intellectual outcome after reduced-dose radiation therapy plus adjuvant chemotherapy for medulloblastoma: A Children’s Cancer Group study. J. Clin. Oncol. 2001, 19, 3470–3476. [Google Scholar] [CrossRef] [PubMed]
- Gessi, M.; Maderna, E.; Guzzetti, S.; Cefalo, G.; Massimino, M.; Solero, C.L.; Finocchiaro, G.; Pollo, B. Radiation-induced glioblastoma in a medulloblastoma patient: A case report with molecular features. Neuropathology 2008, 28, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Madden, J.R.; Addo-Yobo, S.O.; Donson, A.M.; Liu, A.K.; McNatt, S.A.; Kleinschmidt-Demasters, B.K.; Fenton, L.Z.; Foreman, N.K.; Smith, A.A. Radiation-induced glioblastoma multiforme in children treated for medulloblastoma with characteristics of both medulloblastoma and glioblastoma multiforme. J. Pediatr. Hematol. Oncol. 2010, 32, e272–e278. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Hovestadt, V.; Smith, K.S.; Bihannic, L.; Filbin, M.G.; Shaw, M.L.; Baumgartner, A.; DeWitt, J.C.; Groves, A.; Mayr, L.; Weisman, H.R.; et al. Resolving medulloblastoma cellular architecture by single-cell genomics. Nature 2019, 572, 74–79. [Google Scholar] [CrossRef]
- Vladoiu, M.C.; El-Hamamy, I.; Donovan, L.K.; Farooq, H.; Holgado, B.L.; Sundaravadanam, Y.; Ramaswamy, V.; Hendrikse, L.D.; Kumar, S.; Mack, S.C.; et al. Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature 2019, 572, 67–73. [Google Scholar] [CrossRef]
- Zhang, L.; He, X.; Liu, X.; Zhang, F.; Huang, L.F.; Potter, A.S.; Xu, L.; Zhou, W.; Zheng, T.; Luo, Z.; et al. Single-Cell Transcriptomics in Medulloblastoma Reveals Tumor-Initiating Progenitors and Oncogenic Cascades during Tumorigenesis and Relapse. Cancer Cell 2019, 36, 302–318. [Google Scholar] [CrossRef]
- Clifford, S.C.; Lusher, M.E.; Lindsey, J.C.; Langdon, J.A.; Gilbertson, R.J.; Straughton, D.; Ellison, D.W. Wnt/Wingless pathway activation and chromosome 6 loss characterize a distinct molecular sub-group of medulloblastomas associated with a favorable prognosis. Cell Cycle 2006, 5, 2666–2670. [Google Scholar] [CrossRef]
- Kool, M.; Korshunov, A.; Remke, M.; Jones, D.T.; Schlanstein, M.; Northcott, P.A.; Cho, Y.J.; Koster, J.; Schouten-van Meeteren, A.; van Vuurden, D.; et al. Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012, 123, 473–484. [Google Scholar] [CrossRef]
- Northcott, P.A.; Hielscher, T.; Dubuc, A.; Mack, S.; Shih, D.; Remke, M.; Al-Halabi, H.; Albrecht, S.; Jabado, N.; Eberhart, C.G.; et al. Pediatric and adult sonic hedgehog medulloblastomas are clinically and molecularly distinct. Acta Neuropathol. 2011, 122, 231–240. [Google Scholar] [CrossRef]
- Taylor, M.D.; Liu, L.; Raffel, C.; Hui, C.C.; Mainprize, T.G.; Zhang, X.; Agatep, R.; Chiappa, S.; Gao, L.; Lowrance, A.; et al. Mutations in SUFU predispose to medulloblastoma. Nat. Genet. 2002, 31, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Brugieres, L.; Pierron, G.; Chompret, A.; Paillerets, B.B.; Di Rocco, F.; Varlet, P.; Pierre-Kahn, A.; Caron, O.; Grill, J.; Delattre, O. Incomplete penetrance of the predisposition to medulloblastoma associated with germ-line SUFU mutations. J. Med. Genet. 2010, 47, 142–144. [Google Scholar] [CrossRef] [PubMed]
- Zhukova, N.; Ramaswamy, V.; Remke, M.; Pfaff, E.; Shih, D.J.; Martin, D.C.; Castelo-Branco, P.; Baskin, B.; Ray, P.N.; Bouffet, E.; et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J. Clin. Oncol. 2013, 31, 2927–2935. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Pomeranz Krummel, D.; Pomeroy, S. The evolution of medulloblastoma therapy to personalized medicine. F1000Res 2017, 6, 490. [Google Scholar] [CrossRef] [PubMed]
- Garzia, L.; Kijima, N.; Morrissy, A.S.; De Antonellis, P.; Guerreiro-Stucklin, A.; Holgado, B.L.; Wu, X.; Wang, X.; Parsons, M.; Zayne, K.; et al. A Hematogenous Route for Medulloblastoma Leptomeningeal Metastases. Cell 2018, 172, 1050–1062. [Google Scholar] [CrossRef]
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Grobner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A.; et al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017, 547, 311–317. [Google Scholar] [CrossRef]
- Gomez, S.; Garrido-Garcia, A.; Garcia-Gerique, L.; Lemos, I.; Sunol, M.; de Torres, C.; Kulis, M.; Perez-Jaume, S.; Carcaboso, A.M.; Luu, B.; et al. A Novel Method for Rapid Molecular Subgrouping of Medulloblastoma. Clin. Cancer Res. 2018, 24, 1355–1363. [Google Scholar] [CrossRef]
- Batora, N.V.; Sturm, D.; Jones, D.T.; Kool, M.; Pfister, S.M.; Northcott, P.A. Transitioning from genotypes to epigenotypes: Why the time has come for medulloblastoma epigenomics. Neuroscience 2014, 264, 171–185. [Google Scholar] [CrossRef]
- Dubuc, A.M.; Remke, M.; Korshunov, A.; Northcott, P.A.; Zhan, S.H.; Mendez-Lago, M.; Kool, M.; Jones, D.T.; Unterberger, A.; Morrissy, A.S.; et al. Aberrant patterns of H3K4 and H3K27 histone lysine methylation occur across subgroups in medulloblastoma. Acta Neuropathol. 2013, 125, 373–384. [Google Scholar] [CrossRef]
- Jones, D.T.; Northcott, P.A.; Kool, M.; Pfister, S.M. The role of chromatin remodeling in medulloblastoma. Brain Pathol. 2013, 23, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef]
- Schwalbe, E.C.; Williamson, D.; Lindsey, J.C.; Hamilton, D.; Ryan, S.L.; Megahed, H.; Garami, M.; Hauser, P.; Dembowska-Baginska, B.; Perek, D.; et al. DNA methylation profiling of medulloblastoma allows robust subclassification and improved outcome prediction using formalin-fixed biopsies. Acta Neuropathol. 2013, 125, 359–371. [Google Scholar] [CrossRef]
- Hovestadt, V.; Jones, D.T.; Picelli, S.; Wang, W.; Kool, M.; Northcott, P.A.; Sultan, M.; Stachurski, K.; Ryzhova, M.; Warnatz, H.J.; et al. Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature 2014, 510, 537–541. [Google Scholar] [CrossRef]
- Schwalbe, E.C.; Lindsey, J.C.; Nakjang, S.; Crosier, S.; Smith, A.J.; Hicks, D.; Rafiee, G.; Hill, R.M.; Iliasova, A.; Stone, T.; et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: A cohort study. Lancet. Oncol. 2017, 18, 958–971. [Google Scholar] [CrossRef]
- Northcott, P.A.; Nakahara, Y.; Wu, X.; Feuk, L.; Ellison, D.W.; Croul, S.; Mack, S.; Kongkham, P.N.; Peacock, J.; Dubuc, A.; et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat. Genet. 2009, 41, 465–472. [Google Scholar] [CrossRef]
- Parsons, D.W.; Li, M.; Zhang, X.; Jones, S.; Leary, R.J.; Lin, J.C.; Boca, S.M.; Carter, H.; Samayoa, J.; Bettegowda, C.; et al. The genetic landscape of the childhood cancer medulloblastoma. Science 2011, 331, 435–439. [Google Scholar] [CrossRef]
- Northcott, P.A.; Jones, D.T.; Kool, M.; Robinson, G.W.; Gilbertson, R.J.; Cho, Y.J.; Pomeroy, S.L.; Korshunov, A.; Lichter, P.; Taylor, M.D.; et al. Medulloblastomics: The end of the beginning. Nat. Rev. Cancer 2012, 12, 818–834. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Villa, R.; Trojer, P.; Norman, J.; Yan, K.P.; Reinberg, D.; Di Croce, L.; Shiekhattar, R. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science 2007, 318, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.; Parker, M.; Kranenburg, T.A.; Lu, C.; Chen, X.; Ding, L.; Phoenix, T.N.; Hedlund, E.; Wei, L.; Zhu, X.; et al. Novel mutations target distinct subgroups of medulloblastoma. Nature 2012, 488, 43–48. [Google Scholar] [CrossRef]
- Jones, D.T.; Jager, N.; Kool, M.; Zichner, T.; Hutter, B.; Sultan, M.; Cho, Y.J.; Pugh, T.J.; Hovestadt, V.; Stutz, A.M.; et al. Dissecting the genomic complexity underlying medulloblastoma. Nature 2012, 488, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Pugh, T.J.; Weeraratne, S.D.; Archer, T.C.; Pomeranz Krummel, D.A.; Auclair, D.; Bochicchio, J.; Carneiro, M.O.; Carter, S.L.; Cibulskis, K.; Erlich, R.L.; et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 2012, 488, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Wu, J. Epigenetic regulation in medulloblastoma. Mol. Cell Neurosci. 2018, 87, 65–76. [Google Scholar] [CrossRef]
- Lin, C.Y.; Erkek, S.; Tong, Y.; Yin, L.; Federation, A.J.; Zapatka, M.; Haldipur, P.; Kawauchi, D.; Risch, T.; Warnatz, H.J.; et al. Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature 2016, 530, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef]
- Bandopadhayay, P.; Bergthold, G.; Nguyen, B.; Schubert, S.; Gholamin, S.; Tang, Y.; Bolin, S.; Schumacher, S.E.; Zeid, R.; Masoud, S.; et al. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin. Cancer Res. 2014, 20, 912–925. [Google Scholar] [CrossRef]
- Kadoch, C.; Williams, R.T.; Calarco, J.P.; Miller, E.L.; Weber, C.M.; Braun, S.M.; Pulice, J.L.; Chory, E.J.; Crabtree, G.R. Dynamics of BAF-Polycomb complex opposition on heterochromatin in normal and oncogenic states. Nat. Genet. 2017, 49, 213–222. [Google Scholar] [CrossRef]
- Stanton, B.Z.; Hodges, C.; Calarco, J.P.; Braun, S.M.; Ku, W.L.; Kadoch, C.; Zhao, K.; Crabtree, G.R. Smarca4 ATPase mutations disrupt direct eviction of PRC1 from chromatin. Nat. Genet. 2017, 49, 282–288. [Google Scholar] [CrossRef]
- Ho, L.; Miller, E.L.; Ronan, J.L.; Ho, W.Q.; Jothi, R.; Crabtree, G.R. esBAF facilitates pluripotency by conditioning the genome for LIF/STAT3 signalling and by regulating polycomb function. Nat. Cell Biol. 2011, 13, 903–913. [Google Scholar] [CrossRef]
- Spielmann, M.; Lupianez, D.G.; Mundlos, S. Structural variation in the 3D genome. Nat. Rev. Genet. 2018, 19, 453–467. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Lee, C.; Zichner, T.; Stutz, A.M.; Erkek, S.; Kawauchi, D.; Shih, D.J.; Hovestadt, V.; Zapatka, M.; Sturm, D.; et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 2014, 511, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Juraschka, K.; Taylor, M.D. Medulloblastoma in the age of molecular subgroups: A review. J. Neurosurg. Pediatr. 2019, 24, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Shih, D.J.; Remke, M.; Cho, Y.J.; Kool, M.; Hawkins, C.; Eberhart, C.G.; Dubuc, A.; Guettouche, T.; Cardentey, Y.; et al. Rapid, reliable, and reproducible molecular sub-grouping of clinical medulloblastoma samples. Acta Neuropathol. 2012, 123, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Perreault, S.; Ramaswamy, V.; Achrol, A.S.; Chao, K.; Liu, T.T.; Shih, D.; Remke, M.; Schubert, S.; Bouffet, E.; Fisher, P.G.; et al. MRI surrogates for molecular subgroups of medulloblastoma. AJNR Am. J. Neuroradiol. 2014, 35, 1263–1269. [Google Scholar] [CrossRef] [PubMed]
- Wefers, A.K.; Warmuth-Metz, M.; Poschl, J.; von Bueren, A.O.; Monoranu, C.M.; Seelos, K.; Peraud, A.; Tonn, J.C.; Koch, A.; Pietsch, T.; et al. Subgroup-specific localization of human medulloblastoma based on pre-operative MRI. Acta Neuropathol. 2014, 127, 931–933. [Google Scholar] [CrossRef]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Faria, C.C.; Perreault, S.; Cho, Y.J.; Shih, D.J.; Luu, B.; Dubuc, A.M.; Northcott, P.A.; et al. Recurrence patterns across medulloblastoma subgroups: An integrated clinical and molecular analysis. Lancet. Oncol. 2013, 14, 1200–1207. [Google Scholar] [CrossRef]
- Forget, A.; Martignetti, L.; Puget, S.; Calzone, L.; Brabetz, S.; Picard, D.; Montagud, A.; Liva, S.; Sta, A.; Dingli, F.; et al. Aberrant ERBB4-SRC Signaling as a Hallmark of Group 4 Medulloblastoma Revealed by Integrative Phosphoproteomic Profiling. Cancer Cell 2018, 34, 379–395. [Google Scholar] [CrossRef]
- Archer, T.C.; Ehrenberger, T.; Mundt, F.; Gold, M.P.; Krug, K.; Mah, C.K.; Mahoney, E.L.; Daniel, C.J.; LeNail, A.; Ramamoorthy, D.; et al. Proteomics, Post-translational Modifications, and Integrative Analyses Reveal Molecular Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2018, 34, 396–410. [Google Scholar] [CrossRef]
- Quinlan, A.; Rizzolo, D. Understanding medulloblastoma. JAAPA 2017, 30, 30–36. [Google Scholar] [CrossRef]
- Gibson, P.; Tong, Y.; Robinson, G.; Thompson, M.C.; Currle, D.S.; Eden, C.; Kranenburg, T.A.; Hogg, T.; Poppleton, H.; Martin, J.; et al. Subtypes of medulloblastoma have distinct developmental origins. Nature 2010, 468, 1095–1099. [Google Scholar] [CrossRef] [PubMed]
- Oliver, T.G.; Read, T.A.; Kessler, J.D.; Mehmeti, A.; Wells, J.F.; Huynh, T.T.; Lin, S.M.; Wechsler-Reya, R.J. Loss of patched and disruption of granule cell development in a pre-neoplastic stage of medulloblastoma. Development 2005, 132, 2425–2439. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.J.; Ellis, T.; Markant, S.L.; Read, T.A.; Kessler, J.D.; Bourboulas, M.; Schuller, U.; Machold, R.; Fishell, G.; Rowitch, D.H.; et al. Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell 2008, 14, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Spill, F.; Reynolds, D.S.; Kamm, R.D.; Zaman, M.H. Impact of the physical microenvironment on tumor progression and metastasis. Curr. Opin. Biotechnol. 2016, 40, 41–48. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef]
- Pham, C.D.; Flores, C.; Yang, C.; Pinheiro, E.M.; Yearley, J.H.; Sayour, E.J.; Pei, Y.; Moore, C.; McLendon, R.E.; Huang, J.; et al. Differential Immune Microenvironments and Response to Immune Checkpoint Blockade among Molecular Subtypes of Murine Medulloblastoma. Clin. Cancer Res. 2016, 22, 582–595. [Google Scholar] [CrossRef]
- Phoenix, T.N.; Patmore, D.M.; Boop, S.; Boulos, N.; Jacus, M.O.; Patel, Y.T.; Roussel, M.F.; Finkelstein, D.; Goumnerova, L.; Perreault, S.; et al. Medulloblastoma Genotype Dictates Blood Brain Barrier Phenotype. Cancer Cell 2016, 29, 508–522. [Google Scholar] [CrossRef]
- Bockmayr, M.; Mohme, M.; Klauschen, F.; Winkler, B.; Budczies, J.; Rutkowski, S.; Schuller, U. Subgroup-specific immune and stromal microenvironment in medulloblastoma. Oncoimmunology 2018, 7, e1462430. [Google Scholar] [CrossRef]
- Margol, A.S.; Robison, N.J.; Gnanachandran, J.; Hung, L.T.; Kennedy, R.J.; Vali, M.; Dhall, G.; Finlay, J.L.; Erdreich-Epstein, A.; Krieger, M.D.; et al. Tumor-associated macrophages in SHH subgroup of medulloblastomas. Clin. Cancer Res. 2015, 21, 1457–1465. [Google Scholar] [CrossRef]
- Liebner, S.; Corada, M.; Bangsow, T.; Babbage, J.; Taddei, A.; Czupalla, C.J.; Reis, M.; Felici, A.; Wolburg, H.; Fruttiger, M.; et al. Wnt/beta-catenin signaling controls development of the blood-brain barrier. J. Cell Biol. 2008, 183, 409–417. [Google Scholar] [CrossRef]
- Wang, S.S.; Bandopadhayay, P.; Jenkins, M.R. Towards Immunotherapy for Pediatric Brain Tumors. Trends Immunol. 2019, 40, 748–761. [Google Scholar] [CrossRef]
- Iv, M.; Zhou, M.; Shpanskaya, K.; Perreault, S.; Wang, Z.; Tranvinh, E.; Lanzman, B.; Vajapeyam, S.; Vitanza, N.A.; Fisher, P.G.; et al. MR Imaging-Based Radiomic Signatures of Distinct Molecular Subgroups of Medulloblastoma. AJNR Am. J. Neuroradiol. 2019, 40, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Shih, D.J.; Northcott, P.A.; Remke, M.; Korshunov, A.; Ramaswamy, V.; Kool, M.; Luu, B.; Yao, Y.; Wang, X.; Dubuc, A.M.; et al. Cytogenetic prognostication within medulloblastoma subgroups. J. Clin. Oncol. 2014, 32, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Lastowska, M.; Trubicka, J.; Niemira, M.; Paczkowska-Abdulsalam, M.; Karkucinska-Wieckowska, A.; Kaleta, M.; Drogosiewicz, M.; Perek-Polnik, M.; Kretowski, A.; Cukrowska, B.; et al. Medulloblastoma with transitional features between Group 3 and Group 4 is associated with good prognosis. J. Neurooncol. 2018, 138, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Packer, R.J.; Vezina, G. Management of and prognosis with medulloblastoma: Therapy at a crossroads. Arch. Neurol. 2008, 65, 1419–1424. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.M.; Hielscher, T.; Bouffet, E.; Remke, M.; Luu, B.; Gururangan, S.; McLendon, R.E.; Bigner, D.D.; Lipp, E.S.; Perreault, S.; et al. Prognostic value of medulloblastoma extent of resection after accounting for molecular subgroup: A retrospective integrated clinical and molecular analysis. Lancet. Oncol. 2016, 17, 484–495. [Google Scholar] [CrossRef]
- Packer, R.J.; Goldwein, J.; Nicholson, H.S.; Vezina, L.G.; Allen, J.C.; Ris, M.D.; Muraszko, K.; Rorke, L.B.; Wara, W.M.; Cohen, B.H.; et al. Treatment of children with medulloblastomas with reduced-dose craniospinal radiation therapy and adjuvant chemotherapy: A Children’s Cancer Group Study. J. Clin. Oncol. 1999, 17, 2127–2136. [Google Scholar] [CrossRef]
- Merchant, T.E.; Kun, L.E.; Krasin, M.J.; Wallace, D.; Chintagumpala, M.M.; Woo, S.Y.; Ashley, D.M.; Sexton, M.; Kellie, S.J.; Ahern, V.; et al. Multi-institution prospective trial of reduced-dose craniospinal irradiation (23.4 Gy) followed by conformal posterior fossa (36 Gy) and primary site irradiation (55.8 Gy) and dose-intensive chemotherapy for average-risk medulloblastoma. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 782–787. [Google Scholar] [CrossRef]
- Wahba, H.A.; Abu-Hegazy, M.; Wasel, Y.; Ismail, E.I.; Zidan, A.S. Adjuvant chemotherapy after reduced craniospinal irradiation dose in children with average-risk medulloblastoma: A 5-year follow-up study. J. Buon 2013, 18, 425–429. [Google Scholar]
- Gajjar, A.; Chintagumpala, M.; Ashley, D.; Kellie, S.; Kun, L.E.; Merchant, T.E.; Woo, S.; Wheeler, G.; Ahern, V.; Krasin, M.J.; et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): Long-term results from a prospective, multicentre trial. Lancet. Oncol. 2006, 7, 813–820. [Google Scholar] [CrossRef]
- Thomas, A.; Noel, G. Medulloblastoma: Optimizing care with a multidisciplinary approach. J. Multidiscip. Healthc. 2019, 12, 335–347. [Google Scholar] [CrossRef] [PubMed]
- De Braganca, K.C.; Packer, R.J. Treatment Options for Medulloblastoma and CNS Primitive Neuroectodermal Tumor (PNET). Curr. Treat. Options Neurol. 2013, 15, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Remke, M.; Ramaswamy, V. Infant medulloblastoma - learning new lessons from old strata. Nat. Rev. Clin. Oncol. 2018, 15, 659–660. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.W.; Orr, B.A.; Wu, G.; Gururangan, S.; Lin, T.; Qaddoumi, I.; Packer, R.J.; Goldman, S.; Prados, M.D.; Desjardins, A.; et al. Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog-Subgroup Medulloblastoma: Results From Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J. Clin. Oncol. 2015, 33, 2646–2654. [Google Scholar] [CrossRef]
- Gajjar, A.; Stewart, C.F.; Ellison, D.W.; Kaste, S.; Kun, L.E.; Packer, R.J.; Goldman, S.; Chintagumpala, M.; Wallace, D.; Takebe, N.; et al. Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: A pediatric brain tumor consortium study. Clin. Cancer Res. 2013, 19, 6305–6312. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Jones, D.T.; Jager, N.; Northcott, P.A.; Pugh, T.J.; Hovestadt, V.; Piro, R.M.; Esparza, L.A.; Markant, S.L.; Remke, M.; et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell 2014, 25, 393–405. [Google Scholar] [CrossRef]
- Rudin, C.M.; Hann, C.L.; Laterra, J.; Yauch, R.L.; Callahan, C.A.; Fu, L.; Holcomb, T.; Stinson, J.; Gould, S.E.; Coleman, B.; et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N. Engl. J. Med. 2009, 361, 1173–1178. [Google Scholar] [CrossRef]
- Ramaswamy, V.; Taylor, M.D. Medulloblastoma: From Myth to Molecular. J. Clin. Oncol. 2017, 35, 2355–2363. [Google Scholar] [CrossRef]
- Robinson, G.W.; Kaste, S.C.; Chemaitilly, W.; Bowers, D.C.; Laughton, S.; Smith, A.; Gottardo, N.G.; Partap, S.; Bendel, A.; Wright, K.D.; et al. Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget 2017, 8, 69295–69302. [Google Scholar] [CrossRef]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target Ther. 2018, 3, 5. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Recurrent Gene Amplification [7,10,20,21] | Recurrent SNVs [7,10,20,21] | Gain of Chromo-Some [7,10,21] | Loss of Chromo-Some [7,10,21] | Other Recurrent Genetic Events [7,10,21,22] | Signature Transcriptional Markers [23] | Signature Methylation Markers [24] | Cell of Origin [17,18] | |
|---|---|---|---|---|---|---|---|---|
| WNT | NA | CTNNB1, DDX3X, SMARCA4, TP53 | NA | 6 | NA | WIF1, TNC, GAD1, DKK2, EMX2 | LHX6 (cg25542041) USP40 (cg12925355) KIAA1549 (cg01268345) | Progenitors in LRL and dorsal brainstem |
| SHH | MYCN, GLI1 or GLI2 | PTCH1, TERT, SUFU, SMO, TP53 | 3q, 9p | 9q, 10q, 17p | NA | PDLIM3, EYA1, HHIP, ATOH1, SFRP1 | lncRNA2178 (cg02227036) CHTF18 (cg10333416) KIAA1549 (cg01268345) | Granule neurons (infant); GNPs and UBCs (adult) |
| Group 3 | MYC, MYCN, OTX2 | SMARCA4, KBTBD4, CTDNEP1, KMT2D | 1q, 7, 18 | 8, 10q, 11, 16q | Isochromosome 17q; GFI1 and GFI1B enhancer hijacking | IMPG2, GABRA5, EGFL11, NRL, MAB21L2, NPR3 | RPTOR (cg09929238 and cg08129331) RIMS2 (cg12565585) VPS37B (cg13548946) Intergenic region in chromosome 12 (cg05679609) | Nestin positive stem cells |
| Group 4 | SNCAIP, MYCN, OTX2, CDK6 | KDM6A, ZMYM3, KTM2C, KBTBD4 | 7, 18q | 8, 11p, X | Isochromosome 17q; PRDM6, GFI1, and GFI1B enhancer hijacking | KCNA1, EOMES, KHDRBS2, RBM24, UNC5D, OAS1 | USP40 (cg12925355) AKAP6 (cg18849583) lncRNA2178 (cg02227036) | UBCs and GluCNs in URL |
| WNT | SHH | Group 3 | Group 4 | Intermediate 3/4 Group | |
|---|---|---|---|---|---|
| Low Risk (>90% survival) | <16 years (age) | Chromosome 13 loss without neither MYC amplification nor metastasis | Non-metastatic, and whole chromosome 11 loss or whole chromosome 17 gain | All | |
| Average (standard) (75–90% survival) | TP53 wildtype without metastasis and MYCN amplification | Neither metastasis nor MYC amplification | Neither metastasis nor chromosome 11 loss | ||
| High Risk (50–75% survival) | Metastatic, and/or MYCN-amplified | Metastatic | |||
| Very High Risk (<50% survival) | Adult with TP53 mutation | Metastatic or MYC amplification |
| Conditions | Interventions | ClinicalTrials.gov Identifier | Status |
|---|---|---|---|
| WNT | Surgery + Reduced-Dose Radiotherapy + Reduced-Dose Chemotherapy | NCT02066220 NCT01878617 NCT02724579 | Recruiting |
| WNT | Surgery + Chemotherapy, No Radiotherapy | NCT02212574 | Suspended |
| Targeting SHH pathway | Vismodegib (SMO Inhibitor) | NCT00939484 NCT01239316 | Completed |
| Targeting SHH pathway | Vismodegib in combination with Temozolomide | NCT01601184 | Terminated |
| Targeting SHH pathway | Sonidegib (SMO Inhibitor) | NCT01708174 | Completed |
| Targeting SHH pathway | CX-4945 (CK2 Inhibitor) | NCT03904862 | Recruiting |
| Intensified Treatment of Group 3/Group 4 MB | Pemetrexed and Gemcitabine | NCT01878617 | Recruiting |
| MYC-driven Group 3 MB | BMS-986158(Bromodomain (BRD) and Extra-Terminal Domain (BET) Inhibitor | NCT03936465 | Recruiting |
| Group 3 MB | PD-0332991/Palbociclib (CDK 4-6 Inhibitor) | NCT02255461 | Terminated |
| Refractory or Recurrent Group 3/Group 4 MB | Prexasertib (CHK1/2 Inhibitor) and Gemcitabine | NCT04023669 | Recruiting |
| Refractory or Recurrent SHH, Group 3/Group 4 MB | Prexasertib (CHK1/2 Inhibitor) and Cyclophosphamide | NCT04023669 | Recruiting |
| Recurrent MB | Fimepinostat (HDAC and PI3K inhibitor) | NCT03893487 | Recruiting |
| Refractory or Recurrent SHH MB | Ribociclib and Sonidegib | SJDAWN | Recruiting |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zou, H.; Poore, B.; Broniscer, A.; Pollack, I.F.; Hu, B. Molecular Heterogeneity and Cellular Diversity: Implications for Precision Treatment in Medulloblastoma. Cancers 2020, 12, 643. https://doi.org/10.3390/cancers12030643
Zou H, Poore B, Broniscer A, Pollack IF, Hu B. Molecular Heterogeneity and Cellular Diversity: Implications for Precision Treatment in Medulloblastoma. Cancers. 2020; 12(3):643. https://doi.org/10.3390/cancers12030643
Chicago/Turabian StyleZou, Han, Brad Poore, Alberto Broniscer, Ian F. Pollack, and Baoli Hu. 2020. "Molecular Heterogeneity and Cellular Diversity: Implications for Precision Treatment in Medulloblastoma" Cancers 12, no. 3: 643. https://doi.org/10.3390/cancers12030643
APA StyleZou, H., Poore, B., Broniscer, A., Pollack, I. F., & Hu, B. (2020). Molecular Heterogeneity and Cellular Diversity: Implications for Precision Treatment in Medulloblastoma. Cancers, 12(3), 643. https://doi.org/10.3390/cancers12030643